
The National Bioethics Advisory Commission (NBAC) was established by Executive Order 12975, signed by President Clinton on October 3, 1995. NBAC’s functions are defined as follows:
a) NBAC shall provide advice and make recommendations to the National Science and Technology Council and to other appropriate government entities regarding the following matters:
1) the appropriateness of departmental, agency, or other governmental programs, policies, assignments, missions, guidelines, and regulations as they relate to bioethical issues arising from research on human biology and behavior; and 2) applications, including the clinical applications, of that research.
b) NBAC shall identify broad principles to govern the ethical conduct of research, citing specific projects only as illustrations for such principles.
c) NBAC shall not be responsible for the review and approval of specific projects.
d) In addition to responding to requests for advice and recommendations from the National Science and Technology Council, NBAC also may accept suggestions of issues for consideration from both the Congress and the public. NBAC also may identify other bioethical issues for the purpose of providing advice and recommendations, subject to the approval of the National Science and Technology Council.
National Bioethics Advisory Commission
6705 Rockledge Drive, Suite 700, Bethesda, Maryland 20892-7979 Telephone: 301-402-4242 • Fax: 301-480-6900 • Website: www.bioethics.gov
ISBN 1-931022-17-8

Ethical and Policy Issues in Research Involving Human Participants
Volume II
Commissioned Papers and Staff Analysis
Bethesda, Maryland August 2001
National Bioethics Advisory Commission
Harold T. Shapiro, Ph.D., Chair
President Emeritus and Professor of Economics and Public Affairs The Woodrow Wilson School of Public and International Affairs Princeton University Princeton, New Jersey
Patricia Backlar
Research Associate Professor of Bioethics Department of Philosophy Portland State University Assistant Director Center for Ethics in Health Care Oregon Health Sciences University Portland, Oregon
Arturo Brito, M.D.
Assistant Professor of Clinical Pediatrics University of Miami School of Medicine Miami, Florida
Alexander Morgan Capron, LL.B.
Henry W. Bruce Professor of Law
University Professor of Law and Medicine
Co-Director, Pacific Center for Health Policy and Ethics University of Southern California Los Angeles, California
Eric J. Cassell, M.D., M.A.C.P.
Clinical Professor of Public Health
Weill Medical College of Cornell University New York, New York
R. Alta Charo, J.D.
Professor of Law and Bioethics Law School and Medical School University of Wisconsin Madison, Wisconsin
James F. Childress, Ph.D.
Kyle Professor of Religious Studies Professor of Medical Education Director, Institute for Practical Ethics Department of Religious Studies University of Virginia Charlottesville, Virginia
David R. Cox, M.D., Ph.D.
Scientific Director Perlegen Sciences Santa Clara, California
Rhetaugh Graves Dumas, Ph.D., R.N.
Vice Provost Emerita, Dean Emerita, and Lucille Cole Professor of Nursing University of Michigan Ann Arbor, Michigan
Laurie M. Flynn*
Senior Research and Policy Associate
Department of Child and Adolescent Psychiatry Columbia University New York, New York
Carol W. Greider, Ph.D.
Professor of Molecular Biology and Genetics Department of Molecular Biology and Genetics Johns Hopkins University School of Medicine Baltimore, Maryland
Steven H. Holtzman
Chief Business Officer
Millennium Pharmaceuticals, Inc. Cambridge, Massachusetts
Bette O. Kramer
Founding President
Richmond Bioethics Consortium Richmond, Virginia
Bernard Lo, M.D.
Director
Program in Medical Ethics Professor of Medicine
University of California, San Francisco San Francisco, California
Lawrence H. Miike, M.D., J.D.
Kaneohe, Hawaii
Thomas H. Murray, Ph.D.
President
The Hastings Center Garrison, New York
William C. Oldaker, LL.B.
Senior Partner
Oldaker & Harris, L.L.P. Washington, D.C.
Co-Founder and General Counsel NeuralStem Biopharmaceuticals Ltd. College Park, Maryland
Diane Scott-Jones, Ph.D.
Professor
Psychology Department Boston College Chestnut Hill, Massachusetts
*Resigned on May 10, 2001.
CONTENTS
Research Ethics in Australia A-1
Donald Chalmers
University of Tasmania
Location of the Office for Protection from Research Risks Within the National Institutes of Health: Problems of Status and Independent Authority
John C. Fletcher
University of Virginia
B-1
Privacy and Confidentiality in Health Research C-1
Janlori Goldman and Angela Choy
Georgetown University
An Examination of Issues Presented by Proposals to Unify and Expand Federal Oversight of Human Subject Research
C.K. Gunsalus
University of Illinois at Urbana-Champaign
The History, Function, and Future of Independent Institutional Review Boards
Erica Heath
Independent Review Consulting, Inc.
The Danish Research Ethics Committee System—Overview and Critical Assessment
Søren Holm
University of Manchester
D-1

E-1
F-1
Vulnerability in Research Subjects: A Bioethical Taxonomy G-1
Kenneth Kipnis
University of Hawaii at Manoa
Reflections on the Organizational Locus of the Office for Protection from Research Risks
Charles R. McCarthy
H-1
Protectionism in Research Involving Human Subjects I-1
Jonathan D. Moreno
University of Virginia
Federal Agency Survey on Policies and Procedures for the Protection of Human Subjects in Research
National Bioethics Advisory Commission
J-1
Local Institutional Review Boards K-1
Steven Peckman
University of California-Los Angeles
Institutional Review Board Assessment of Risks and Benefits Associated with Research
Ernest D. Prentice and Bruce G. Gordon
University of Nebraska Medical Center
L-1
Oversight of Human Subject Research: The Role of the States M-1
Jack Schwartz
Office of the Maryland Attorney General
v
Privacy and Confidentiality: As Related to Human Research in Social and Behavioral Science
Joan E. Sieber
California State University, Hayward
Unfulfilled Promise: How the Belmont Report Can Amend the Code of Federal Regulations Title 45 Part 46—Protection of Human Subjects
Harold Y. Vanderpool
University of Texas Medical Branch, Galveston
The Ethical Analysis of Risks and Potential Benefits in Human Subjects Research: History, Theory, and Implications for U.S. Regulation
Charles Weijer
Dalhousie University
Charles Weijer of Dalhousie University, Halifax, Nova Scotia, Canada, prepared a paper for NBAC on the topic of protecting communities in research. That paper was published in 1999 in the journal Cambridge Quarterly of Healthcare Ethics. The reader can find the article at the following citation:
Weijer C. 1999. Protecting Communities in Research: Philosophical and Pragmatic Challenges. Cambridge Quarterly of Healthcare Ethics 8:501–513.
The papers included in this volume were edited to conform to minimal stylistic consistency. The content and accuracy of the papers are the responsibility of the authors, not the National Bioethics Advisory Commission.
N-1
O-1
P-1

vi
RESEARCH ETHICS IN AUSTRALIA
Commissioned Paper Donald Chalmers University of Tasmania

A-1
Preface
Australia has had a comparatively creditable record of ethical research involving humans. The litany of criticism about shoddy medical research documented in the epochal article by Professor Beecher (Beecher 1966, 1968; Levine 1986) has not occurred in this country. Comparatively fine as the Australian record may be, that record is not unblemished. A report commissioned by the Commonwealth Government in 1994 by Professor Margaret Allars into unsatisfactory aspects of the collection, manufacture, and injection of human growth hormone (Allars 1994) recommended that aspects of the research structure had to be reassessed. In particular, the Allars Report recommended a review of the National Health and Medical Research Council (NHMRC) Statement on Human Experimentation and the Supplementary Note on Reproductive Technology Procedures. Similarly, the Commonwealth Minister for Health (now called the Commonwealth Minister for Health and Aged Care) referred ethical concerns about two postwar procedures and one multi-center clinical trial in the 1990s to the Australian Health Ethics Committee (AHEC). The two postwar procedures involved first, the inclusion of orphans and State wards in vaccine trials conducted in the postwar years and, second the experimental use of estrogen to reduce the height of “tall girls” in the 1950s. The multicenter trial involved the so-called “morning after pill” (RU486).
Research and experimentation has been a major issue, at least for the research community, in the last two decades in Australia. This “age of skepticism” (pace Eric Hobsbawn) has seen continuing demands for open government and greater public accountability, demands for expanded civil liberties, and demands for privacy protection rights. This wide debate has translated into debate about the protection of subjects in medical research (Laufer 1990; Darvall 1993), its major focus being the maintenance and improvement of ethical standards. This focus of concern is reflected in much of the work of the peak national health ethics body, the AHEC. In particular, the AHEC has conducted two series of National Workshops for Institutional Ethics Committees, a major review of the ethics review system in Australia (Chalmers 1996), and a major revision of the guidelines on research ethics published as the National Statement on Ethical Conduct in Research Involving Humans in mid 1999 (National Statement 1999). Ethical standards in human research and experimentation have not been static. The Australian research ethics community conducted a debate on improving and professionalizing the ethics review system during the late 1980s and 1990s. Researchers, institutions, trial sponsors, academic and professional critics, and changing attitudes to accountability have all contributed to an improvement in the practices and culture of research involving humans in this country.
The AHEC has come far since the Finn Report amalgamated the National Bioethics Consultative Committee (NBCC) and the Medical Research Advisory Committee to form the AHEC. Professor Finn stated in his report that “until the HEC (AHEC) concept is more fully developed and particularized, until the Council addresses more directly the burden of the ethics function...one cannot surmise with any confidence as to the extent to which those differences between the two bodies in their areas of mutual interest are likely to recede or be perpetuated” (Finn 1990 at 14). Considerable advances were made in the first three triennia toward this “evolutionary” change.
The Australian research ethics review system continues to evolve. The system could be described as a hybrid or intermediate system in contradistinction to entirely legislatively regulated systems or voluntary self-regulated models. There is no Australian equivalent of the National Research Act 1974. However, there is greater regulation of the system since the pre-1982 Australian voluntary system. Human Research Ethics Committees (HRECs), which conduct ethics review are not established by specific Commonwealth legislation, but they are recognized within the NHMRC Act 1992. In this major respect, research ethics review in Australia is not a voluntary system; it is better classified now as a regulated system.
A-3
Comparisons between HRECs in Australia and Ethics Committees in the United States are misleading. Some HRECs in Australia may perform some of the functions of Ethics Committees, but the comparable institution in the United States is an Institutional Review Board (IRB). As well as the infamous Tuskegee Study (Furrow et al 1995 at 548–550), a number of questionable human experiments were disclosed before the U.S. Congress in the early 1970s. Disclosures were made particularly about dubious research conducted in prisons and mental hospitals and on human fetuses. Following these events, the National Research Act 1974 was introduced which required each institution conducting federally supported research involving human subjects to establish an IRB. These IRBs are required to review the ethical aspects of all research protocols within the institution. The general standards for the composition, operation, and responsibility of IRBs are contained in federal regulations (Code of Federal Regulations 1992).
In order to fulfill the requirements of the federal regulations, each IRB is required to follow written procedures for the conduct of initial and continuing review of research and for reporting findings and actions to the investigator and the institution. An IRB determines which projects require review more often than annually and which projects need verification from sources other than the investigator. Changes in approved research may not be initiated without IRB review and approval, except where there are apparent immediate hazards to the human subjects. In addition to reporting to the IRB, there are other safeguards in the system. Both institutional officials and the Food and Drug Administration (FDA) must be told of any unanticipated problems involving risks to human subjects or others. Similarly, any instance of serious or continuing noncompliance with federal regulations or the decisions of the IRB (or any suspension or termination of IRB approval) must be reported to the institution or FDA. There are IRB procedural requirements aimed at ensuring proper consideration of the research. Except when an expedited review procedure is used, a research proposal must be reviewed by a majority of the members of the IRB. On review, at least one of the IRB members must be primarily concerned with nonscientific areas, and the proposal must receive the approval of a majority of those members present at the meeting.
American Ethics Committees continue to evolve and are not settled in their functions (Annas 1984; In Re Quinlan 1976; President’s Commission 1983). Ethics Committees in the USA include the following roles:
In effect, American Ethics Committees are patient care committees and are often referred to by this title. Some Australian hospital HRECs may perform some of the same functions as American Ethics Committees.
Comparisons are also sometimes made with Research Ethics Committees in the United Kingdom, but, again, their functions do not compare precisely with those of Australian HRECs. The United Kingdom Research Ethics Committees are diverse in their functions and do not directly relate to Australian HRECs in that they operate within the National Health Service. A United Kingdom Department of Health circular of 1989 (HSC (IS) 153) requires that each district health authority appoint a “...properly constituted Local Research Ethics Committee (LREC), which meets regularly, to register, review and approve (or not approve) the research conducted by its staff, or using its premises or facilities, including access to personal health information held by the authority (and research undertaken by general practitioners within its boundaries).” Research Ethics Committees in the United Kingdom are locally established and formally constituted as subcommittees within the health authority system. It has been noted that an “Ethics Committee acts for and on behalf of the Authority” (Brazier 1990).
A-4
The growth of ethics committees has followed diverse paths, and a number of other ethics committees have been established beyond the terms of the Department of Health Circular Guidelines (Rawbone 2000). Brazier particularly notes that a number of fertility units have established advisory committees to assist practitioners in making decisions about the admission of individual patients to the program (Brazier 1990).
This report presents background information on the ethics review system in this country, defines the current ethical system, and provides some background information on the new National Statement on Ethical Conduct in Research Involving Humans. This paper considers the current operation of the AHEC and the system of ethical review of research involving humans by HRECs in Australia. The paper also addresses some specific questions posed by the National Bioethics Advisory Commission (NBAC), namely the following:
| 1. |
What are the strengths and weaknesses of nonregulatory systems of protection? |
| 2. |
What features of these systems, if any, should be incorporated in the U.S. system? |
| 3. |
What are the strengths and weaknesses of models that are comprehensive, those that encompass private and government sectors, and nonbiomedical and biomedical research? 1. Introduction |
1.1 Three Tiers: Researcher Ethics Committee and National Body
A three-tier system of ethics review operates within Australia:
At the first level, the researcher continues to carry ethical responsibilities toward research participants. The National Statement begins with a reference to the researcher and states that the “…guiding value for researchers is integrity…” (National Statement 1999, Principle 1.1 at 11). The National Statement continues that “the guiding ethical principle for researchers is respect for persons…” (Principle 1.2) and that “… the ethical principle of beneficence is expressed in researchers’ responsibility to minimize risks of harm or discomfort to participants in research projects” (Principle 1.3). Researchers are also required to design their protocols to ensure respect for the dignity and well-being of the participants (Principle 1.4). Researchers should not discriminate in the distribution of benefits and burdens of participation in research or in the selection of research participants (Principle 1.5). Researchers have great responsibility in ensuring participant consent is obtained (Principles 1.7–1.12). Researchers must conduct research that has merit and balance the risks and likely benefits to be gained. Only people with the required experience, qualifications, and competence should conduct the research (Principles 1.13–1.15). These General Principles are bolstered throughout the National Statement with specific contextual duties of researchers to research participants in relation to the project. For example, in a clinical trial the researcher must declare any conflicts of interest through involvement in business or other similar association (Principle 12.5 at 36). It was a deliberate policy in drafting the National Statement to recognize and reinforce the ethical responsibilities of researchers.
HRECs, which, until 1999 were referred to as Institutional Ethics Committees (IECs), conduct the second level of ethical review. The Australia HRECs compare closely with the U.S. IRBs established under federal regulations. Some HRECs were already operating before the system was formally established in 1982 by amendments to the Statement on Human Experimentation. The NHMRC issued the Statement on Human Experimentation, which was the predecessor to the current National Statement on Ethical Conduct in Research Involving Humans,
A-5
promulgated in 1999. The NHMRC was a nonstatutory body until 1992. In that year the NHMRC became a statutory authority when the Commonwealth Parliament passed the National Health and Medical Research Council Act, 1992 (Cth.). Although HRECs are not statutory bodies, institutions cannot receive research funding from public bodies unless consideration had been given to the research proposal by a properly constituted HREC. Originally, HRECs only considered medical and health research projects. Later, the Australian Research Council (ARC) (the major funding agency for nonmedical research) introduced a similar requirement that, in effect, expanded the jurisdiction of HRECs to all research involving humans.
The third level in the system is the AHEC. This body is established under § 35 and § 36 of the National Health and Medical Research Council Act 1992 (Cth.). The AHEC is required to oversee the operation of the HREC system and receives annual Compliance Reports from every registered HREC (National Statement 1999 Principles 2.46–2.48). In addition, the AHEC has the sole authority to publish medical research guidelines. In so doing, the AHEC is required to follow § 11–14 of the National Health and Medical Research Council Act 1992, which provides a unique procedure of two stages of public consultation before such guidelines may be issued.
1.2 The National Statement: Changes in the Research Environment
The National Statement reflects a number of significant changes in the ethics of human research. First, the National Statement includes a wider and more comprehensive view about research involving humans, going beyond medical experimentation and extending to all research involving humans. The first Australian guidelines in relation to research, the Statement on Human Experimentation, followed the Declaration of Helsinki and applied ethical standards to medical research involving human subjects. Gradually, the Statement on Human Experimentation was applied not only to medical research but other research involving humans particularly in the social and behavioral sciences. The new National Statement recognizes this evolution. Second, the National Statement recognizes the evolution of community and research community acceptance that now “…all kinds of research involving or impacting upon humans should conform to the highest standards academic integrity and ethical practice” (National Statement 1999 at 2).
Third, legislation is now more common place in the once self-regulated area of research ethics. Increasingly, Commonwealth and State legislation is impacting on and becoming more relevant to any consideration of research ethics. The regulation of Australian research is no longer a voluntary regulatory system of protection for research participants. Many Commonwealth and State Acts apply directly or indirectly to research. In particular, the NHMRC was brought under a statutory framework with the enactment of the National Health and Medical Research Council Act by the Commonwealth Parliament in 1992. Fourth, in a number of countries there have been efforts to identify a better definitional understanding of what is meant by research. The National Statement notes that:
There are many definitions of research. These include a systematic investigation to establish facts, principles or knowledge a study of some matter with the objective of obtaining and confirming knowledge. A defining feature of research is the validity of its results….
An alternative approach to finding a definition of research is to list examples for what constitutes research, such as:
or
| n |
the administration and analysis of data in response to surveys or questionnaires, interviews or opinion polling” (National Statement 1999 at 6). |
A-6
It is accepted that it is difficult to find an agreed-upon definition of research. The National Statement accepts that problems may arise from “…including activity that would not normally be included, like quality assurance activities or audits and excluding activity that probably should be included, such as research conducted as part of a course of education…[and]…omitting newly emerged genres of research, of which various kinds of multi-disciplinary research are examples” (National Statement 1999 at 6). The definitional problem of research has been considered seriously in Australia. The issue of the appropriate boundary between research and innovative therapy in practice arose in the inquiry conducted by Professor Margaret Allars in relation to innovative hormone treatment (Allars 1994; Giesen 1995).
Fifth, debates about the protection of subjects in research have expanded from concerns about physical protection to modern concerns about personal information privacy. Public concern about individual privacy is a major emerging challenge. Moves to store medical records on computer (rather than hard copy) have increased fears that privacy will be threatened. In respect of privacy, the federal Privacy Act 1988 (Cth.) was a watershed. The Privacy Act, particularly § 95 dealing with privacy in public research and the Information Privacy Principles (NHMRC 2000) has had a significant impact on public health (Cth.). The Privacy Commissioner has also extended the protections available to individuals in relation to their personal information held in the public sector under the Privacy Act 1988 (Cth.) to the private sector with amendments to this Act.
Sixth, peer review and declining funding to research generally and medical research in particular cannot be discounted as an influence on changing research culture. It is far more difficult to obtain research funding. For example, the NHMRC funds only 20 percent approximately of research applications. Finally, moves to encourage private industry to contribute more funds to national research efforts, particularly in the area of genetics, has introduced increasing commercial considerations into the research environment.
All of these developments are leading to a more regulatory environment in Australia but still without specific legislation for the HRECs. Legislation, in the form of the National Health and Medical Research Council Act 1992 (Cth.), establishes a national supervisory committee (the AHEC) and recognizes the HREC system. All public research-funding bodies require ethics approval before research can be undertaken. The Commonwealth statutory authority, the Therapeutic Goods Administration (TGA), regulates clinical trials of drugs and devices in the same fashion as the FDA in the United States. Finally, although private institutions and organizations are not obliged to follow NHMRC guidelines, there is a high degree of voluntary compliance on the part of private research organizations.
2. A Brief Background to the Development of Ethical Review in Australia
A brief background is presented of the developments leading to the current system of ethical review in Australia. The primary purpose for the introduction of both codes of research practice and committees to review research has been and remains the protection of the welfare and rights of participants in research. It is axiomatic that the foundation of any system of ethical protection for the welfare and rights of participants depends on the integrity of the researchers themselves. The new Australian National Statement recognizes the centrality of the researcher as the first level of review. The National Statement states that:
| 1. |
The guiding value for researchers is integrity, which is expressed in a commitment to the search for knowledge, to recognize principles of research conduct and the honest and ethical conduct of research and dissemination and communication of results. |
| 2. |
When conducting research involving humans, the guiding ethical principle for researchers is respect for persons which is expressed as regard for the welfare, rights, beliefs, protections, customs and cultural heritage both individual and collective, or persons involved in research. |
A-7
| 3. |
In research involving humans, the ethical principle of beneficence is expressed in researchers’ responsibility to minimize risks of harm or discomfort to participants in research projects (National Statement 1999 at 11). |
Ethics review committees conduct the second level of review. These were gradually introduced during the 1970s and formally so in the 1980s. HRECs grant ethical approval to researchers for their research and, in so doing, aim to protect the welfare and rights of research participants. However, they are not funded to or capable of acting as a policing agency for the work of researchers (Chalmers and Pettit 1998). Finally, in the early 1990s Australia introduced a third level, with the establishment of a national bioethics committee, the AHEC.
2.1 Toward National Ethical Standards in Research: The First Period—1973–1982
Until 1965, the prime responsibility for ethical standards in human experimentation rested with the integrity of the individual researcher subject to the oversight of that researcher’s institution and colleagues. Australia ratified the Declaration of Helsinki in 1965. This was an important symbolic act that was later realized by the introduction of committees to review the ethical aspects of research experiments on humans. During the same decade, there was awareness of the concerns for ethical standards in the United States, but it is not clear how far this awareness influenced developments toward the establishment of ethics committees to review research (Editorial 1976). Some institutions in Australia already operated ethics committees in the 1960s, and these influenced the development of the ethics review system. These early ethics committees in Australia predated American developments and may account for differences in the ways in which the Australian system has developed. Australia was essentially proactive in developing standards for ethical conduct in research rather than reactive to revelations or incidents of research impropriety.
A major response to the Declaration of Helsinki was the drafting of Australia’s first guidelines on human experimentation, which were prepared by an ad hoc committee of the Medical Research Advisory Committee and adopted by the NHMRC. This first NHMRC Statement on Human Experimentation was amended in 1973 and again in 1976. This latter amendment was important as it provided that applications to the NHMRC for research grants were required to be submitted to a medical ethics review committee for ethical approval, and that medical ethics research committees were required to be established by institutions conducting medical research and experimentation (Jonas 1969; Fletcher 1973; Gillespie 1988 at 3). Funding was therefore made conditional upon ethical approval. The intention was to ensure peer review. There was only one minimal stipulation in relation to the composition of these committees, namely that one person not connected with the institution was to be appointed.
This marked the first major step toward developing a systematic structure of ethical review by IECs, which in 1999 became known as HRECs in Australia. In an important sense this marked the end of the era of the self-regulation “closed shop.” This development was contemporaneous with demands for open government and greater public accountability, demands for expanded civil liberties, and demands for consumer rights. It was also in the mid-1970s that the public was beginning to hear reports of recombinant DNA research, genetic engineering, and the possibilities of IVF.
2.2 Toward IECs and the Medical Research Ethics Committee of the NHMRC: The Second Period—1982–1989
The next significant steps in the development of ethical review were the revisions to the NHMRC Statement on Human Experimentation in 1982 and the establishment of the Medical Research Ethics Committee (MREC) in 1983.
IECs were established formally in 1982. There were already many ethics committees in operation, particularly in the teaching hospitals before 1982. The NHMRC issued a new and substantially revised Statement on Human Experimentation that included four Supplementary Notes (these Supplementary Notes dealt in detail
A-8
with the following specific topics: IECs; research on children, the mentally ill, those in dependent relationships (including unconscious patients); clinical trials; and IVF and embryo transfer). Supplementary Note 1 provided an expanded statement of the membership and functions of IECs, which were to be composed of men and women reflecting different age groups and including a person not associated with the institution. The minimum composition was a minister of religion; a lawyer; a medical graduate with research experience; and a lay woman and a lay man (NHMRC 1993b; McNeill 1993).
In broad terms, IECs were concerned with the approval of research activities. In this respect a primary concern was ensuring effective consent on the part of subjects in research projects. The IEC reviewed copies of relevant consent forms, the research protocol, relevant past research, the selection criteria for research participants, the scientific method to be employed, the risks and benefits to subjects in the research program, and the perceived benefits of the research. The Supplementary Note established the functions of the IECs that were, in summary, to:
a) Consider ethical considerations of all proposed research projects; b) Maintain surveillance of approved research; c) Maintain a register of projects; and d) Establish and maintain communication with the MREC.
In carrying out the functions defined in Supplementary Note 1, IECs were required to
The MREC, which replaced the Medical Research Advisory Council, was established as one of the standing advisory committees to the NHMRC. It was commissioned to keep under review and make recommendations to the council on ethical principles in relation to human experimentation. In addition, the MREC was required to keep under review the work of IECs. The MREC thus created a third level of ethical consideration, and it was directly related to the systematic development of IECs in Australia.
In 1984 it was decided that the MREC should review the operation of IECs throughout Australia and, in particular, consider the performance and effectiveness of the Supplementary Note on IECs in relation to their composition and function. During 1984 and 1985 a series of workshops were held in the major State capitals dealing with the constitution and functions of IECs (NHMRC 1985). A further round of workshops was held in the late 1980s.
A-9
2.3 Toward a National Ethics Committee: The Third Period—1988 to the Present
2.3.1 MREC
The MREC of the NHMRC was a major step in the evolution toward a national ethics body. The original remit of the committee was to make recommendations to the council on ethical principles in relation to human experimentation, and this the committee did with distinction during the 1980s. For example, the MREC updated the Statement on Human Experimentation in 1982 and included notes on IECs, research in children, the mentally ill, and those in dependant relationships or comparable situations; therapeutic trials; and IVF and embryo transfer (ET). In 1983 the NHMRC produced Ethics in Medical Research Involving the Human Fetus and Human Fetal Tissue which became Supplementary Note 5 to the Statement of Human Experimentation, and, in 1985 the NHMRC produced the Report on Ethics and Epidemiological Research, which was added as a new
Supplementary Note 6.
At the same time as the revisions to the NHMRC Statement on Human Experimentation in 1982 and the establishment of the MREC, the controversial area of reproductive technology was considered by the NHMRC.
Supplementary Note 4 – In-vitro Fertilisation and Embryo Transfer, adopted by the NHMRC at its 94th session in October 1982, was the “first official, Government-approved regulatory code for the practice of in-vitro fertilisation in this country (or, for that matter, anywhere)...” (Scott 1984 at 3). This Note described IVF as a “justifiable means of treating infertility” (NHMRC 1992 at 14). The note went on to say, however, that “…much research remains to be done and the NHMRC Statement on Human Experimentation and Supplementary Notes should continue to apply to all work in this field.” Accordingly, any institution offering IVF was required to have all aspects of its program approved by an IEC with a register being kept detailing parentage, treatment cycles, and records of success. The programs were to “normally involve” the ova and sperm of married partners (NHMRC 1992 at 14). Research remained “... inseparable from the development of safe and effective IVF and ET” and so embryonic development “...beyond the stage at which implantation would normally occur is not acceptable” (NHMRC: 1992 at 15). Finally, with some prescience, cloning experiments were declared ethically unacceptable (NHMRC 1984).
2.3.2 The Short-Lived National Bioethics Committee
An avalanche of Australian government reports followed this NHMRC Supplementary Note on IVF and embryo transfer (Waller 1982–1984; Demack 1984; Chalmers 1985; Cornwall 1984; Michael 1986; NSW Law Reform Commission 1980–1989; Family Law Council 1985; Senate Select Committee 1986). Reports on artificial conception from some States recommended State regulatory bodies; other States recommended that voluntary adherence to NHMRC guidelines was adequate without the need to introduce further regulatory schemes.
There were essentially inconsistent recommendations in relation to regulation of embryo experimentation. Then the Commonwealth Senate set up a Select Committee that presented a report on Human Embryo Experimentation in Australia in 1985 (Senate Select Committee 1986). The report made recommendations on the regulation of embryo experimentation. The committee recommended that voluntary adherence to nationally promulgated guidelines monitored by IECs was not adequate (Senate Select Committee 1986, Chapter 4, para. 4.17). Instead, the Select Committee envisaged a national body, issuing research protocols and research licenses that should be required before experimentation of any kind was undertaken on human embryos. The license was to be for a limited time and subject to conditions (Senate Select Committee 1986, Chapter 4, para. 4.25). The committee recommended that a Commonwealth Statute, preferably in company with the States and the Northern Territory, should set down a broad declaration of the principle banning nontherapeutic embryo experimentation that frustrated the development of the embryo and should establish a licensing scheme.
Importantly, in relation to the development of a national ethics committee, the report recommended the national body be controllable through administrative proceedings, where licenses may be issued outside its
A-10
powers or where the body acted in any way outside its charter. This national body would report to Parliament (Senate Select Committee 1986, Chapter 4, para 4.42), consult with the public (Senate Select Committee 1986, Chapter 4, para 4.43), and
Formulate guidelines, consider research protocols, and monitor research procedures...and initiate prosecution or injunction against those carrying out prohibited experimenting. Such a body would supersede the NHMRC with its MREC (Senate Select Committee 1986, Chapter 4, para 4.46).
The report by the Family Law Council (a statutory council set up under the Commonwealth Family Law Act 1975 to advise on the development of federal family law) also recommended establishing a National Body (Family Law Council 1985). This report recommended a National Council on Reproductive Technology, which was to take a national approach to research and practice in reproductive technology in Australia (Family Law Council 1985, recommendations 30, 31).
Both the report of the Senate Select Committee and the Report of the Family Law Council echoed the call in 1982 by Justice Michael Kirby, who had promoted some form of institution to tackle questions of ethics and experimentation, particularly in the area of IVF:
Otherwise, it will be the judgment of history that the scientists of our generation brought forth most remarkable development of human ingenuity—but the lawyers, philosophers, theologians and law-makers proved incompetent to keep pace (Kirby 1983 at 12).
Following the publication of the Senate’s Select Committee Report, the federal government decided to establish the NBCC. In 1988 the Federal Minister for Health in conjunction with the other Australian State Health Ministers announced that, in view of rapid advances in biotechnology creating bioethical issues, a new body would be established. The NBCC was established by the Health Ministers of Australia (with approval of the States’ Attorneys-General), but it was not invested with executive functions and only had advisory powers. The NBCC was limited to issues of artificial conception and was requested to consider and made recommendations in the area of human embryo experimentation.
The committee was multidisciplinary, with representatives in areas of philosophy, moral theology, social science, women’s health, law, medical research, nursing, and gynecology. It was effectively and ably led by Ms. Robyn Layton QC of the South Australian Bar. The aim of the NBCC was to search for a
more coordinated, national approach to this issue [reproductive technology]...and the National Bioethics Consultative Committee will play an important part in formulating such an approach“ (Senate Select Committee 1986).
The NBCC met for the first time in August 1988. During its brief and at times turbulent period, the NBCC produced a number of major reports including the following:
By mid-1990 the NBCC was gearing down as proposals were being considered to incorporate it into the NHMRC structure (Finn 1990).
A-11
2.3.3 The AHEC Established
Before the introduction of the National Health and Medical Research Council Act, 1992 (Cth.), in 1991 formal discussions began between the Chair of the NBCC, Robyn Layton QC of the South Australian Bar, and the Chair of the NHMRC, Dr. Di Horvath, with a view to amalgamating the MREC and the NBCC. The then Minister for Community Services and Health, The Hon. Mr. Brian Howe MHR, had commissioned a report on the advisability of concentrating advice to government on health ethics matters within a principal committee of the NHMRC (Finn 1990). The NBCC was established to handle specific references from the Australian Health Ministers Advisory Committee (AHMAC). As such, the NBCC could never have been a permanent standing committee. By the time of the publication of its Report on Surrogacy (NBCC 1990), the NBCC had completed the review of the key issues in reproductive technology. In a similar vein, the MREC was not the sole repository of ethical advice within the NHMRC.
The Minister for Community Services and Health decided to establish a new committee within the NHMRC to advise on health ethics. The new committee was to take up many of the responsibilities of the NBCC and the MREC as well as the ethical advice, which could flow, from the other principal committees of the NHMRC. In early 1991 it was decided that the new committee would be a principal committee of the NHMRC and was to be tentatively called the Health Ethics Committee (HEC). At early meetings, the broad terms of reference and focus of the new amalgamated HEC were established. These were:
| 1. |
To focus upon the social, legal, and ethical dilemmas arising from the fields of medical research, health care practice, and public health; |
| 2. |
To pursue an agenda within the broad priorities of NHMRC; |
| 3. |
To provide advice on particular ethical situations by linking people within the networks of the NHMRC; and |
| 4. |
To respond to issues identified by the principal committees of the NHMRC. |
The issue of the continued independence of the proposed HEC was the subject in some of these earlier discussions. It should be noted that the early Terms of Reference specified that the HEC was neither to have the role of providing an ad hoc ethics advisory service to the NHMRC nor to be used as a clearinghouse for reports from other principal committees of the NHMRC. Early discussions conceived of a committee of ten people covering many disciplines, with a national representation and balanced gender mix. It was agreed that the expertise of the NBCC could be broadened with the possible inclusion of a further clinician, health economist, and epidemiologist. Most importantly the expertise of the NBCC had to be supplemented with expertise from the MREC, particularly in relation to the operation of IECs. The success of these negotiations were quickly realized with the presentation of a work program to the June 1991 Council Meeting of the NHMRC.
The processes of the new HEC were discussed in some detail. The new principal committee was to enjoy a fair degree of independence within the structure of the NHMRC with power to set its own priorities. Matters could be referred by the NHMRC, other principal committees of the NHMRC, or from Commonwealth and State ministers. In addition, the new committee:
A-12
Finally, it was felt that the NBCC had achieved a high international profile and a style and quality of consultation that was important to maintain. For this reason the title “Australian” was to be added to the original suggested title of HEC to form the new AHEC.
3. The Commonwealth Review of Ethics Committees 1995–1996
3.1 Background to the Ministerial Review
Under the National Health and Medical Research Council Act 1992 (Cth.), the AHEC was made responsible for the administration of the national system of HRECs. While the system was generally recognized as working well during the 1990s, a number of areas of improvement were frequently mentioned in correspondence to AHEC, in the Medical Journal of Australia, and at public seminars, particularly the AHEC sponsored workshops in 1993 and 1995. Some of those included:
In 1995, the Commonwealth Minister for Health, the Hon. Dr. Carmen Lawrence MHR, announced an inquiry into the ethics review system. The review was requested in the context of two events. First, there was the controversy surrounding Family Planning Australia trials of the abortifacient RU486 in 1994. Second, in the same year, the Report of the Inquiry into the Use of Pituitary Derived Hormones in Australia and Creutzfeldt-Jakob Disease by Professor Margaret Allars (hereafter referred to as the Allars Report) (Allars 1994) was released. The Ministerial Review Committee was to inquire into the operation of HRECs with particular reference to the problems which have been identified following the Allars Report (Allars 1994) and the RU486 trials. RU486 was the so-called Morning After Pill which was counter-trialed in both Sydney and Melbourne. These trials formed part of an international multicenter study to determine the effectiveness of various doses of the drug and were sponsored by the World Health Organization (WHO). Although much of the controversy surrounding the trials related to ideological differences and concerns as to the appropriateness of the drug importation procedures, issues regarding the adequacy of the ethics committee review process were also raised. A separate and independent review on the RU486 trials (chaired by Professor John Funder) was conducted. That committee reported that ethics committee review had been adequate and recommended, following some modifications to the consent forms, that the trials recommence.
The 1995 Ministerial Review was not required to address the science or ethics of the RU486 trials but was requested to comment on issues relating to consent and the adequacy of HREC operation and review
A-13
procedures (including issues of membership and decisionmaking). The Allars Report (Allars 1994) also raised fundamental issues relevant to the Ministerial Review relating to monitoring of ongoing research, the distinction between treatment and research, and the importance of consent by, and the duty to warn, research participants. The pituitary hormones program, which was the subject of the Allars Report, had been initiated at a time before the establishment of the ethics review system. In addition, the use of these hormones was considered to be treatment that had already been tested and adopted overseas. Many of the issues raised in the Allars Report concerned poor practice in relation to the collection and use of damaged pituitaries and were beyond the scope of the Ministerial Review. The Review’s Terms of Reference required that it have “special regard to issues of concern to women particularly in trials relating to reproductive technology” and to “examine and report on recommendation 10 of the Allars Report” which stated:
| 10. |
That the NHMRC n review the Statement on Human Experimentation to ensure that n it provides guidance with regard to decisions as to whether treatment in a therapeutic setting constitutes an experiment; n a procedure is developed by which such decisions are scrutinized and not left entirely to the treating medical practitioner. n issue a Supplementary Note on Reproductive Technology Procedures which ensures that new procedures, including the use of drugs in new treatment regimes, are: n registered with the Health Ethics Committee of the NHMRC; and n approved by the institutional ethics committee of the institution in which the procedure is carried out; and n consent is made in on the basis of full information regarding risks and outcomes as defined in the Supplementary Note 2 on Research on Children, the Mentally Ill and Those in Dependent Relationships or Comparable Situations” (Allars 1994). |
3.2 Matters Addressed by the Ministerial Review
A number of issues, summarized below, were addressed in the Ministerial Review and presented in the Report of the Review on the Role and Functions of Institutional Ethics Committees (Report on IECs) (Chalmers 1996). These issues provide a background to the consultation and led to the publication of the revised National Statement on Ethical Conduct in Research Involving Humans. A list of the actual recommendations is included in Schedule 1. The Report on IECs noted the heavy and increasing workload of IECs, their lack of resources, their limited expertise in dealing with some types of research, difficulties with monitoring and with multicenter trials, and the dominance of scientists on the committees. The following are some of the main areas addressed.
Multicenter Research. There was no system of formal regional or national ethics review. Each IEC gave approval to research conducted in the institution. The practice had developed for individual IECs to communicate and exchange views with other IECs, particularly in relation to research projects carried out at different centers. The AHEC received numerous requests urging the establishment of a single national research ethics committee to consider multicenter trials involving humans. Researchers raised difficulties experienced in conducting multicenter trials where ethics approval must be obtained from a number of different IECs which may reach different conclusions in relation to the ethical acceptability of the trial. Different procedures, different meeting times, and different IEC membership often resulted in considerable delay in mounting a trial.
The Report on IECs proposed that it was appropriate for one Australian IEC to accept the scientific assessment or reasons for ethical approval of another IEC. There was no reason in principle why this other committee need be Australian based; it could be an approved overseas committee.
A-14
Multicenter Clinical Trials. Until 1991 all pharmaceutical and device trials were conducted under the auspices of the centralized Commonwealth TGA. Following the Baume Report (Baume 1991) a deregulated Clinical Trials Notification Scheme (CTN) was introduced which allowed IRCs to participate in organized clinical trials of pharmaceutical drugs and devices by notification only to the TGA (AHEC 1992). As a result of the CTN scheme, only a self-selecting group of IECs (now known as HRECs), with appropriate infrastructure support, mainly based in major teaching hospitals, participates in this scheme. This issue is dealt with in Section 6 of this report.
Adequacy of Compensation and Insurance Arrangements. The AHEC considered the issues of compensation, indemnity, and insurance in relation to the introduction of the deregulated CTN scheme for clinical trials of drugs and devices. The concerns of IECs were twofold. First, IECs were concerned that the individual members of the committee might have attracted legal liability from the decisions giving ethical approval to a CTN application (Capron 1985). Second, there were concerns that the institutional arrangements for insurance cover for participants in a clinical trial might not have been clear in relation to existing institutional insurance arrangements.
In relation to the first concern, a number of legal decisions were widely discussed causing concern in the Australian research ethics community. The High Court of Australia decision in Rogers v Whitaker established that a medical practitioner has not only a duty to exercise reasonable care in the diagnosis and treatment of a patient’s condition, but also a duty to disclose material risks inherent in any proposed treatment. A risk is material if in the circumstances a reasonable person is likely to attach significance to it, and the medical practitioner knows or should know that the particular patient is likely to attach significance if warned of the risk (this is consistent with U.S. and Canadian case law Canterbury v Spence and Reibl v Hughes). In this respect there is a higher duty of disclosure in the case of research projects: Halushka v University of Saskatchewan. There is further direct authority on the liability for nondisclosure of risks to research participants in the Canadian decision in Weiss v Solomon. This case also excited much critical comment (Freedman and Glass 1990). A number of other American cases have established the liability of hospitals in relation to decisions by Ethics Committees (see, for example, Davis v Rodman; Bouvia v Glenchur; Merritt 1987 at 1250–1252).
In relation to the concerns some institutions questioned the compensation limits, which were included in the documentation supporting some protocols for multicenter clinical trials. The AHEC reviewed a number of research compensation arrangements, which included limits on the amount of any claim for compensation by a research subject in a trial. These limits were clearly inadequate in comparison with Australian insurance payouts for injuries. The AHEC had addressed these concerns earlier in a report that required institutions to review their compensation indemnity and insurance arrangements with their insurer and to put in place appropriate compensation cover for research participants (NHMRC 1994). A major national insurer introduced a specific no-fault liability cover for clinical trials, which was taken up by a number of institutions participating in multicenter clinical trials.
Workload and Resource Support for IECs. This issue was clearly identified through the 1993 Survey of IECs and the Workshops for IECs (AHEC 1993). There was an expansion in workload because of a failure to sufficiently define the distinction between clinical practice and human experimentation. The result was that additional projects were referred to IECs, which would be more properly described as clinical practice and not experimentation. The other major growth in workload arose from referrals of health related and social science research projects to IECs.
Monitoring of Projects. Under the NHMRC Guidelines (NHMRC 1992), IECs were required to monitor research. A variety of methods were reported by IECs, mainly taking the form of reports by the investigator. Very few IECs reported systematic methods for monitoring, and only a handful reported the use of “site” visits.
A-15
Composition. There were concerns that the decisionmaking process was influenced too heavily by those with research interests. The original idea of an IEC was that it should have a majority of outside members. Surveys confirmed that clinicians and medical researchers dominated most IECs in Australia. The NHMRC Statement on Human Experimentation provided a minimum membership (NHMRC 1992). In fact, the majority of IECs were in the range of 10 to 15 members (16 or more members – 5 percent; 10 to 15 – 55 percent; 10 or fewer – 40 percent) with the majority represented by researchers. Paul McNeill has been a strident critic of this (McNeill 1993). Much of this diversity was due not only to the purpose of the institution and the nature of the research, but particularly to the authority, power, and responsibility given to, or accepted by, or assumed by IECs. In some institutions, the IECs had a broader function providing an advisory, policy and educational role relating to matters of clinical practice and management. Such committees may only rarely consider research proposals.
Procedures. Many of the IECs reported that they were not well resourced. This had the consequence, in some cases, of inadequate official record keeping. IECs make decisions that can have a direct effect on the reputation or standing of the researcher, the rights of the research subject, and the interests of the institution. The question which arises is whether these decisions ought to conform with the accepted standards of good administrative practice requiring that decisions are recorded and that reasons should generally be given. There is some authority for the proposition that an IEC’s decisions are reviewable (R v Ethical Committee of St Mary’s Hospital ex-parte Harriott), and it is probable that professional members in an IEC are answerable to the disciplinary authorities of their profession.
3.3 Comment
The Report on IECs (Chalmers 1996) was accepted by the Council of the NHMRC during 1996, and its various recommendations were steadily introduced culminating in the introduction of the National Statement on Ethical Conduct in Research Involving Humans in 1999. The report recommended that the original NHMRC Statement on Human Experimentation (NHMRC 1992) required a thorough revision taking into account parliamentary references to the AHEC, issues of public interest, and new ethical questions raised by technological advances.
It is interesting to note the similarities between this Australian Report and a review in the United States by the Office of the Inspector General of the Department of Health and Human Services. This review noted concerns that the IRBs in the United States have generally been doing “too much, too quickly with too little expertise.” The steady move toward more formal, regulated, and professional processes of ethics review of research is, no doubt, a common theme in most countries.
4. The Current System of Ethical Review in Australia
4.1 The National Health and Medical Research Council of Australia
Since its creation in 1937, the National Health and Medical Research Council has been the peak Australian funding body for health and medical research. One of the original aims of the NHMRC was to promote consistency in the health and public health policies of the individual State governments within the federal system. The NHMRC, having been established by Order-in-Council in 1937, was placed under a new statutory framework with the passage of the National Health and Medical Research Council Act 1992. The NHMRC remains the principal independent advisory body on health under the Act. Importantly, it is the principal national body for the provision of advice on matters of health ethics. Under the National Health and Medical Research Council Act, the council is charged with a number of functions including inquiring and issuing guidelines on the improvement of health; the prevention, diagnosis, and treatment of disease; the provision of health care; public health research and medical research; and ethical issues relating to health.
A-16
The Act confers four obligations on the NHMRC:
4.2 The AHEC Function and Relationship with the Commonwealth Parliament
The ethics advisory function is carried out by the AHEC, a principal committee of the NHMRC.
The AHEC was established under the National Health and Medical Research Council Act 1992 (Cth.) (see particularly § 35 and § 36). It is a multidisciplinary committee which, under the Act has the following Terms of Reference:
| 1. |
To advise the Council on ethical issues relating to health. |
| 2. |
To develop and give the Council guidelines for the conduct of medical research involving humans. |
| 3. |
Such other functions as the Minister from time to time determines. |
The Minister made such a determination at the time of the Act and conferred further functions on the AHEC as follows:
3.1 To develop and give the Council guidelines for ethical conduct in the health field, additional to those required for function 2 above, and for the purposes of the Privacy Act 1988;
3.2 To promote community debate and consult with individuals, community organizations, health professions and governments, on health and ethical issues;
3.3 To monitor and advise on the workings of institutional ethics committees (now HRECs);
3.4 To monitor international developments in relation to health ethical issues and files with relevant international organizations and individuals.
The NHMRC had some initial challenges in becoming fully acquainted with the expectations of the Senate-initiated AHEC that replaced the MREC (Commonwealth Parliamentary Debates: 1991 at 1089–1092). A short time after the passage of the National Health and Medical Research Council Act, it was decided that there should be an external review of the NHMRC. A Canadian academic was commissioned, and a report was presented in December 1993 (Bienenstock 1993). This report recommended that the NHMRC improve its planning processes for developing and setting priorities and strategies; improving the advisory processes of the NHMRC Committees; improving and simplifying the research funding allocation processes; and, finally, recommending substantial changes to the administrative support of the NHMRC.
AHEC was the subject of specific comment in the Bienenstock Report, which is worth quoting at length:
AHEC is the most recently established of the Principal Committees of the NHMRC, having been in operation for two and a half years at the time of this review. It evolved from the former Medical Research Ethics Committee of NHMRC and the National Bioethics Consultative Committee (NBCC) of the Australian Health Ministers’ Conference.
It has continued the work of monitoring and supporting around 150 institutional ethics committees through activities such as workshops, introducing a newsletter and providing
A-17
advice and speakers on request. AHEC has also developed the broader ethics role, conducting some preliminary work into the ethics of health resource allocation, guidelines to promote ethical conduct in the health field, and issued various discussion papers on health ethics issues....
It is apparent that AHEC has had some difficulty in coming to grips with its role and function in what is undoubtedly a complex and extraordinarily wide ranging area. It has attracted considerable criticism from some quarters for failing to provide concrete advice on practical issues relating to research, particularly those relating to the operations of Institutional Ethics Committees (IECs), though some progress appears to have occurred in this area at the most recent Council meeting. It is seen by some people as being dominated by the members of the former NBCC, which was concerned with broader ethical, social and legal aspects of health care, and as having insufficient expertise and involvement by practicing researchers to deal with concrete ethical problems relating to research. On the other hand, some members of AHEC have felt that the Committee has been too occupied with the agendas of subcommittees, particularly the IEC Subcommittee, to be able to define its broader role and activities.
Consideration of the legal and ethical aspects of health will grow in importance in the future. The NHMRC will play a vital part in this development. A balanced approach to this issue must involve recognition by health practitioners that ethical considerations are crucial in their work, and by the NHMRC that health practitioners and researchers must be an integral part of the development of appropriate guidelines. To separate ethical considerations from the practice of health and research is to invite irrelevance rather than independence” (Bienenstock 1993 at 23–24).
Professor Bienenstock recommended that AHEC should integrate its activities and priorities with those of the NHMRC as a whole, focus its energies on issues of highest practical and immediate priority, and be accountable to Council for its work. In so doing AHEC was to be restructured to more fully integrated activities with the principal committees of NHMRC (Bienenstock 1993, Recommendation 11). AHEC was to operate as any other principal committee of the NHMRC, but with the unique guideline development function under § 8 of the Act.
4.3 The AHEC Composition and Role
Only two of the principal committees of the NHMRC, namely the Research Committee and the AHEC, were specifically mentioned within the terms of the National Health and Medical Research Council Act 1992. By § 35 of the Act, the Minister must establish principal committees called the Medical Research Committee (now the Research Committee) and the AHEC. During the parliamentary debate and particularly those in the Senate, the composition and independent role of the AHEC was established.
n § 36 of the National Health and Medical Research Council Act 1992 provides that AHEC is to have the following membership:
A-18
The organizational and structural changes recommended by the Bienenstock Report (Bienenstock 1993) were put into place during the first half of the 1990s. By the second triennium of the AHEC (1993–1996) the Council of the NHMRC had a clear appreciation of the role and function of the AHEC. In particular, the Council recognized that the guideline development function of the AHEC was neither an advisory role nor a role which could be interfered with by the Council.
4.4 Guidelines of AHEC and Consultation
The AHEC, in its role as one of the principal committees of the NHMRC, is responsible for developing guidelines for the conduct of medical research involving humans, other advice relating to health, and for providing assistance to HRECs.
The guideline development function of AHEC is critical. Under § 8 of the National Health and Medical Research Council Act 1992 (Cth.), the NHMRC issues guidelines for the conduct of medical research involving humans. However, the guidelines for the conduct of medical research are developed by the AHEC and must be issued by the NHMRC precisely as developed by the AHEC (§ 8(2)). It should be noted that guidelines promulgated by the NHMRC do not have the same legal effect as legislation. However, the NHMRC is a creature of
A-19
statute (National Health and Medical Research Council Act 1992) (Cth.), and the Act provides that the NHMRC may promulgate guidelines. NHMRC guidelines relating to ethics are laid before Parliament before they come into force. It is therefore not accurate to describe the guidelines as voluntary. Guidelines have two specific legal aspects. First, they establish standards of reasonable practice. HRECs must follow these guidelines and in so doing act with fairness. Rules of administrative law deal with the standards of fairness required of committees. In this way HRECs are probably subject to administrative review which looks to standards of natural justice and procedural fairness. Second, and more importantly, the guidelines could be used and admitted as evidence in court proceedings to demonstrate that the deliberations and actions of a HREC are reasonable and fair and provided that the guidelines themselves are reasonable and that the HREC acted within their scope.
This rather unusual guideline-making function was inserted by the Commonwealth Parliament. It appears from the Senate Debates in relation to the Act (Senate Debates 1992, at 1089–1092) that this was inserted to ensure that the guidelines were a product of the public consultation process rather than the individual, and possibly medically biased, views of the Council of the NHMRC itself. In this respect the AHEC is a part of the NHMRC but is independent in the development of national guidelines in relation to medical research.
A complex consultation procedure was established under § 11–14 of the Act. Concerns that guidelines were “in-house” rather than public products resulted in the introduction of a unique two-stage consultation system. At the first stage, there is an advertisement of the intention to consider and develop guidelines in a particular area. In most cases, the AHEC circulated an information package or Issues Paper on the topic proposed for the guidelines. At the second stage the draft guidelines themselves were circulated for further advice and comment. Through these means it was intended that ex cathedra opinions by AHEC were to be avoided. Later, a decision by the Federal Court of Australia placed additional responsibilities on the NHMRC in relation to public consultation. In the case of Tobacco Institute of Australia Ltd v National Health and Medical Research Council and Others,
Justice Finn considered the specific terms of section 12 of the National Health and Medical Research Council Act. This section requires that the NHMRC have “regard” to the submissions presented to consultation and give “genuine consideration to the material.” The appellant, Tobacco Institute, had presented copious material to a consultation in relation to a draft Report on the Effects of Passive Smoking and Health (The report contained guidelines and was therefore subject to the two stage consultation requirements of the Act). The working party on the report decided to divide this material among the various members for reading and comment. Accordingly, each member read only part of the material. Justice Finn concluded that the obligation to have regard to the submissions required the NHMRC in its working parties preparing any report to give “positive consideration” to the contents of the submissions as this was a fundamental element of decisionmaking. As a result of this decision, the AHEC introduced lengthy minute taking of all consideration of submissions. AHEC developed a system of recording the acceptance or rejection (with reasons) of particular points raised. The minutes of AHEC in relation to public consultation were always treated as public documents available under the Freedom of Information Act 1982 (Cth.).
The AHEC is also required to promote community debate and consults with individuals, community organizations, health professionals, and governments on health and ethical issues.
4.5 Accountability of AHEC
The AHEC is subject to the normal organizational accountability procedures. The AHEC is required to present a work plan to the Council of the NHMRC. In addition, the AHEC is subject to financial and internal audits, presents reports (through the Chair) to meetings of the full Council and prepares a final report that is included in the publicly available Annual Report of the NHMRC (an example is included in Schedule 2).
Public accountability is perhaps best achieved by the public consultation provisions of the National Health and Medical Research Council Act. As described above, the AHEC is required to conduct public consultation,
A-20
and the guidelines which issue must have proper “regard” and pay positive consideration, to the contents and views expressed in the submissions. As a national organization, it is also subject to professional comment and criticism in the press and academic literature.
The AHEC is also answerable through the political processes. First, the relevant Commonwealth Minister may refer matters for consideration by the AHEC. For example, in late 1997 the Commonwealth Minister for Health and Aged Care referred the issue of human cloning to the Committee for advice (AHEC 1998). Importantly, the Commonwealth Parliament of Australia Senate was modeled on the United States Senate and enjoys the strong investigator committee system of the United States (the Lower House of Representatives reflects the Westminster Parliamentary system, and the Upper House Senate reflects the American Senate; as such the Parliamentary system is often referred to as a “Washminster” Parliamentary system). The Senate Estimates Committee has regularly interrogated the Executive Secretary of the AHEC on its works and finances. This was a deliberate consequence of placing the NHMRC under a Commonwealth statutory framework.
4.6 Australia’s System of Ethics Committee Review
Number of Committees. HRECs are the foundation of the ethical review system in Australia. (Breen 1997; Bennett 1997; Skene 1998; Freckelton and Petersen 1999). There are some 217 HRECs operating in Australia and registered with the AHEC. HRECs rely on the voluntary contribution of members, a degree of self-regulation, and modest financial support. The HRECs are responsible for the protection of research participants and ensure that research protocols are considered in conjunction with NHMRC and other applicable guidelines, with support and advice from AHEC.
At the time of this writing there are now 217 registered HRECs in Australia with the following approximate proportional distribution:
There continues to be variation among the HRECs. There are several aspects to this variation, which can be identified. There are a number of different types of institutions within which HRECs operate, ranging from large teaching hospitals to small regional universities, and from research institutes to small, special purpose organizations. Health institutions for example, range from the large teaching hospitals associated with the major medical schools to small rural base hospitals. There are also repatriation (for ex-defense force personnel) hospitals, area health services (in NSW and Queensland), specialist organizations such as the Red Cross and the Bone Marrow Donor Registry, as well as the specialist medical colleges. A third level of variation among HRECs, which can be identified, is the regional differences that arise from the variation in State legislation. For instance, HRECs in different States face different issues when considering a specific type of research (such as embryo experimentation) when State legislation is inconsistent. Therefore, it should be borne in mind that the HRECs in Australia are not entirely homogeneous, though much standardization is under way.
Review by and Role of HRECs. The Preamble to the National Statement clarifies its purpose as a whole and the role of HRECs in particular as the protection of the welfare and rights of participants involved in research. Some submissions to the public consultation in relation to the new National Statement expressed the view that Research Ethics Committees should “facilitate” research. While it is to be hoped that the HREC is not deliberately obstructive, the National Statement clearly places the protectory role on HRECs. Members of a HREC do
A-21
not have many representative responsibilities to the constituency from which they are appointed. The members do not in any sense represent the constituency. The National Statement again clarifies that the HREC members’ responsibility is to decide independently whether conduct of the research proposal involves the proper protection of the welfare and rights of research participants (see, for example, Bennetts v Board of Fire Commissioners of New South Wales). Importantly, HRECs consider all research involving humans and are not confined to the consideration of medical research only. HRECs are required to consider a large number of protocols ranging from drug trials and gene therapy to behavioral or social science research. All research involving clinical trials, regardless of the funding source, are assessed. To date, the review system has managed to cope adequately with the increasing number of clinical trials and research projects. In 1997 around 1,400 clinical trials were approved under the CTN, not to mention those trials under way and being monitored.
Membership of the HREC. The National Statement has increased the core membership of HRECs with a view to ensuring that the HREC responds to its protectory role rather than the institutional interests in promoting research. The membership now consists of:
If, at any stage, further members are added to the HREC, the institution is required to retain the balance and diversity of the institutional/noninstitutional members.
Procedures. The National Statement has introduced a number of new requirements to ensure proper discussion, contributions from members, and recording of decisions (this is discussed more extensively in Section 5 of this report).
4.7 Accountability of HRECs
Annual Compliance Requirements to AHEC. Under the previous Statement on Human Experimentation, IECs were required to present a minimal report confirming compliance with the guidelines at the end of the calendar year. There was no formal system of certification or accrediting of the committees. Under Principles 2.46–2.48 of the new National Statement on Ethical Conduct and Research Involving Humans, the compliance reporting requirements have increased considerably. The AHEC audits the activities of the HRECs to ensure compliance through a detailed Annual Report that seeks responses on issues of membership, meetings, agendas, approvals, rejections of projects, difficulties, and complaints. A failure to present an acceptable compliance report may, after investigation, lead to a removal of external funding from the institution. In this respect, HRECs are required to register with the AHEC as a precondition to being able to submit research projects for funding to the major public bodies.
Complaints Mechanisms. Before the National Statement, many of the long-standing Research Ethics
Committees had established complaints mechanisms. The National Statement now requires that any institution that establishes a HREC must also establish an independent complaint mechanism to handle complaints from research disciplines. In the first instance, it is expected that a research protocol should include a reference to a person nominated by the HREC to receive complaints. If this initial procedure cannot resolve the complaint
A-22
from the research participant, the HREC must formally refer the complaint to the institution’s complaint handling processes. The HREC is also required to ensure that information about pursuing complaints is made known to the research participants at the time of consenting to entering the research protocol.
Independent of these National Statement complaint mechanisms, all States and Territories have established administrative procedures for making complaints about the health system. The Health Complaints Commissioners in the States and Territories receive complaints about medical practitioners and the delivery of medical services. Where these complaints relate to research by a medical practitioner or medical research carried out in the health system, these complaints may be referred to the Health Complaints Commissioner. Very few complaints concerning research have been referred to the Health Complaints Commissioners among the many thousands of general complaints. This may indicate an absence of complaints about the research system or, alternatively, problems in the making and reporting of complaints.
4.8 The Work of the AHEC: 1991–2000
A brief outline of the references, work, and guidelines produced by the AHEC is presented. This illustrates the manner in which the AHEC has established functions both within the NHMRC and nationally within the research ethics committee system.
AHEC met for the first time at the end of August 1991. During its first two triennia (1991–1996), AHEC undertook work on a case study of the legal and ethical implications of HTLV-I; information papers on the legal liability of institutional ethics committees, ethical considerations relating to health care resource allocation decisions, nature of qualitative research, human gene therapy, workshops for institutional ethics committee members; monitoring and supporting the HRECs through workshops, newsletters and advice, and, guidelines relating to IVF and embryo transfer, privacy in medical research, and the use of patient tissue samples for research.
The third triennium of the AHEC was marked by a substantial revision on the Statement of Human
Experimentation and a formal reference of work on human genetics by the Commonwealth Minister for Health and Aged Care. The work was as follows:
n The National Statement on Ethical Conduct in Research Involving Humans
The Statement is discussed below at Section 5 of this report.
n The Genetics Program
The ethics of human genetic research was the major focus of the work of the AHEC during this period. A specific Working Party was convened, and developed and finalized two sets of guidelines are as follows:
Guidelines for Genetic Registers and Associated Genetic Materials and Guidelines for Ethical Review of Research Proposals for Human Somatic Cell Gene Therapy and Related Therapies. The former gave guidance on all aspects of the operation of genetic registers on the collection, use, and access to this material. The guidelines also deal with aspects of recruitment and storage of genetic material. The latter is intended to give guidance to the select HRECs that deal with gene therapy applications. In addition to the two sets of guidelines, the AHEC has published an Information Paper addressing issues of equity, resource allocation, commercialization, and counseling and testing of children in a document entitled Ethical Aspects of Human Genetic Testing: An Information Paper.
Finally, for the first time, the National Statement included a specific set of principles of human genetic research (National Statement 1999, Principles 16.1–16.21).
A-23
n HREC Operating Manual
A HREC Operating Manual is in preparation, which will be in the form of annotations to the Statement providing explanation and procedural information to HREC members. It is important to note that the use of the operating manual will not be mandatory and will not prevent HRECs from developing their own operating manuals or varying any published national standard operating manual. It is anticipated, though, that an operating manual at the national level will assist the decisionmaking processes of HRECs, contributing to consistency and predictability in the operation of HRECs in Australia. The manual will be developed in consultation with HRECs and other key stakeholders and is based in part on the Kings College Manual in the United Kingdom.
n 1999 HREC Workshops
The AHEC held workshops for HRECs in 1993 and in 1995. There were many calls for another series of workshops as a means of imparting information, discussing issues, and networking for the HRECs. The fifth series of National Workshops were held in 1999 to launch the National Statement on Ethical Conduct in Research Involving Humans.
n Guidelines for the Protection of Privacy in Medical Research (1995)
The Guidelines for the Protection of Privacy in Medical Research were revised and issued under § 95 of the Commonwealth Privacy Act 1988 and provide a framework for the protection of privacy in medical research involving personal information obtained from Commonwealth agencies. The purpose of the guidelines is to ensure that such personal information is protected against unauthorized collection or disclosure.
| n |
Ethical, Legal, and Social Implications Program for the HUGO Human Genome Meeting 1999, Brisbane, Australia |
The AHEC was invited to develop the ethics program for the HUGO Human Genome Meeting (HGM 1999) held in Brisbane, Australia in March 1999.
n Cloning of Human Beings
In January 1998, the Commonwealth Minister asked AHEC to provide advice on the ethical issues and the need for further pronouncement or possible legislation regarding the cloning of human beings.
This advice was published in a report to the Minister entitled Scientific, Ethical and Regulatory Consideration Relevant to Cloning of Human Beings (AHEC 1998).
n Xenotransplantation
Given the national and international interest in the possibility of xenotransplantation, the AHEC was asked to consider issuing ethical guidelines on the subject. In view of the risk of rejection and possibility of transmission of unknown infectious agents from animals through immuno-compromised hosts into the general community, the AHEC sought scientific advice from the Research Committee of the NHMRC to clarify the potential risks and benefits before considering necessary action.
5. The National Statement on Ethical Conduct in Research Involving Humans
5.1 Background to the National Statement
The report on IECs (Chalmers 1996) recommended that the AHEC should redraft the Statement on Human Experimentation and “…change its title so that all health investigation involving humans (including non-biomedical research and innovative practice) was encompassed” (Recommendation 5.3.1).
A-24
The review process incorporated not only the advice in the submissions made but also a number of developments, documents, and practices that may be briefly summarized as follows:
There was a perceived need to take into account international considerations in introducing the new National Statement. Submissions received by AHEC during the public consultation processes included increasing references by researchers, organizations, and community groups to overseas research guidelines, international conventions
A-25
and treaties, and international practices. The Australian Government, with the consequent implementation obligations had signed some of these Conventions and Treaties. The most notable of these international developments were as follows:
5.2 The National Statement and Its Nationwide Application
In 1999 the NHMRC concluded its public consultation on a new National Statement for Ethical Conduct in Research Involving Humans (National Statement 1999). The report on IECs had recommended that “The NHMRC in conjunction with other peak bodies responsible for research and clinical practice (Australian Research Council, Australian Vice-Chancellors’ Committee, Australian Medical Council) should promulgate guidelines representing a national statement for the ethical conduct of research” (Recommendation 5.2.2.). This was achieved during late 1998 and the first half of 1999, and the Statement was also endorsed by all the national funding agencies, universities, and the learned academies. This is the first time that all research funding agencies, universities, and learned academies have subscribed to a single national code of conduct for the ethical conduct of research involving humans. Importantly, this statement has continued to include a section on clinical trials, which was the subject of considerable comment by researchers, the Australian Pharmaceutical Manufacturers Association, institutions, and consumer organizations. The predecessor Statement on Human Experimentation 1992 contained a Supplementary Note 4 that dealt summarily with key elements of clinical trials.
The National Statement applies universally to all disciplines of research involving humans. The guidelines includes new sections on human genetics research, use of human tissue samples, emergency care research, and some additional guidance in relation to multicenter trials and modified composition of HRECs. This is a significant step in promoting a uniformly high ethical standard for all research involving humans.
A-26
5.3 The Contents of the National Statement
Comments are made in this section in relation to parts of the National Statement to provide some background rationale for the Principles.
Purpose of the Statement. The Preamble to the National Statement notes that the purpose of the Statement is to provide a national reference point for ethic consideration relevant to all research involving humans. All major bodies involved with human research have endorsed the National Statement. Not only symbolically, but also actually, the National Statement will serve the major national reference point in the future development of research ethics involving humans in this country. If this goal is achieved, it is hoped that not only will there be simplification in place of many differing codes but also improvement in the quality of ethical consideration through uniform standard setting.
The Principles to be Applied. The National Statement includes a more detailed summary of the Principles of ethical conduct than the former Statement of Human Experimentation (Brody 1981; Engelhardt 1986; Beauchamp and Childress 1994; Pellegrino and Thomasa 1996). It is intended that the General Principles (Principles 1.1–1.21) will assist in the interpretation of the other parts of the National Statement. Integrity of the researcher is placed at the forefront of these principles. Respect for persons and beneficence are expressed in traditional forms, but the well-being of the participants takes precedence over expected benefits to knowledge, and researchers have a responsibility to minimize risks of harm or discomfort to research participants. For the first time, the principle of justice is included and requires fair distribution of benefits and burdens of participation in research; avoidance of unfair burden by participation; fair recruitment of research participants; and avoidance of discrimination in the selection of participants. The Operating Manual when published will explain the intention of these important principles, which are intended to address concerns about over-researching of particular groups, questionable recruitment practices for participants, and applying selection criteria for the participants which may, in effect, discriminate. The focus, in this respect was on the process of research rather than the results of the research. The National Statement focuses on the dissemination of such findings but does not oblige researchers, sponsors, or others to actually distribute research benefits among the participants.
HRECs (Principles 2.1–2.5). The National Statement attempts to achieve further development of the established ethics review system. A clear responsibility is established for institutions to establish and properly resource HRECs. Institutions are now required to set clear Terms of Reference for the HREC. If, for example, the HREC is to undertake policy or educational tasks as well as the primary research review function, these additional functions must be provided for in the Terms of Reference. In addition, the institution is required to accept expressly the legal responsibility for the members of the HREC while they are acting within the scope of their approved functions. Where researchers are not affiliated with a particular institution, institutions are encouraged to accept these projects for consideration by the HREC. The aim of this provision is to try to ensure that all research conducted in this country is under the umbrella of the protectory research ethics review system.
Membership of the Committees (Principle 2.6–2.9). No longer are medical graduates required to form the core membership of a HREC. There is now provision to appoint a person with knowledge of and current experience in the research that is regularly considered by the HREC. Thus, if the research considered by the HREC is social science, then the person appointed should be knowledgeable and experienced in social science research. Second, the core membership has been expanded by the inclusion of a person with knowledge of or current experience in professional care, counseling, or treatment. This person was seen as offering additional insights into the way in which research participants may view a research project and the way in which it impacts upon them. This does not have to be a doctor, but can extend to a psychologist, nurse, social worker, or the like depending on the type of research considered by the HREC.
A-27
From time to time suggestions have been made that some members of a HREC should be appointed solely for the purpose of representing the research participants (McNeill 1993). This view is misconceived in the sense that all members of a HREC are required to protect the welfare and rights of the research participants.
In addition, the National Statement includes a requirement that institutions should be mindful of institutional and noninstitutional balance in the membership of their HRECs. The National Statement requires that any increase in the core membership of the HREC should retain this balance. This provision was included to address difficulties that have arisen under the old IEC system. Many of the old IECs had, in addition to the five core members, a membership of between 12 to 15 members (See Section 3.2 of this report). Many of these additional members were appointed for research expertise resulting in lay members being in the minority. Institutions must, if membership is increased, maintain the balance of core membership to new members. For example, if another researcher was to be appointed the institution may very well wish to appoint a further lay person.
HREC Meetings (Principles 2.15–2.24). A number of new provisions are included in relation to meetings for HRECs. The HREC may now invite a researcher to attend to provide advice to the HREC. This formalizes the procedure, which had developed in some IECs. Importantly, a HREC must proceed to deliberate without any conflict of interest by any member. It is the responsibility of HREC members to announce any conflict of interest, which may affect the independence of their decisionmaking. HRECs may seek expert scientific advice on a research protocol. This procedure was introduced to address concerns by many researchers that HRECs were spending too much time deliberating on the scientific rather than ethical aspects of research protocol. As there is no neat division between scientific aspects and ethical aspects of research, the National Statement directs the HRECs’ attention to their ethical function but recognizes from time to time that research protocol may require explanation to illuminate the ethical issues involved. Researchers are now required to disclose any funding or any financial interest, which they may have which may be related to the project. A HREC must then decide whether its disclosure in any way affects any relevant ethical considerations in the protocol.
Monitoring (Principles 2.33–2.38). The previous Statement on Human Experimentation was amended in 1992 to recognize the responsibility of ethics committees or monitoring research. The new National Statement includes Principles requiring the HREC to monitor research. The Principles also recognize that the primary responsibility rests with the institution. In addition, the frequency and type of monitoring which is carried out in relation to research protocol should reflect the relative degree of risk to the participants. In this way, HRECs are encouraged to concentrate on riskier protocols. HRECs are required to receive reports of anything that might warrant review of the original ethical approval or anything which may result in the early discontinuance of the research.
These Principles were intended to address growing concerns among members of ethics review committees that they have neither the expertise nor the resources to conduct effective and timely monitoring of research. Many institutions and ethics committees had, during the 1990s, developed “tailored” monitoring mechanisms, which, as a matter of fact, reflected the degree of risk involved. The National Statement reflects this development and requires HRECs to implement appropriate monitoring mechanisms dependent on the risk involved in their research protocol.
Expedited Review and Multicenter Research (Principles 3.3–3.7). For the first time the National Statement has formalized expedited review for minimal risk research. Recognizing the growing burden on HRECs, the National Statement permits a HREC to nominate classes of research, which may be reviewed in an expedited fashion by the Chairperson and later ratified by the full Committee. However, the National Statement does not permit risky or ethically controversial research to be subjected to expedited review.
The National Statement for the first time sets up two procedures for handling multicenter research. First, HRECs are now permitted to communicate with other HRECs; to accept scientific assessments of other HRECs;
A-28
to adopt the reasons and ethical decisions of other HRECs; to adopt any procedures of another HREC with a view to avoiding duplication; and to agree on common monitoring responsibilities. Second, there is now a formal procedure, which allows HRECs and institutions to agree before the start of a multicenter research project to nominate the “primary, ethical and scientific assessment process subject to the approval of the other participating institutions and HRECs.” These informal and formal multicenter research procedures are intended to address complaints by researchers about delays and inefficiencies in ethical review. Frequently, researchers complained that HRECs were more engaged in difficulties about procedure or documentation rather than points of ethical substance. These procedures are intended to facilitate multicenter research without in any way compromising proper ethical safeguards. In both New South Wales and Victoria efforts are now in progress to develop common application forms and systems to allow multicenter research procedures to be implemented (Kelly and Boyages 1999).
Special Categories for Protection (Principles 4–7). The National Statement includes specific Principles intended to protect participants who are either vulnerable or at greater risk. In the case of children and young people, research should only be permitted where their participation is indispensable and the physical, emotional, and pathological safety of the children and young people are ensured. As with other like categories, a HREC should not approve the research where it is contrary to the child or young person’s best interests. Similar provisions apply to research projects that involve participants with an intellectual or mental impairment.
The National Statement recognizes that those in highly dependent medical care situations (emergency, intensive, neo-natal intensive and terminal care) may be unconscious or otherwise impaired in their capacity to communicate. In such cases, it may not be possible for the researcher to obtain consent to the research. However, in these circumstances there may be greater risk of coercion and undue burdens from involvement in research. HRECs, in these cases, may allow the research to be conducted provided it is generally not contrary to the patient’s interests; the research is therapeutic; the risks are no greater than those involved in accepted treatment; and there is a reasonable possibility of benefit over standard care. In addition, the patient, guardian, or family is informed as soon as possible of the option to withdraw.
Recognizing the pressures that can be brought to bear in the workplace, education, or in institutions, the National Statement recommends that HRECs should exercise extra care when considering research where there are dependent or unequal relationships. In these cases, the HRECs should be satisfied that the consent is, in fact, voluntary and that no discrimination should follow where a person refuses.
Research on Collectivities (Principle 8). The National Statement includes principles to cover research involving collectivities. Collectivities are defined to include those with common cultural, customary, social organization but not extending to clubs or associations. The term was proposed by the Canadian Tri-Council Code and was considered a helpful contribution to understanding research among the multicultural communities of Australian society. In essence, research in collectivities requires, as well as individual consent, consent by the collectivities recognized legally. In addition, researchers must satisfy a HREC that the customs and beliefs of that collectivity will be respected.
Aboriginal and Torres Strait Islander (Research Principle 9). Interim Guidelines were introduced by the NHMRC in 1991 before the establishment of the AHEC. During the public consultation, differences were expressed in this area. Some submissions expressed satisfaction with the existing Interim Guidelines, others suggested new Guidelines and others suggested that the proposed principles on research involving collectivities were sufficient to include Aboriginal and Torres Strait Islander people. The Interim Guidelines have been continued in force, and the Interim Guidelines will be reviewed in the future.
Clinical Trials (Principles 12.1–12.13). This topic is discussed in greater detail in Section 6 of this report.
A-29
Innovative Therapy. Innovation is a major part of good clinical practice. The medical practitioner is given freedom to vary standard treatments to find the best and most appropriate treatment regime for his/her patient. The integrity and professional responsibility of the medical practitioner define the limits to the use of this clinical freedom. The Ministerial Review recommended that a guideline be introduced to regularize practice in this area (Chalmers 1996). The National Statement includes a principle that any systematic investigation or innovation to determine efficacy should be considered as clinical research and referred to a HREC for approval. The purpose of this guideline is to permit and encourage clinicians to seek HREC advice and approval where researcher innovation is in fact, being conducted (see Section 3 of this report).
Epidemiological Research (Principles 14.1–4.13). The National Statement includes a number of new principles to facilitate epidemiological research while maintaining proper protections for research participants particularly in relation to privacy. First, the National Statement distinguishes epidemiological research from conventional public health surveillance of public health records by authorized public servants. This definition was included to address concerns by State and Territory government departments in increasing requests for access to records under their control.
Second, the National Statement includes 3 categories of data:
Confusion has arisen in recent years in Australia with access to “coded” information. On the one hand, researchers complained that HRECs set unrealistic and impractical consent requirements in relation to their projects. On the other hand, HRECs are reflecting growing community concerns about privacy and access to personal records. This schema of identified, potentially identifiable, and de-identified data aims to assist HRECs to focus on projects that involve identified or potentially identifiable information. The first category is straightforward. “Potentially identifiable information” refers to information that is coded and may easily be translated into identified information. In addition, the term “potentially identifiable” refers to small population groups (by region or by disease indications) which may be identified by reference to other sources, e.g., post code.
Third, where potentially identifiable data is used by a HREC, the HREC should generally require that once the linkage has been established, the information should be coded and placed in secure storage.
Fourth, these principles permit a HREC to approve access to data without consent when the consent process is likely to cause unnecessary anxiety or prejudice to scientific value and there is no disadvantage to participants. The HREC may also grant access without consent where it is impossible in practice to gain consent because of the numbers involved or accessibility to them. In either of these cases the HREC must again be satisfied that the research interest outweighs to a substantial degree interest in privacy. This expression is used in the Commonwealth Privacy Guidelines in relation to research conducted using Commonwealth data. The expression is also to be used in the new public sector guidelines produced by the Commonwealth Privacy Commissioner. It is used in these Guidelines to develop a consistent approach to personal privacy and research.
The privacy principles were included to address directly researchers’ concerns about HRECs setting unrealistic consent requirements in relation to large data sets. The principles also require any new use of the data for a new research project to be resubmitted to a HREC for a new approval. In addition, if clinical knowledge is disclosed to researchers during the research project that information should be made available to health authorities and where possible to participants or their medical practitioner.
Finally, the general principle that research results be disseminated is qualified by further requirement that the results should not identify the participants and should respect cultural sensitivities.
A-30
Human Tissue (Principles 15.1–15.9). For the first time the National Statement includes principles for the use of human tissue in research. The use of human tissue samples in medical research raises compliance issues with both ethical and legal standards (Magnusson 2000). Samples are defined to include diagnostic, statutory (e.g., Coroner’s Inquiry), and research samples not including fetal, reproductive, or autopsy tissue. Institutions are requested to develop policies for research on tissues related to the source, nature, cultural sensitivity, and reason for collection in the purpose for the research. Generally, consent is required for the use of a person’s tissue. Where there is follow-up research, the new research should be presented for new approval by a HREC. Consistent with the principles in epidemiological research and genetic research, a HREC may waive consent having regard to the following considerations:
These principles are expressed in relatively general terms. They represent the first step in setting a direction for the more regulated use of human tissue and research. This is a sensitive area where there are public concerns about coronial powers to dispose of human tissue, commercial access to samples, and retention of samples without an individual’s knowledge or consent.
Genetic Research (Principles 16.1–16.16). A special Working Party was convened to prepare these principles which were developed in close consultation with community groups, professionals, and the Human Genetics Society of Australia. After outlining the special aspects of genetic information and its capacity to stigmatize, HRECs are requested not to approve research with contestable or dubious scientific merit. HRECs are reminded that much genetic research at this stage will be more likely to contribute to knowledge rather than products and treatment. For this reason research proposals must be balanced against the potential for risk to individuals. Research results are to be carefully stored to ensure privacy, and researchers are required to state whether the information will be kept in an identified, identifiable, or de-identified form. Generally, the consent of participants will be required and researchers are required to inform them:
A-31
Consistent with the principles on epidemiological research and human tissue, a HREC may waive consent having considered a number of matters (essentially the same considerations as above under Human Tissue). There are also requirements that the institution conducting research has access to current genetic counselling services for the benefit of the participant.
Deception (Principles 17.1–17.2). The National Statement recognizes that some research; for example, psychological research involves deception pursuant to purpose or covert observation of individuals. HRECs should approve such research only as an exception where the research cannot be conducted without deception. In these cases a HREC may approve if it is satisfied that:
Privacy (Principles 18.1–18.5). The Commonwealth Privacy Act 1998 includes a number of Information Privacy Principles defining the proper collection, detection, use, access, challenge, and amendment of privacy information. This Commonwealth Act only refers to information held by a Commonwealth Department or its agency. However, for the last ten years many HRECs have used the Information Privacy Principles as standards for privacy protection for research involving information held by agencies other than the Commonwealth. In this respect the highly unsatisfactory patchwork of Australian law in this area has been remedied to a degree by the practice of some HRECs. The National Statement sets very general Guidelines for the protection of privacy. This is clearly an area, which will require further legislative and guideline development in the future. The privacy of personal information is to be protected using the Information Privacy Principles as a standard.
There is a specific Section 95 in this Act that requires Commonwealth agencies to report to AHEC where the HREC has released information without the consent of the individuals concerned (and in breach of any Information Privacy Principle) but is satisfied that the public interest in the research outweighs, to a substantial degree, the public interest in the protection of privacy (NHMRC 2000).
6. Some Matters for the Future and the New National Statements
Ethical review in this country remains, as elsewhere in the world, in a revolutionary stage. Ethical standards in the review of research were never envisaged as constant. For example, in the introduction to the Declaration of Helsinki it was stated that the guidelines should “…be kept under review in the future.” The Declaration was adopted by the 18th World Medical Assembly, Helsinki, Finland, June 1965 and amended in Tokyo 1975, Venice 1983, Hong Kong 1989, and the Republic of South Africa 1996. The Declaration is currently under review (Bulletin of Medical Ethics 1996) The Australian research guidelines have been regularly reviewed. This section briefly outlines a number of matters, which are likely to command attention in the near future. These matters are clinical trials, the development of a clinical trial register, multicenter research, expedited review, and monitoring of research.
A-32
6.1 Clinical Trials
Clinical trials are likely to command greater international public attention. In recent years there has begun a steady stream of media and academic revelations about certain trials.
Clinical Trials in Australia: Some Background. Before examining the new clinical trial guidelines, some background may be useful (AHEC 1992). Until 1983, sponsors of all clinical trials involving imported products were required to obtain Federal approval prior to the initiation of the trial. Pharmaceutical chemistry, preclinical, and clinical data were required in the same detail as that required to support applications to market a new chemical entity. In February 1983, review times were changed to 45 working days for early phase trials (Phase I and IIa) and 80 working days for later phase trials. In addition, a degree of deregulation was introduced in that sponsors were permitted to undertake additional trials without Federal review of the subsequent protocols, provided that the trial was within the approved dosage range and duration of treatment. Each trial required approval by the IEC of the host institution, and sponsors were required to notify the Federal agency at the time of approval by a HREC.
The TGA is a Commonwealth organization responsible for the registration of therapeutic goods including drugs and devices. The TGA conducts monitoring of licensed manufacturers who must comply with the Code of Good Manufacturing Practice; in addition, the TGA tests drugs and devices, reports and acts on problems, and ensures fair and truthful advertising (Therapeutic Goods Act). The scheduling of drugs is usually conducted under the various drug legislation of the States and Territories. In August 1987, revised procedures for review of clinical trials were introduced incorporating the concepts of a Clinical Trial Exemption (CTX) scheme under which the trial was permitted to proceed if no objection was raised by the TGA within a given time frame.
A-33
Under these arrangements, consideration of the essential safety aspects of a product proposed for use in a clinical trial remained a Federal responsibility, and consideration of the inter-related protocol was the responsibility of the HREC at the institution(s) at which the trial was to be conducted. The scientific validity of the study and the ability of the researcher and institution to effectively carry out the particular study were to be included in the HREC’s consideration of ethical aspects of the trial.
The CTN Scheme. In the early 1990s following the publication of the Baume Report the centralized system of approval for drug trials was replaced with a devolved approval system. HRECs were given the option of approving drug trials under the CTN. At first, there were considerable concerns about the implementation of this new scheme particularly in relation to potential legal liability (Day 1993). However, the implementation of the scheme has been realized through a process of self-selection under which only HRECs in large hospitals are now undertaking significant involvement in the CTN process. In the early 1990s it was recognized that there had been a major increase in the workload of those IECs that had undertaken this type of work (Chalmers 1996). In May 1991, links between clinical trials in Australia and marketing applications were severed. This allows clinical trials to be conducted while an application for registration for marketing is under review and vice versa.
The introduction of the CTN Scheme at the same time allows for drugs to be released for clinical trial purposes, provided authorities are notified of the trial beforehand and the trial is approved by the ethics committee of the hospital or university where it is to be conducted. Only HRECs complying with the National Statement (National Statement 1999), particularly Principles 2.1–2.48 on HRECs, are able to participate in these arrangements.
The main impact of the deregulation of clinical trials, from the point of view of HRECs, has been an expansion of their tasks and responsibilities to include the assessment of toxicological and safety data for trials submitted under the CTN Scheme. This was the subject of a specific review of the introduction of the CTN Scheme which was completed in 1993 (Day 1993). HRECs expressed particular concern over possible legal liability in administering these schemes and the need for appropriate indemnity. Of particular concern was the fact that some HRECs did not have the expertise to assess pharmacology or toxicology data. The responsibility of HRECs was reflected in the Therapeutic Goods Regulations as amended by the Therapeutic Goods Act. This provides that the institution which is responsible for conducting the trial must take advice from the IEC (now HREC) on the conduct of the trial, give approval to the trial (the institution may be responsible for more than one site), set terms of approval for the trial which are no less restrictive that the ethics committee’s advice, and withdraw approval for the trial if the ethics committee advises that continuation of the trial is not appropriate.
The move to using the CTN Scheme has been steadily increasing. By mid-1999, the TGA reported that some 1,500 were proceeding under CTN and only 10 under the CTX (information provided by Manager of TGA to AHEC, July 1999). In essence the CTN is a deregulated system where all responsibility for the trial rests with the institution, and notification only is given to the TGA about the conduct of the trial. On the other hand, under the CTX Scheme the TGA remains responsible for the safety aspects of the product and charges fees for this service.
The National Statement Principles. The new Australian National Statement (National Statement 1999) is a comprehensive and uniform set of guidelines which includes general principles and sections (Principles) on many aspects of research (e.g., epidemiological research, genetic research, use of human tissue, psychological research, and multicenter research). The National Statement includes more detailed guidelines of the establishment, composition, operation, functions, and duties of HRECs.
The National Statement includes a section dealing with clinical trials, which are defined to apply to natural therapies and other interventions. The previous Statement on Human Experimentation included a supplementary note on clinical trials but in considerably less detail than the National Statement. The introduction to Principles 1212.1–12.13 states:
A-34
A clinical trial is a study involving humans to find out whether an intervention, including treatments or diagnostic procedures, which it is believed may improve a person’s health, actually does so. A clinical trial can involve testing a drug, a surgical or other therapeutic or preventive procedure, or a therapeutic, preventive or diagnostic device or service. Any intervention, including so-called ‘natural’ therapies and other forms of complementary medicine, can be tested in this way. Other related disciplines also conduct research, which involves similar ethical considerations to those raised in clinical trials.
In pharmaceutical and medical device trials there are established codes of good clinical research practice which define clearly what is meant by a clinical trial for those purposes.
12. Clinical Trials has principal application in the context of biomedical clinical trials but should also apply to any other intervention claiming therapeutic benefit, wherever provided or conducted (emphasis added).
The trial must be properly designed and conducted and be approved by a HREC. The HREC that considers the clinical trial is not required to judge the actual science involved. Rather the HREC must ensure that it is “…sufficiently informed on all aspects of a research protocol, including its scientific and statistical validity” (National Statement 1999, Principle 2.8). Principle 12.1 goes on to state:
The aims of every trial must be precisely stated in a protocol presented to and approved by a Human Research Ethics Committee (HREC) and every trial must be conducted by researchers with suitable experience, qualifications and competence and, where applicable, adequate training in relevant procedures including the use of any device being trialed.
See also Principle 12.2, which gives details on scientific hypothesis and methodology.
A HREC, before granting approval to a clinical trial, must be satisfied that the protocol conforms to a number of international obligations in addition to the National Statement as well as relevant Australian laws. The Code of Good Manufacturing Practice issued by the TGA is broadly similar to many equivalent documents in other countries (TGA 1991). In addition, it is recognized that Australian researchers may be involved in multi-center international trials. Indeed, in the case of American trials, Australian researchers are required to comply with American regulations promulgated by the FDA. There was a quite deliberate intention in the revision of the National Statement to ensure consistency with established international guidelines. In this regard, Principle 12.3 of the National Statement provides:
An HREC, before granting approval to a clinical trial, must be satisfied that the protocol conforms to:
| (a) |
this Statement; |
| (b) |
the World Medical Association Declaration of Helsinki; |
| (c) |
where relevant, the CPMP/ICH Note for Guidance on Good Clinical Practice (CPMP/ICH-135/95) and the ISO 14155 Clinical Investigation of Medical Devices and the requirements of the TGA; |
| (d) |
any requirements of relevant Commonwealth or State/Territory laws. |
Principles 12.12 and 12.13 also refer to relevant standards.
The National Statement also includes a specific guideline on the acceptable uses of placebos in clinical trials and, essentially, outlaws their use where there is an effective treatment available (National Statement 1999, Principle 12.4). There was considerable discussion in relation to this particular guideline. In the end the AHEC, in publishing the guideline, preferred the view that it is difficult to create a research project (testing a
A-35
hypothesis when there is a treatment available which has been clearly shown to be effective). To ignore a proven effective treatment breaches the medical practitioner’s duty to provide best available treatment to the patient.
12.4
The use of a placebo alone or the incorporation of a non-treatment control group is ethically unacceptable in a controlled trial where:
| (a) |
other available treatment has already been clearly shown to be effective; (emphasis added) and |
| (b) |
there is risk of significant harm in the absence of treatment. |
If there is genuine uncertainty about the net clinical benefit of treatment, a placebo controlled trial or a trial with a no-treatment arm may be considered.
Apart from general guidelines against conflict of interest, (National Statement 1999, Principles 1.1 and 2.20) researchers are required to declare financial or business interests in relation to the clinical trial presented for approval before the HREC (National Statement: 1999, Principles 12.5 and 12.6). A researcher is not required to disclose every interest to research participants; rather, a HREC is required to examine the budget of the clinical trial and consider aspects of the budget that raise ethical issues. The HREC then decides whether any information in relation to the financial aspects of the trials should be declared to participants.
12.5
A researcher must inform an HREC of any business or other similar association which may exist between a researcher and the supplier of a drug or surgical or other device to be used in the trial.
An HREC must examine those aspects of the budgets of clinical trials which raise ethical issues, including capitation fees, payments to researchers, institutions or organisations involved in the research, current and consequential institutional or organisational costs and costs which may be incurred by participants. It should be satisfied that:
| (a) |
payment in money or kind would not cause researchers to apply pressure to individuals so as to obtain their consent to participate; |
| (b) |
payment in money or kind could not influence the findings of the research; |
| (c) |
there will be disclosure to the research participants of relevant aspects of those budgets; and |
| (d) |
funding is sufficient to conduct and complete the trial so that participants are not disadvantaged by premature cessation. |
12.6
Since the early 1990s the NHMRC has published guidelines requiring HRECs to review the compensation arrangements for the trial (NHMRC 1994). Principle 12.7 of the National Statement provides that compensation arrangements must be in place for participants who may be injured in the trial.
12.7
An HREC must be satisfied, before approving a clinical trial, that arrangements exist to ensure adequate compensation to participants for any injury suffered as a result of participation in the trial.
There are, finally, guidelines about the reporting of all serious or unexpected adverse events, review of the trial, suppression of the trial, and privacy of findings (National Statement 1999, Principles 12.8–12.11).
The new Principles have deliberately aimed to put greater responsibility on the HREC that approves a trial, the reality being that the preponderance of Australian clinical trials of drugs and devices are performed under
A-36
the terms of the CTN Scheme. In summary, the HREC must be satisfied that the trial is properly designed (including methods of recruitment and statistical significance). The HREC must also decide whether the trial conforms with the international standards where relevant (CPMP/ICH 1995). Placebos should not be used where they are already proven effective available treatment. In addition, conflicts of interest must be declared, funding arrangements reviewed; compensation arrangements put in place; all serious or unexpected adverse events reported by the researcher; the trial monitored and reviewed; and information on the trials kept in a durable form to protect privacy. The monitoring of trials and research generally has been a continuing difficulty in Australia (Chalmers 1996).
Again, the new Principles are only a start and further questions remain for consideration for the further development for ethical clinical trials. For example, should the same rules apply where the trial involves an entirely new procedure, e.g., malaria vaccine, where new knowledge is being developed and the risks attaching to long-term effects are quite unknown or unpredictable at this early stage? Should there be different rules for autologous immuno-therapies and certain types of oncological gene therapies where the patients are usually suffering from terminal illnesses? Should there be a separation of drug trials conducted in the public institutions as opposed to those conducted in private institutions? Should special rules apply to trials conducted by the doctors in general practice whose primary duties to the patient may conflict with any research protocol in which the doctor is involved? Should different rules apply where the trial involves blood or tissues on which genetic information is to be gathered? This is not a comprehensive list but illustrative only (Mant 1999)
6.2 Other Matters
Development of a Clinical Trial Register. The report of the Review of the Role and Functioning of Institutional Ethics Committees supported the implementation of a clinical trial register in Australia. The report stated that a national register of statistics and data would enable the effectiveness of particular interventions to be monitored over time and would facilitate the effective monitoring of clinical trial operations. This database will be a useful information resource for HRECs and will reduce duplication of efforts. The proposal has appeared from time to time in the pages of the Medical Journal of Australia and was part of the official submission of the AHEC to the Wills Review (Wills 1999). A central Clinical Trial Register would track the results of all trials, not simply the results that are later published in official journals. In this way the poor as well as the best results would be recorded and a proper assessment of the level of clinical trials could be maintained.
The NHMRC Clinical Trials Centre is an NHMRC funded center at Sydney University, with Professor John Zynes as director. At present it has a voluntary system of registration for cancer research only. The benefits of expanding this role to include all clinical trials would significantly add to community confidence and support for research. Data from these clinical trials would significantly assist the long-term follow-up of participants of clinical trials.
Training. Training and continuing education are key elements in the effort to increase the responsiveness of the ethical review system. The continuing professionalising of HRECs requires the introduction of formal accredited courses. For a number of years the Monash Bioethics Centre ran annual residential seminars for HREC members. In recent years other course providers have advertised in their programs. The AHEC has not begun to formally accredit these courses.
HRECs are becoming increasingly concerned about legal aspects of protocols. Often protocols cross legislative boundaries and HRECs must be sufficiently versed in areas such as privacy, guardianship, and other matters addressed in Commonwealth and State legislation. The AHEC workshops, conducted in 1993, 1995, and 1999 provided a forum for networking and information sharing but should not be seen as substitutes for certified, professionally conducted training programs.
A-37
A major contract was tendered by AHEC for the preparation of a HREC Operating Manual that will consist of explanatory, textual and reference annotations to the National Statement on Ethical Conduct in Research Involving Humans. The HREC Operating Manual is intended as a resource and reference for all members of HRECs, especially new members.
Centralized System of Scientific and Ethical Review for Streamlining Clearance of Multicenter Clinical Trials. There has been an ongoing debate in a range of forums that the scientific assessment of clinical trials be undertaken centrally to streamline the process of review and to assist HRECs in focusing their deliberations on the “ethical” issues of the protocol (Cohen 1998; Clarke 1998; Henman et al. 1998; O’Brien et al. 1998; Gandevia et al. 1998). In effect, the TGA undertakes this “centralized” scientific assessment under the CTX Scheme. This debate has also raised the problem of accreditation of ethics committees. The NHMRC does not currently have authority over State institutions to allow a system of HREC accreditation unless there was the necessary referral of power from the States and Territories to the Commonwealth Parliament.
There have been many debates about a form of centralized approval for research; particularly multicenter research is desirable. Suggestions have ranged from the establishment of a “peak” national HREC to the establishment of regional HRECs akin to the United Kingdom LRECs or the New Zealand Regional Ethics Committees. As a matter of practice, there has been considerable and developing cooperation and collaboration between existing HRECs. The process of ethical review of multicenter trials can become complex and protracted, particularly when a number of centers are involved.
The National Statement proposes two options to streamline the ethical review process for multicenter trials (National Statement 1999, Principles 3.1–3.8). First, when a project is under way, HRECs are permitted to communicate with each other; accept the scientific assessment of another body; adopt the ethical reasoning for another body; or adopt any other procedure from that body to avoid unnecessary duplication (National Statement 1999, Principle 3.4). Second, there is for the first time in Australia a formal system for initially setting up multicenter research. Under this system institutions may agree before the start of the research that “…the primary ethical and scientific assessment be made by one agreed institution or organisation…” (National Statement 1999, Principle 3.5). There have already been some efforts in some regions of Australia to streamline the scientific and ethical review of protocols (Kelly and Boyages 1999).
Any system for centralized HREC decisionmaking must preserve local HRECs. Ethical considerations concerning the safety and scientific validity of a proposal may not differ substantially from one HREC to another; however, there may be important local issues. For example, certain institutions may be involved in research with subjects from a particular ethnic, social, or minority group, which might involve special consideration of local cultural, moral, religious, and/or ethical values. In addition, the particular institutional mission will need to be observed. This consideration would apply, for example, for hospitals of religious affiliation.
Expedited Review and Efficiency. The recommendations elsewhere in this report to introduce expedited review will assist the HRECs in concentrating on approval and monitoring of research projects involving higher risk. Under these procedures a HREC can determine classes of research which may be subject to expedited review and confer authority on the Chair of the HREC to approve the research subject to later ratification by the HREC (National Statement 1999, Principles 2.27–2.29). Expedited review is not suitable for research projects with the potential for harm or where there may be some departure from ethical standards in the Statement. In these cases the full Committee must consider the project.
The Report of the National Council on Bioethics and Human Research in Canada (Canada 1995) encourages Research Ethics Committees considering fewer than 50 research protocols to amalgamate with another or other Research Ethics Committees. In Australia there has been a substantial increase in HREC numbers. There have been suspicions expressed in some submissions that some HRECs may have been established with the researcher interests rather than the subjects in mind. The Canadian approach of amalgamation where a
A-38
Research Ethics Committees considers less than 50 protocols was not included in the final draft of the National Statement. The Second Consultation Draft included a section inviting small HRECs to amalgamate. This was dropped from the final National Statement in the light of submissions received. Provided a HREC was properly and independently constituted, there were good reasons for the continuation of certain specialized HRECs. For example, the National Red Cross HREC considers few protocols but most are complex requiring considerable discussion by the Committee.
Monitoring of Research. Monitoring responsibilities are constrained by resources. Recognizing this, the National Statement has recommended a strategic approach to monitoring where “the frequency and type of monitoring determined by a HREC should reflect the degree of risk to participants in the research project” (National Statement 1999, Principle 2.33). The National Statement includes minimum reporting and proposes that the HREC adopt “…any additional appropriate mechanism for monitoring…” provided that researchers immediately report any “…serious or unexpected adverse effects on participants; changes to the protocol; and unfit foreseen events” (National Statement 1999, Principles 2.36 and 2.37). The National Statement followed the recommendations of the Ministerial Review Committee and the submissions at the Second Stage Consultation. The National Statement did not introduce a system of public monitor-officials as recommended in the United Kingdom (Neuberger 1992) or as operates in the United States with the Office of the Inspector General of the Department of Health and Human Services.
Monitoring by a HREC is only one aspect of the overall strategy for the protection of the interests of research participants. Peer review, institutional supervision, ethical integrity of researchers, and effective information and complaints mechanisms should all be promoted to facilitate the earliest possible detection of potential harm in the course of research projects.
7 The Questions of the National Bioethics Advisory Council
7.1 What Are the Strengths and Weaknesses of Nonregulatory Systems of Protection?
The philosophical debates in bioethics rarely operate in a legislative or legal vacuum (Englehardt 1981; Pellegrino and Thomasma 1996). In most areas debated by bioethicists, governments have played a role either in the form of policy development or legal regulation (Breen 1997; Bennett 1997; Skene 1998; Freckelton and Petersen 1999). As examples, mental institutions have been governed by legislation for over a century; marriage laws have to an extent established rules about reproduction; hospitals are legally regulated and within them research is conducted and resources allocated; euthanasia has remained under the fiat of the criminal law; mass screening was a cornerstone of the public health movement and population genetics and the discredited eugenics movement have, at different times influenced governments. There is established case law in relation to doctrines of informed consent and the duty to warn in the doctor/patient relationship. Where children, the aged, the disabled, or the mentally impaired are treated the rules of consent are varied in the circumstances, the courts have a protective jurisdiction. Specific guardianship legislation may apply also in these circumstances. Finally, debates about artificial conception have led to the introduction of specific status of children legislation and restrictions on experimentation either in the form of legislation or guidelines.
Australia has moved gradually from a self-regulatory system of research ethics review to a more regulated system. HRECs in Australia are not directly established by statute but rather, AHEC was given the responsibility for monitoring and advising on the workings of HRECs (see Section 4.2 of this report). The Australian ethical review system has the following regulatory features:
| n |
The NHMRC is established by Commonwealth Act. The NHMRC is responsible for health and medical research funding, research guidelines and standards setting. (See section 4.1 of this report.) This Act also establishes the Council, the Research Committee and the AHEC. |
A-39
The Australian ethics review system has the following strengths and weaknesses.
7.1.1 Strengths
(a) A National System of Review of Research Ethics. Since the formal decision to establish research ethics committees in 1982, there has been a steady development toward an integrated national system of research ethics review (see Section 2 of this report). HRECs are established within institutions under the oversight and guidance of the AHEC. AHEC is the statutory national apex to the research ethics review system. The Report on IECs (Chalmers 1996) (Schedule 3 of this report provides a summary of recommendations from that report) did not recommend that specific legislation be enacted to regulate HRECs. The report considered that the HREC system was operating satisfactorily under the legislative supervision of AHEC. The report further accepted that the AHEC and the HRECs could adapt to meet future demands on the system.
The development of the national research ethics system was particularly prominent during the 1990s (see Sections 1.2, 2 and 3 of this report). A number of events contributed to the accelerated development of the national research ethics system during this decade. Included in these events were the enactment of the National Health and Medical Research Council Act 1992 (Commonwealth), the establishment of the AHEC under this Act, the Commonwealth Ministerial determination to confer responsibility on AHEC for monitoring and advising on
A-40
HRECs (see Section 4.2 of this report), three rounds of national workshops to consider the operation of HRECs, and the decisions by funding bodies, other than the NHMRC, to require ethics approval for human research projects (see Sections 1.1 and 4.6).
(b) Ownership. The ethics review system was not imposed but rather recognized by government. The system was introduced through the NHMRC and evolved over a number of years, the members of the HRECs and the institutions themselves have developed a sense of ownership and responsibility for the system. The accelerated development toward an integrated national system of ethics review in Australia was driven largely by those involved in the system. The National Health and Medical Research Council Act 1992 (Cth.) gave detailed prescriptions about the composition and operation of the AHEC but left the “…monitoring and advising on HRECs” to be developed by the AHEC in consultation with the HRECs. This sense of ownership was built up during the 1990s in the following ways:
A-41
Institutional Ethics Committees (AHEC 1993) confirmed that institutions had the responsibility to ensure that proper compensation arrangements were in place for research participants and that HREC members were indemnified for decisions made in the course of their work.
| n |
HRECs and Their Advisory Role. The National Statement (National Statement 1999, Principle 2.2) provides that the institution must set up the Terms of Reference for a HREC including the scope of its responsibilities. The HREC therefore advises an institution and is not directed by the AHEC or other organization. In addition, the institution is responsible for adequately resourcing the HREC (Principle 2.1). |
| n |
HREC Membership. HRECs were originally and continue to be established by institutions. Many members of some HRECs have served as members for a number of years. These long-serving members have knowledge, experience, and expertise and are assets to the system. |
| n |
Organizational Developments. One State health authority has appointed public servants to coordinate and facilitate the work of HRECs in their area (NHMRC Annual Report 1997 at 71–73). In addition, some hospitals have developed collaborative networks with other hospitals in their region. These developments were initiated by the States and hospitals themselves with the knowledge of the AHEC. |
| n |
HREC Responsibilities. The AHEC refused to take on the role as final arbiter in ethical review. From time to time during the 1990s the AHEC was called on to give advice to HRECs on difficult ethical research projects or to intervene where there were disagreements about research approval within a HREC. The AHEC consistently declined to act as a final “Court of Appeal.” Rather, the AHEC continued to follow a policy decision made in the first year of its establishment that “AHEC’s role should be to give guidance as to what is ethically relevant (in a particular decision by a HREC) allowing IECs to make their own decisions” (NHMRC Annual Report 1993 at 23). In such cases the AHEC always attempted to provide relevant information but declined to offer an actual opinion in relation to the project. (c) Public Consultation. One of the strengths of the AHEC has been the two-stage statutory public consultation |
requirement (see Section 4.4 of this report). The first-stage consultation operates in the same manner of any other public consultation, namely advertisements are placed seeking submissions on the subject under consideration by the AHEC. The second-stage consultation is conducted in relation to the draft guidelines prepared by AHEC in response to the submissions received at the first-stage consultation. This second stage has the following advantages:
A-42
| n |
Third, with draft guidelines, specific submissions can be requested from specialists in given areas. It has been the experience of AHEC that many experts may not have the time to prepare an extensive submission to a public enquiry but are happy to comment on specific draft guidelines. These specialists are particularly willing to provide specialist information on parts rather than all draft guidelines. |
This second stage has added invariably to the quality of the published guidelines. The National Statement is a very good example of quality improvement. The Working Group at First Stage Consultation was persuaded that it should build on the former Statement on Human Experimentation rather than copying or adopting an available international Code. The draft circulated at the second stage was substantially rewritten in response to the extraordinary number of submissions received. Importantly, the National Statement “…significantly altered many aspects of research involving humans. These changes ranged from research involving deception through to the membership and operating requirements for HRECs” (NHMRC Annual Report 1999 at 70).
Accountability. Although neither Commonwealth nor State legislation create HRECs, there are a number of ways in which the system is publicly accountable (see Section 4.5 of this report).
(d) Accountability. Researchers are at the first tier of ethical review. Researchers must present all publicly funded research for ethics approval. In addition, a substantial amount of privately funded research (e.g., within private hospitals) is also subject to the ethics review system. Almost all funding bodies now require annual progress reports including reports on any difficulty with the ethical conduct of the project. Importantly, the National Statement clarifies the various circumstances in which it is the responsibility of the researcher to report adverse events during the course of the project or to discontinue the research (National Statement 1999, Principles 1.4, 1.15, 1.17, 1.21, 2.35, 2.44, 245 and 12.8). In addition, researchers must avoid conflicts of interest and, in the case of clinical trials, are required to declare any conflict of interest to the HREC as a condition for approval. (National Statement 1999, Principle 12.5) (see Section 1.1 of this report).
HRECs conduct the second level of ethical review and are also accountable in a number of ways within the system. HRECs are advisory and are accountable within the structures of the institution in which they are established (National Statement 1999, Principle 2.2). The HRECs are also required to report annually to the NHMRC (National Statement 1999, Principle 2.48). These HREC reports are consolidated by the AHEC, which then presents a report to the Council, which is later included in the NHMRC Annual Report presented to Parliament (see Section 4.7). The institutions which establish HRECs carry considerable responsibilities under the National Statement. The institution is required to properly resource the HREC (National Statement 1999, Principle 2.1) and must set out the HREC Terms of Reference including the scope of its responsibilities (National Statement 1999, Principle 2.2). The institution must accept legal responsibility for decisions and advice received from the HREC and indemnify its members (National Statement 1999, Principle 2.3). The institution should ensure that adequate compensation arrangements are in place for research conducted under its supervision. The institution is also required to set up proper complaints handling mechanisms for receiving and promptly dealing with complaints and concerns about the conduct of an approved research project (National Statement 1999, Principles 2.39–2.43).
The AHEC constitutes the third tier in the review system. It was the express intention of the Commonwealth Parliament, particularly the Senate, to ensure that the NHMRC was an open and accountable public institution. The openness and transparency of the AHEC processes to public scrutiny arise from the following:
| n |
A Federal Court decision, Tobacco Industry Australia v National Health and Medical Research Council, has confirmed that the AHEC is required to have “regard” to all submissions and must pay “positive consideration” to those submissions by all members of the AHEC (this ruling applies equally to all other committees and parts of the NHMRC). |
A-43
| n |
All proceedings, including submissions to the AHEC during the process of public consultation, are public documents and obtainable under Freedom of Information legislation (this does not apply when the submission is marked “confidential”). |
| n |
A Complaints Commissioner has been appointed under the terms of the National Health and Medical Research Council Act. The Commissioner may hear complaints in relation to any of the operations of NHMRC. In fact, to date the small number of complaints have consisted of requests for review of decisions by the Research Committee, which is responsible for funding applications for research grants. No complaint has ever been lodged in relation to the work of the AHEC. Realistically, complaints about research or research outcomes are more likely to be referred to the institution or to the HREC directly. In fact, under the National Statement formal complaints structures must be introduced by every institution establishing a HREC or handling complaints (National Statement 1999, Principles 2.3 9–2.43). |
| n |
The AHEC has been ready to provide public information and presentations about any reference before it and is willing to engage in debate on wider issues. The NHMRC has a media officer to handle relations with the media, and organizations tend to approach the AHEC directly. In 1998, at the height of the preparation of the National Statement, approximately 200 speeches, radio interviews, or major national newspaper interviews were conducted by the Chair or other members of the Committee. |
| n |
The AHEC as with other Committees of the NHMRC, is required to prepare an Annual Report, which is included in the overall NHMRC Annual Report that is laid before the Commonwealth Parliament. |
| n |
The AHEC is established under Commonwealth legislation and is subject to the investigatory powers of the Commonwealth Parliament. As a statutory authority, the AHEC is open to interrogation by the Committees of Commonwealth Parliament. The Senate Estimates Committee has interrogated the senior Secretariat of the AHEC and of the NHMRC in relation to its activities (see Section 4.5 of this report). |
| n |
AHEC is a statutory body within the portfolio of the Commonwealth Minister for Health and Aged Care. As such, the Minister may be questioned in Parliament in relation to the activities of the AHEC or the HRECs. (e) National Guidelines. Under the terms of § 8(1) and (2) of the National Health and Medical Research Council |
Act 1992 (Cth.), the AHEC has sole responsibility for the development of guidelines for the ethical conduct of medical research. This authority combined with the two-stage consultation process has resulted in the production of the series of guidelines with national application. In a federal system, it is difficult to achieve uniformity in legislation and policy in some areas within State and Territory authority. Similarly, uniformity in guidelines is more difficult and elusive in a largely self-regulatory medical research environment. During the early period of efforts by the NHMRC, through the Medical Research Ethics Committee, to establish a national ethics system, many organizations produced guidelines. The NHMRC had an influential but not exclusive function in producing guidelines for health and medical research. Guidelines were frequently published by a variety of funding authorities, medical colleges, and associations. It is difficult to gainsay the importance of the work by the NHMRC in moving toward national uniform guidelines. This process was finally realized and consolidated by the National Health and Medical Research Council Act. Two examples may assist in illustrating the strengths of having a central national committee with authority to publish national guidelines:
| n |
Assisted Reproductive Technology. The Australian competence in the science of reproductive technology was not matched with equal competence in its regulation. Australian governments produced a Babel of reports in the area (Waller 1982–1984; Demack 1984; Chalmers 1985; Cornwall 1984; Michael 1986; NSW Law Reform Commission 1980–1989; Family Law Council 1985; Senate Select Committee 1986). The reproductive |
A-44
technology debate in Australia as elsewhere raised fundamental social, ethical, and legal questions about the very essence of personhood and humanness; the debate saw the clash of science and religion. There was considerable uniformity in the various Commonwealth, State, and Territory reports with respect to Status of Children (Status of Children Act 1978 (Qld)); Access to Programmes; Keeping of, and Access to, Information and Records; Counselling; Use of Donor Gametes; and Surrogate Motherhood.
In two major areas, there were substantial differences in the conclusions in the reports. These were Research and Experimentation on Embryos and Control and Regulation. Three States in Australia introduced committees to deal with decisions in the area of reproductive technology. These States were in order, Victoria, South Australia, and Western Australia. The Victorian Parliament passed the Infertility Treatment Act 1995 (successor to the Infertility [Medical Procedures] Act 1984), but the Act was not proclaimed for some years afterwards. The relevant legislation in South Australia is the Reproductive Technology Act 1988 and in Western Australia, the Artificial Conception Act 1985.
When the AHEC was set up in 1992, a reference was reserved by the Commonwealth Senate that required the AHEC to consider the publication of guidelines in the area of reproductive technology. The NHMRC published specific guidelines entitled the Ethical Guidelines on Assisted Reproductive Technology 1996 (AHEC 1996). These Guidelines applied uniformly and were later accepted by the Reproductive Technology Accreditation Committee (RTAC). The RTAC is a voluntary organization funded by the Fertility Society of Australia, which accredits centers offering such services. Once the RTAC accepted the AHEC Ethical Guidelines on Assisted Reproductive Technology, they formed part of its Code of Practice for centers using IVF and related reproductive technologies.
In effect, therefore, the nonlegislation States were practically and uniformly covered by the AHEC
Guidelines.The Reproductive Technology Councils in South Australia and Western Australia also approved the AHEC Ethical Guidelines on Assisted Reproductive Technology, thus achieving new uniformity in approach to research in the area.
| n |
National Research Guidelines. The National Statement on Ethical Conduct in Research Involving Humans has been endorsed by the other major public research funding organizations, the Australian Research Council, the Australian Vice Chancellors’ Committee representing all universities, and the Learned Academies (the Australian Academy of Humanities; the Australian Academy of Science; and the Academy of the Social Sciences in Australia and supported by the Academy of Technological Sciences and Engineering. In addition, a number of other associations are in the process of replacing their particular guidelines with the National Statement. This has been a most significant advance in the path toward uniformity in guideline development. (f) A National Committee. The AHEC also has a representative function for Australian medical research ethics |
in overseas forums. Following the initial invitation of NBAC, the Summit of National Bioethics Commissions was convened in San Francisco in 1996 and again in Tokyo in 1998. Many countries have appointed national bioethics commissions, although there is far from being comparability in jurisdiction, terms of reference, resourcing, status, and guidelines. The meeting in Tokyo agreed that there were matters of common interest between the various commissions. In particular, it was noted that clinical trials (discussed elsewhere in this report) were an area likely to command public international attention. Developments in the last two years have proved this view to be prophetic. The issue of clinical trials has commanded further public attention with the debates within the World Medical Association to revise the current wording of the Declaration of Helsinki. The amendments proposed by the American Medical Association would include a new Article 18, Access to Health Care, in the following terms:
A-45
In any biomedical research protocol, every patient-subject, including those of a control group, if any, should be assured that he or she will not be denied access to the best programme, diagnostic, prophylactic therapeutic method which would otherwise be available to him or her. This principle does not exclude the use of a placebo on non-treatment control groups with such a justified or scientifically and ethically sound research protocol.
Arguably, the proposed changes to wording may lead to “ethical export” where developing countries may be used for the conduct of clinical trials where lesser ethical standards are applied than in developed countries (Nuffield Council on Bioethics 1999; Healy 1999; Bulletin of Medical Ethics 1999b). This would not replace but complement the work which is currently under way with the development of international standards represented in the CPNC/ICH Note for Guidance on Good Clinical Practice (135–95).
The AHEC has, on behalf of the NHMRC, sent comments to the World Medical Association consultation. Equally, national bioethics commissions are in the position to liaise with other national bodies to provide information to contribute to the development of improved ethical trials.
7.1.2 Weaknesses
A number of weaknesses can be identified within the current ethical review system in Australia as follows:
(a) Enforcement. § 8(1)(ii) of the National Health and Medical Research Council Act authorizes the AHEC to develop medical research guidelines and for the Council to issue those guidelines in the form developed by the AHEC. Infringement of any Principle in the National Guidelines does not constitute a prosecutable legal offence. The sanctions for infringement of the Principles involve the loss of access to or withdrawal of research funds. In practice, this has been threatened on a number of occasions and is treated most seriously by institutions. For example one major metropolitan hospital was noncompliant for part of a year of report. Senior officers from the hospital were granted time to reconsider and ratify noncompliant decisions by the HREC. This particular incident resulted in the review of the sanction procedures of the NHMRC. In particular, a “show cause” opportunity was introduced into the procedures. In another example a major national research institute is required to reconvene with a compliant HREC and reconsider de novo decisions dealing with a noncompliant period. With the statutory requirement for the NHMRC to report annually to Parliament, the NHMRC could name guideline infringers in the report tabled before the Parliament (this has never been done to date).
At one time there was a deal of criticism of the NHMRC for being “in-house” and lacking any “teeth” to prosecute. In defence of the NHMRC, this view confuses police-style prosecutions for anti-social criminal behaviour with the promotion and maintenance of ethical standards in an otherwise orderly research community. It is the difference between as police person patrolling on the assumption that crime is breaking out as opposed to the fire service, which attends when the unexpected fire breaks out (Chalmers and Petit 1998). It is the latter analogy that is more applicable to health and medical research. Nevertheless, the enforceability question is raised frequently by the medical and in the public forum.
(b) Uniformity and Complimentarity. In some areas the AHEC has produced national guidelines with national remit. In other areas, the guidelines have not applied uniformly. For example, as noted above the Ethical Guidelines on Assisted Reproductive Technology form a de facto national code in all States except Victoria, where the Infertility Treatment Act 1999 (Vic) overrides the Ethical Guidelines. However, the legislation in the three States (Victoria, Western Australia, and South Australia) have different provisions in relation to human cloning. This will be a barrier to uniform legislation or AHEC guidelines.
In late 1997 and with the benefit of the substantial work done by NBAC (NBAC 1997), the Commonwealth Minister for Health and Aged Care requested a report on cloning from the AHEC. The issue of human cloning was not confined to ethical questions; the issue overlapped substantially with existing regulations in three
A-46
States. The report from the AHEC (AHEC 1998) has now been referred on for consideration by the
Commonwealth House of Representatives Standing Committee on Constitutional and Legal Affairs with a view to introducing uniform or complementary regulation. This lengthy and complex process may be seen as a weakness in the AHEC structure and authority with respect to guidelines. On the other hand the AHEC is essentially advisory only when requested to give a report to a Commonwealth Minister. Admittedly, guidelines would suffer the same lack of force in three States with legislation. In recognition of this, the AHEC produced a recommendation that the Parliament consider legislation. An extract from Chapter 4 of the AHEC Report is included in Schedule 3 to illustrate this jurisdiction limitation in relation to legislation and guidelines relevant to cloning in Australia at the relevant period.
(c) Private Institutions. As a matter of law the provisions of the National Health and Medical Research Council Act 1992 (Cth.) do not apply directly to privately funded research (see also comments in Section 7.3 below). So far Australian private institutions have generally complied with NHMRC and other public standards. Some of these institutions informed the AHEC (in the consultation process for the National Statement) that compliance was observed because NHMRC guidelines represented best practice; private institutions were conscious of avoiding possible negligence claims, and all universities, the AVCC, the ARC, and all the Learned Academies had endorsed the National Statement.
Nevertheless, the AHEC recognized in its Report on Cloning (AHEC 1998) that commercial pressures are increasing in this country, and there is no guarantee that the current regulatory and part self-regulatory system of self-restraint will continue. Certainly, in the case of human cloning, it was considered for ethical and commercial reasons that uniform national legislation was required to bolster existing guidelines.
(d) Second-Stage Consultation. The second-stage consultation process has proved to be a lengthy and costly exercise. The AHEC has profited from the quality and depth of input at the second stage consultation. However, other principal committees of the NHMRC, especially the Health Advisory Committee (HAC), have questioned the value of the process. Many of the reports prepared by the HAC are developed in draft by other major specialist health organizations, and the second-stage consultation is of less value as the specialist input has already been given. For example, the HAC received a report from the Victorian Anti-Cancer Council on Familial Cancers. This report had been prepared over a period of three years and involved the Australian Cancer Network. One stage of consultation was arguably sufficient to inform the public and seek their views on a complex and technical area. In fact, two stages had to be conducted under the terms of the NHMRC Act. In fact very few submissions were received at the second stage.
The NHMRC decided in 1999 to propose amendments to its Act to allow the possibility of one-stage public consultation in most cases rather than exceptional cases. One-stage consultation was previously permitted in exceptional cases under the NHMRC Act 1992 (Cth.). The amendments to the Act were passed by the Commonwealth Parliament in 1999 (NHMRC Annual Report 1999 at 9). The AHEC is most likely to continue to apply the full two stages of public consultation.
7.2 What Features of These Systems, If Any, Should Be Incorporated in the U.S. System?
At a general level, there is much commonality between the research community in basic ethical principles. There would be little dispute that among the essential values for research is the integrity of the researchers. The Australian National Statement did not invoke any autochthonous principles but referred to the classic U.S. Belmont Report for a statement of the three basic ethical principles for the ethical evaluation of human action (Belmont 1979). These are respect for the person, beneficence, and justice (Beauchamp and Childress 1994; National Statement 1999 at 4). On the other hand, institutions are not so easily transplanted. Committee structure, which operates successfully with refinements, subtleties, and technicalities, may not be suited to
A-47
the conditions of another country. Adaptation and pruning will always be required (Nyali Ltd. v the Attorney-General per Lord Denning at 16–17).
With the cautionary remark about ethical institutional transplants, the following features of the Australian system may be worthy of some consideration by the members of NBAC.
(a) A National Committee/Commission. It may seem inconceivable to the international ethics community that the engine-room of modern biomedical research does not have a permanent standing committee considering ethical issues. The reports of the present NBAC, like the Belmont Report (Belmont 1979), remain profound reference points and rich sources for ethical discussion. NBAC contributed significantly to the global debate with its report on Cloning of Human Beings.
There is a lacuna if the NBAC or some other appropriate nationally based ethics body is not operating to organize and encourage the development of international collaboration between national bioethics commissions. NBAC has already fulfilled this role with distinction at the inaugural meeting in San Francisco and the second meeting two years later in Tokyo in 1998. Obviously, NBAC or an equivalent body would be concerned principally with the preparation of national guidelines, reports, or advice on specific matters.
Nevertheless, relations with other national bioethics commissions can be a smaller but highly important roles for a national body. The AHEC has devoted a small but not insignificant percentage of its time dealing with other nations’ bioethics commissions. In fact, many of these dealings have involved the collection of reports of documents or seeking advice on specific regulations, guidelines, or procedures from a national bioethics commission.
(b) Reporting to Parliament. Under the terms of the National Health and Medical Research Council Act 1992 (Cth.) the NHMRC is required to prepare a plan of work which is presented to the Parliament. In each subsequent year the NHMRC including the AHEC present a report to Parliament. This not only provides an essential and important line of accountability; it requires the NHMRC and AHEC in particular to establish work programs to complete reports in a timely and orderly fashion. As both the Strategic Plans and Annual Reports are presented to Parliament they form public documents which are accessible to the public and interested bodies. The process of reporting to Parliament is recognition of the status of the NHMRC and AHEC.
(c) Public Consultation. The two-stage public consultation has been a complex and weildly process. Nevertheless, it has provided an authentic and transparent opportunity for public comment and for that comment to be integrated into the body of the report and guidelines. As noted earlier in this report the second-stage consultation where the draft guidelines are presented for comment has proved to be successful. At this stage, detailed comments on the specific draft guidelines have invariably led to improvement in the content as well as the wording of the final guidelines. Some 200 submissions were received at each of the stages of consultation for the National Statement on Ethical Conduct in Research Involving Humans. In a small population of 20 million this number may be magnified so much in the more populous United States as to present very considerable challenges to the management of the information presented.
(d) Aspects of the National Statement. NBAC may wish to consider the current principles in the National Statement in relation to epidemiological research, human tissue, and genetic research, which are noted in Section 5.3 of this report. These particular Principles are internally consistent and may offer a modest contribution in these difficult areas.
A-48
7.3 What Are the Strengths and Weaknesses of Models That Are Comprehensive, Those That Encompass Private and Government Sectors, and Nonbiomedical and Biomedical Research?
Fears of High Medical Dominance by Nonbiomedical Researchers. During the period of the Ministerial Review (the IEC Report) and also during the consultation for the National Statement, comments were made and submissions received expressing concerns that some forms of social science research were not appropriate for consideration by HRECs. In essence, many of these concerns centered on the composition of the pre-National Statement HRECs. Until recently, the former Statement on Human Experimentation required a medical graduate as one of the core members. Under the terms of the new National Statement a HREC should be composed of a person with experience in the research considered by the Committee. This has removed some of the concerns. Nevertheless, there has been in Australia for a number of years some tension between the nonbiomedical and biomedical researchers. It is too early to tell whether the comprehensive revisions in the new National Statement will assuage these concerns.
Creating a Universal Research Culture. The consensus of opinion supported the move to establish a single National Statement as a means to achieving the goal of a universal research culture in this country. Universities in submissions to the public consultation particularly promoted this universal view for the new National Statement. In particular, these submissions stressed the continuing blurring of distinctions between private and publicly funded research and growing of distinctions between medical, health, health-related, and social science research. Many submissions noted that Australia, in line with other countries, was developing research policies to encourage private investment in research. For this and other reasons, it was more appropriate to consider a single research code. Similarly, a researcher has a number of common obligations and ethical duties to the research participant, which are common to research generally.
That Research Can No Longer Be Assumed to Be of Value to the Community. There is an assumption expressed in the new National Statement that the development of the recognition of human rights and the ethical standards of respect of persons preclude conducting research without the knowledge and voluntary consent of the participant. In this respect, an assumption can no longer be made validly that research is automatically a value to the community. Research, whether privately or publicly funded and whether nonbiomedical or biomedical, must be disclosed to the research participants. The National Statement requires disclosure, information, and voluntary consent. More critically, the Preamble recognizes that the researcher is required to justify the research and that the community expects that research will be conducted in an equitable, professional, and ethical fashion.
Risk Minimization. The idea of expanded human rights protections in the late 20th century extends far beyond the protection of the physical body of the individual. The doctrines of human rights extend to rights to the protections of law, rights of freedom of speech, rights to nondiscrimination, and equitable treatment as examples. In this sense, the ethical and legal requirements for the respect for persons extends to respecting the privacy of the individual as well as the bodily protection. The National Statement throughout places responsibilities on researchers and HRECs to ensure that risk is minimized and that if risk exists there is a careful balancing of those risks against the potential benefits to be gained within the research project.
International Research. Australia conducts research outside of its national borders. The National Statement places responsibilities on researchers to conform not only to the standards within the National Statement but to also conform to any local ethical standards in the country in which the research is conducted. With more research being conducted as part of international multicenter trials, the National Statement recognizes that there are national responsibilities to regulate and supervise research conducted outside Australian borders in overseas countries. The existence of a comprehensive National Statement conveys clearly to all researchers be they non-biomedical or biomedical that the high standards of research integrity expected of researchers conducting research in Australia applies equally to overseas research. There is a responsibility on national governments in
A-49
their international relations to maintain appropriates standards. In this respect the recognition that trade and commerce standards probably extend to aspects of international research.
Private Institutions. There are no compulsory or mandatory powers in the National Health and Medical Research Council Act or in the AHEC to make private institutions comply with the standards of ethical review. The Australian research review system is essentially compulsory in the public arena. Major public institutions including universities and hospitals and research centers have endorsed the National Statement. These bodies recognize that funding from the major public funding organizations (NHMRC and ARC) require approval by a HREC. On the other hand, private companies are essentially complying voluntarily. If they wish to access public funds they are required to comply. In addition, many private companies comply because they are conducting the research in public institutions. Finally, many private companies comply because approval by a registered HREC is considered a prudent step in reducing risks of complaints or possible litigation. As there is an approved national standard for ethical approval from a registered HREC many private companies use the HREC system to ensure that in the event of misadventure a failure to receive ethics clearance would not be seen as a negligent act.
The National Statement applies to de facto private institutions for the following reasons:
As stated, in practice, many private institutions follow the principles of the National Statement. It is not clear at this early stage whether this voluntary compliance will continue. It is equally unclear whether the increasing movement toward greater private funding of research will affect this process of voluntary compliance. For example, will private funders expect ethics clearance as part of the “service” provided by the research organization? If the HREC refuses clearance will the private funder simply go to the “market” and seek approval elsewhere? In practice, privately funded clinical research under the national CTN scheme has not followed this path. In practice, the institution conducting the research has required clearance from its own HREC. No “market” in ethics approval has arisen.
A-50
Schedule 1
Report of the Review of the Role and Functioning of Institutional Ethics Committees AGPS Canberra (1996) Summary of Recommendations
1. To National Health and Medical Research Council
The NHMRC in conjunction with other peak bodies responsible for research and clinical practice (Australian Research Council, Australian Vice-Chancellors’ Committee, Australian Medical Council) should promulgate guidelines representing a national statement for the ethical conduct of research. Recommendation 5.2.2 The Review Committee endorses the moves by the NHMRC to implement a clinical trials register in Australia. Recommendation 5.6.1
2. To Australian Health Ethics Committee
AHEC should redraft the Statement on Human Experimentation and change its title so that all health investigation involving humans (including nonbiomedical research and innovative practice) is encompassed.
Recommendation 5.3.1
AHEC should re-draft the Statement on Human Experimentation to include reference to research on distinct cultural groups to the effect that these groups have specific needs that must be addressed. In particular, the guidelines should address the need for an IEC to:
AHEC should re-draft the Statement on Human Experimentation to:
The redrafted Statement should cover all research on humans and not be restricted to NHMRC-funded research. Recommendation 6.1.3 To improve communication and networking between IECs generally and in particular in relation to multi-center trials, AHEC should prepare an IEC directory which includes the names and contact addresses for the Chairs and Secretaries of all Australian IECs. Recommendation 5.5.4 The annual IEC compliance report to AHEC should require details of monitoring arrangements for high risk projects. Recommendation 5.7.3 A checklist for researchers detailing the requirements for the collection and storage of research data and results should be developed by AHEC, and IECs should be made responsible for monitoring compliance with the checklist on privacy guidelines. Recommendation 5.8.2 AHEC should coordinate the preparation of a national standard form of Application for Approval of a research project before an IEC. Recommendation 6.4.1
A-51
AHEC should supervise the preparation of a Manual of Procedures for IECs following the completion of the re-drafting of the Statement on Human Experimentation and Supplementary Notes, and AHEC should be allocated adequate resources to fund this project. Recommendation 6.5 AHEC should maintain a clearinghouse function, and be responsible for coordinating, collecting, and disseminating information as well as monitoring IECs in line with its statutory requirements. As well, education of IECs researchers and institutions should form a part of the role of AHEC. Recommendation 7.3.1 AHEC should be funded for the appointment of an IEC officer. This officer is required as a matter of priority to coordinate the development of a resource kit (educational package) for ethics committees. Following the development of the kit this officer should remain responsible for ongoing duties relating to the administration and education of IECs. Recommendation 7.3.2 AHEC through its Research Ethics Working Committee should identify appropriate stakeholders in the ethics committee system and consider appropriate means to facilitate their contribution to the system.
Recommendation 8.2
AHEC should examine the issue of appropriate levels of administration fees for IEC approval. Recommendation 8.5
AHEC should revise its current compliance information form to include the following information from IECs:
| n |
Membership details |
|
| n |
Number of meetings |
|
| n |
Confirmation of full participation by minimum required members |
|
| n |
Confirmation of due procedures |
|
| n |
record of decisions has been kept |
|
| n |
promulgate procedures and ensure they have been followed |
|
| n |
number of rejections and reasons for rejections/amendments |
|
| n |
monitoring procedures in place and any problems encountered |
|
| n |
no member had an apparent or actual conflict of interest |
|
| n |
no financial profit by members |
|
| n |
Complaint procedures, number of complaints handled |
|
| n |
That an annual report has been produced. Recommendation 9.1.2 3. To Institutional Ethics Committees |
|
Institutional Ethics Committees which do not consider more than 50 research protocols should consider amalgamating their IEC with another IEC or IECs. Recommendation 5.5.1 The Review Committee does not recommend the establishment of regional Institutional Ethics Committees. Recommendation 5.5.2 Institutional Ethics Committees should consider procedures for improving the consideration of multi centre research protocols such as communication between chairs of IECs and the acceptance of another IECs scientific assessment of a project where appropriate. Recommendation 5.5.3 An IEC has the responsibility when approving a research protocol to ensure that appropriate and adequate monitoring arrangements are in place consistent with the level of risk involved in the project to research subjects. Recommendation 5.7.1
A-52
An IEC must ensure that appropriate and adequate procedures for monitoring are in place prior to the commencement of the project. Recommendation 5.7.2 An IEC should put in place good administrative and record keeping practices. Recommendation 6.1.1 Where an IEC has grounds for concern about a research protocol, the IEC should initiate consultation with the researcher, and where a protocol is rejected by an IEC, reasons for the rejection should be recorded and made available to the researcher. Where a researcher is unhappy with the decision the complaint should be referred to the institution. Recommendation 6.1.2 An IEC should consider the introduction of a system of expedited of review allowing IECs to grant approval to research projects not involving significant risk to the research subjects. Such expedited review have the following features:
Institutional Ethics Committees should not approve a research project unless they are satisfied that an acceptable Consent Form will be administered to the subjects of the research project. Recommendation 6.4.3 An IEC should have in place appropriate grievance/complaints procedures for participants and these procedures should be included as part of an information sheet provided prior to involvement in the research. This information should include both internal and external contact names and numbers of available participant advisors. Recommendation 6.6 IECs should produce an annual report or contribute to the annual report of their institution. This report should include the compliance information forwarded to AHEC and a listing of all research approved by the committee. Recommendation 9.1.1
4. To Institutions Which Have an Established IEC
An institution should appoint members to the IEC with attention to the following:
| (a) |
The selection of members. The selection of members should be subject to advertising and an open selection process. The selection process may vary between institutions; however the institution is responsible for recording details of the process. |
| (b) |
Attributes of members. In addition to their particular knowledge/skills, all members should have good judgment, the ability to function in a committee, and a commitment to the research subject. |
| (c) |
Independence of the IEC from the institution. The committee must be capable of acting independently. The ethics committee should be considered a part of, but independent within, the institution, performing an advisory function for the institution. Recommendation 7.1.1 |
A-53
An institution should maintain its IEC with the following minimum required membership:
| n |
Chairperson |
| n |
Person with knowledge of and experience in research involving humans (medical, social, epidemiological, as appropriate) |
| n |
Medical practitioner with current/previous involvement with direct patient care |
| n |
Minister of religion or equivalent (e.g., aboriginal elder) |
| n |
Layman |
| n |
Laywoman |
| n |
Lawyer and, in the case of a hospital IEC |
| n |
Nurse Recommendation 7.1.2 An institution should promulgate the following additional guidelines for the operation of their IEC: 1. Due regard should be paid to age and gender balance of committee representation. 2. Due regard should be paid to the appointment of lay members with appropriate ethnic backgrounds where the research reviewed by the committee is predominantly focused on a particular ethnic group. 3. Members will not fill more than one category. 4. The responsible institution (university, hospital) will formally appoint members of the IEC after receiving appropriate advice. The members should receive a formal notice of appointment which includes a guarantee that the institution will provide legal protection for the member. 5. The duration of membership should be determined by the relevant institution. It is desirable, however, that the members are appointed for an appropriate period to allow the members to acquire and apply new ethical knowledge and decision-making skills. A period of between three and five years is suggested. 6. Where additional members are appointed an appropriate balance between institutional/non-institutional -medical/non-medical must be maintained. Specifically, not less than half the committee should consist of non-institutional, non-medical members. 7. The 7 (8) required members must participate in all decisions (NB it is not necessary for all required members to be present at all meetings, however, all should be involved in the decision-making process). 8. With regard to participant representation it is the view of the Committee that no one person could be representative of all participant groups. All IEC members are appointed to represent participants in research. Consequently, it is the objective of all committee members to use their particular knowledge/skills to anticipate the rights, needs, and expectations of participants. As a result there should be no need for a separate patient advocate or participant representative on the committee. Recommendation 7.1.3 |
Members of an IEC should be reimbursed for expenses incurred in the conduct of their duty (e.g., parking, additional child care expenses) but should not ordinarily receive a fee for service. In exceptional circumstances a fee for service may be appropriate; however, care should be taken to ensure that this does not result in an apparent or actual conflict of interest for the member(s) concerned. Recommendation 7.2 An institution should make available sufficient (ongoing) funding to enable its IEC members to avail of opportunities leading to improved performance of the IEC (e.g., attendance at seminars/conferences; support for IEC network meetings). Recommendation 7.3.3
A-54
Each institution is responsible for ensuring that adequate resources are made available to its IEC for the assessment and ongoing monitoring of approved research protocols. Recommendation 8.1 An institution should not establish an IEC unless the institution can assure AHEC that there are adequate means for resourcing the committee. Recommendation 8.3
5. To Researchers
The UK, MRC distinction between innovative therapy/treatment and research should be adopted by AHEC and the Statement on Human Experimentation modified to reflect that the systematic use of an innovative treatment or therapy be considered as research and consequently be subject to assessment by an IEC.
| (a) |
Where a particular experimental treatment/intervention is expected to benefit an individual patient it may be considered as innovative practice rather than research. Where this is the case, the treatment should be governed by doctor/patient ethics considerations. |
| (b) |
Where any innovative therapy/intervention undergoes systematic investigation (i.e., is trialed on a number of patients) it should be subject to the same ethical assessment as any research protocol. Recommendation 5.2.1 |
Researchers should endeavour to simplify all Consent Forms for research subjects and should aim to achieve a form of words which is understandable by a student with Grade 8 schooling. Recommendation 6.4.2
6. Further Recommendations
Funded positions should be created in each State for an “area liaison” officer whose duties will involve coordination of liaison between AHEC and IECs and fostering communication/networking between IECs. Recommendation 8.4
Schedule 2
AHEC Chair’s Report for the NHMRC Annual Report 1999
I am pleased to report that 1999 has been a very productive year for the Australian Health Ethics Committee (AHEC). Some significant documents have been finalised by AHEC namely the National Statement on Ethical Conduct in Research Involving Humans, Guidelines for Genetic Registers and Associated Genetic Material, and Guidelines for Ethical Review of Research Proposals for Human Somatic Cell Gene Therapy. A number of other documents are close to being completed. One of the highlights for AHEC in 1999 was its organisation of the Ethical, Legal and Social Implications program of the prestigious Human Genome Organisation meeting held in Brisbane.
Objective IV of the NHMRC Strategic Plan 1997–2000. ‘To continue to provide high quality ethical advice with respect to health research and health care’ concerns the Australian Health Ethics Committee. The documents produced by AHEC in 1999 will allow Council to continue to provide high quality advice about health from an ethics perspective.
Research Standards/Protection of Research Participants
To support a strong and well-managed research sector, the Australian Health Ethics Committee completed its revision of guidelines relating to the ethical conduct of research. The National Statement on Ethical Conduct in Research Involving Humans was presented to NHMRC in June 1999, following an intensive period of development.
The National Statement was developed by the Australian Health Ethics Committee and endorsed by the Australian Vice-Chancellors’ Committee, the Australian Research Council, the Australian Academy of the Humanities, the Australian Academy of Science and the Academy of the Social Sciences in Australia. The
A-55
Academy of Technological Sciences and Engineering also gave the National Statement its support, as did the Ministers for Health and Aged Care, Industry, Science and Resources, and Education and Youth Affairs.
The significance of this level of support for the National Statement should not be underestimated, as it will ensure a very high standard of protection for participants in all areas of research. All research involving human participants conducted in Australian universities, funded by NHMRC or the Australian Research Council, or involving the learned academies, will now have to be conducted in accordance with these guidelines.
National Workshops
In August 1999, the National Statement was the focus of a series of workshops convened in the capital cities of each State and Territory, and including Alice Springs. These workshops were designed to facilitate the use and understanding of the National Statement by those directly responsible for the maintenance of ethical standards of research in Australia. They were attended collectively by approximately 1,000 representatives of Human Research Ethics Committees from around the country.
Human Genetics
A further major achievement for AHEC has been the finalisation of two guidelines in the field of genetics: Guidelines for Genetic Registers and Associated Genetic Material and Guidelines for Ethical Review of Research Proposals for Human Somatic Cell Gene Therapy and Related Therapies.
Guidelines for Genetic Registers and Associated Genetic Material covers all aspects of register operation and provides guidelines in such difficult areas as gathering, using and releasing register data and associated genetic material; recruiting people to genetic registers and obtaining their consent; and security and storage of genetic material. The revised document has a wider focus than the original guidelines.
Human somatic cell gene therapy remains experimental. Guidelines for Ethical Review of Research Proposals for Human Somatic Cell Gene Therapy and Related Therapies provides guidance to Human Research Ethics Committees that are asked to review and approve research proposals involving somatic cell gene therapy, and assists researchers to prepare their submissions for ethical review. The document identifies bodies other than Human Research Ethics Committees from which approval may need to be obtained. An information paper on human somatic cell gene therapy, that provides background information to the Guidelines, is included with the Guidelines.
A third genetics document is expected to be finalised early in 2000. Ethical Aspects of Human Genetic Testing—an information paper addresses issues of equity, access and resource allocation; commercialisation; geneticisation; counselling; and genetic testing of children. Although not formal guidelines, this information paper has been the subject of wide consultation—a feature which has strengthened the document.
Genetics is an ever-changing field of research and the guidance and guidelines developed by AHEC will play a crucial role in protecting individuals whilst encouraging a high standard of research.
Human Research Ethics Committees
Compliance by Human Research Ethics Committees with NHMRC ethics guidelines is reported annually to the Research Committee and NHMRC. This process ensures consistent application of the guidelines as well as providing an auditing mechanism to support quality research.
In 1999, AHEC continued to provide support to Human Research Ethics Committees by acting as a focal point for queries and concerns as well as preparing guidelines on issues that are likely to be raised during the conduct of research. A major thrust to this end was the 1999 Workshop series which introduced the new National Statement and gave representatives from the research, academic and HREC sectors an opportunity to discuss issues of concern.
AHEC is developing an operating manual for Human Research Ethics Committees, which is expected to be finalised in 2000. When completed, the manual will form a “how to” guide addressing common questions and
A-56
providing procedural advice on the application of the National Statement on Ethical Conduct in Research Involving Humans.
Section 95 Privacy Guidelines
Stage two of the public consultation process for the privacy guidelines was conducted in 1999. The Privacy Act 1988 (Commonwealth) authorises the NHMRC to issue guidelines for the protection of privacy in the conduct of medical research. The Federal Privacy Commissioner is also involved in this process. The existing guidelines, Aspects of Privacy in Medical Research, were issued in 1995.
The revision of these guidelines is a result of a number of changes in the environment in which the guidelines operate, namely the introduction of the NHMRC Act 1992 and the National Statement on Ethical Conduct in Research Involving Humans, and developments in privacy regulation.
The guidelines provide a framework in which medical research involving personal information obtained from Commonwealth agencies should be conducted, to ensure that such information is protected against unauthorised collection or disclosure.
The revised Guidelines under Section 95 of the Privacy Act were developed in collaboration with the Federal Privacy Commissioner. Two stages of public consultation were conducted as required by the NHMRC Act, and AHEC endorsed the revised guidelines at its November 1999 meeting. They will be tabled at Council and in the Federal Parliament in early 2000.
Aboriginal and Torres Strait Islander Guidelines
AHEC has reaffirmed its commitment to the protection of Indigenous Australians participating in research by planning a revision of the ‘Interim guidelines for ethical matters in Aboriginal and Torres Strait Islander health research.’ Recognising that the revision must be a transparent and inclusive process, AHEC is committed to full consultation.
Ethical, Legal and Social Implications Program
AHEC organised the Ethical, Legal and Social Implications (ELSI) program of the Human Genome Organisation’s 1999 meeting. The meeting was a vehicle by which AHEC was able to showcase its own work, as well as contribute to the national and international debate on ethical issues.
The ELSI program included a debate, chaired by the Hon. Justice Michael Kirby, that “Too much is expected of human genetics research and the human genome project.” It was judged a great success by participants.
Three workshops were chaired by AHEC members and were part of the ELSI program. These were: ‘Commercialisation and benefit-sharing’; ‘Religious and cultural perspectives in contemporary genetics’; and ‘Genetic susceptibility testing.’ The financial and intellectual contributions made by the Australian Health Ethics Committee were duly acknowledged. The ELSI program was highly praised by participants and the President of HUGO, and was considered to be one of the best prepared and attended.
Conclusion
This is the third year of the triennium and, in doing my report, I would like to pay tribute to the dedicated and hard-working members of AHEC who have given unstintingly of their time. The Committee’s success is due to the combined efforts of members.
It has been my pleasure to chair this Committee for a second triennium. The challenges for AHEC in the future are increasing, especially as a result of the increased use of technology and the improvements in health care testing and information collection.
Professor Donald Chalmers
Chairman
A-57
Schedule 3
Recommendations to the Commonwealth Minister for Health and Aged Care
Recommendation 1
The Commonwealth Government, through the Minister for Health and Aged Care, should reaffirm its support for the UNESCO Declaration on the Human Genome and Human Rights, in particular Article 11, which states that:
Practices which are contrary to human dignity, such as reproductive cloning of human beings, shall not be permitted. States and competent international organisations are invited to cooperate in identifying such practices and in determining, nationally or internationally, appropriate measures to be taken to ensure that the principles set out in this Declaration are respected.
Recommendation 2
Noting that Victoria, South Australia and Western Australia have legislation regulating embryo research and prohibiting the cloning of human beings, the Minister for Health and Aged Care should urge the other States and Territories to introduce legislation to limit research on human embryos according to the principles set out in Sections 6 and 11 of the NHMRC Ethical Guidelines on Assisted Reproductive Technology.
Recommendation 3
Noting that there are statutory authorities established in Victoria, South Australia and Western Australia which consider and may approve human embryo research under strict conditions, the Minister for Health and Aged Care should urge the remaining States and Territories to establish similar statutory authorities with power to regulate research on human embryos according to the principles set out in Sections 6 and 11 of the NHMRC Ethical Guidelines on Assisted Reproductive Technology.
Recommendation 4
The Minister for Health and Aged Care should encourage and promote informed community discussion on the potential therapeutic benefits and possible risks of the development of cloning techniques.
Resolutions of the Australian Health Ethics Committee Pending State and Territory Legislation
Resolution 1
The AHEC proposes that, until legislation is introduced in the remaining States and Territories, the AHEC will collect information from institutional ethics committees (IECs) in these States and Territories on IEC research approvals of projects involving the application of current cloning techniques to human embryos. This information will be obtained in the course of the IEC annual compliance reporting system that is currently in place.
Resolution 2
The AHEC proposes that, until legislation is introduced in the remaining States and Territories, the NHMRC should consider the establishment of an expert advisory committee to assist IECs which seek advice on the scientific aspects of research projects involving the application of current cloning techniques to human embryos.
A-58
Chapter 4 - Australian Legislation and Guidelines Relevant to Cloning in Existence at November 1998
Introduction
4.1
This chapter discusses current State legislation and NHMRC ethical guidelines governing research which deal directly or indirectly with human cloning. The Reproductive Technology Accreditation Committee (RTAC) of the Fertility Society of Australia also issues a Code of Practice for accreditation of all IVF clinics.
The chapter evaluates the adequacy and effectiveness of the current legislation and research guidelines to deal with current and likely future technological processes with human cloning projects.
The definition of cloning in the three States which have relevant legislation is not consistent. The importance of clearly defining this term will be of great importance in ensuring adequate regulation of this expanding area of science.
4.2
4.3
Embryo Experimentation
4.4
4.5
Some of the work in cloning research may involve human embryos. In this case, the current legislation and ethical guidelines on human embryo experimentation will apply directly to such research proposals.
State and Territory governments established Committees of Inquiry which produced a succession of Australian reports on IVF during the 1980s. These reports also dealt with the difficult and controversial issue of embryo experimentation. There continues to be a tension between views that the embryo is, if not a human being, certainly deserving of respect, and that some experimentation ought to be allowed to uncover information relevant for the purposes of: (a) improving IVF techniques; (b) understanding male infertility; (c) understanding chromosomal abnormalities; (d) understanding gene defects; and (e) improving contraception.
Most reports recommended that no experimentation could be carried out either on embryos produced specifically for research or on embryos excess to IVF requirements.
4.6
Victoria
4.7
4.8
Victoria was the first state and the first jurisdiction in the world to introduce legislation to regulate infertility treatment. Legislation was later introduced in both Western Australia and South Australia.
The Victorian Infertility Treatment Act 1995 explicitly prohibits certain research which involves the “formation or use of a zygote if the research proposed that the zygote continue to develop to syngamy” amongst other prohibited practices is altering the genetic constitution of a gamete intended for use in a fertilisation procedure.
Western Australia
4.9
The Western Australian Human Reproductive Technology Act 1991 contains a list of offences which include conducting unapproved research or diagnostic procedures with an egg in the process of fertilisation or an embryo, and maintaining an embryo outside the body of a woman after fourteen days from the time of mixing of the gametes.
Ministerial Directions under the Human Reproductive Technology Act 1991 (WA) include regulations which would apply if research involving human cloning were to be carried out. Where approval is sought for any research or diagnostic procedure to be carried out involving an embryo, the intention must be that the procedure will be therapeutic and unlikely to have any detrimental effects.
4.10
A-59
South Australia
4.11 The Reproductive Technology Act 1988, together with the Reproductive Technology (Code of Ethical Clinical Practice) Regulations and the Reproductive Technology (Code of Ethical Research Practice) Regulations, prohibit, except in accordance with a licence, experimenting with “human reproductive material” (meaning a human embryo, human semen or a human ovum).
New South Wales
4.12 In October, 1997, the New South Wales Government issued a discussion paper titled “Review of the Human Tissue Act 1983.” In the Foreword to this paper, the New South Wales Minister for Health, the Hon. Dr Andrew Refshauge stated that
In response to community concern the Government has decided to introduce a law to ensure that two procedures do not develop in New South Wales. The Government has announced the banning of human cloning and trans-species fertilisation involving human gametes or embryos.
NHMRC Ethical Guidelines on Assisted Reproductive Technology (ART)
4.13 The NHMRC has published specific guidelines dealing with ART which include reference to cloning of human beings. The Ethical Guidelines were tabled in Parliament prior to their release in 1996. These guidelines were accompanied by a recommendation that they form a basis for complementary legislation in the States and Territories which had not yet introduced legislation.
4.14 The NHMRC Act authorises the Council to issue guidelines for the conduct of health research and of other purposes related to health. Although infringement of their provisions is not a legal offence, sanctions for infringement usually involve loss of access to research funds from the fund managed and administered by the Council or publication of the names of infringers in Parliament. The guidelines are regarded as national standards of acceptable practice.
4.15 The NHMRC Ethical Guidelines include a number of guidelines relating to embryo experimentation. A practical requirement of note is that “the recognition that any experimentation and research involved in these technologies should be limited in ways which reflect the human nature of the embryo, acknowledging that there is a diversity of views on what constitutes the moral status of a human embryo, particularly in its early stages of development.”
4.16 The NHMRC Ethical Guidelines contain restrictions on research relevant and specifically prohibit certain practices.
Comment
4.17 In Australia, substantial limits are placed on research involving embryos. Statutory approval for embryo experimentation is required in three States. The effect of the NHMRC Statement on Human Experimentation and the specific NHMRC Ethical Guidelines which deal with embryo experimentation allow research in this area only in exceptional circumstances. In the other States and Territories an institutional ethics committee (IEC) is required to grant approval for such research in accordance with the NHMRC Ethical Guidelines on Assisted Reproductive Technology.
Assisting in Reproductive Technology Programs
4.18 Cloning techniques of nuclear transfer or embryo splitting could have applications in assisted reproductive programs. One commentator has noted that the nuclear transfer process may have applications in assisted reproductive programs to overcome male infertility problems. An infertile husband could benefit from the asexual nuclear transfer process by contributing his genetic material to the enucleated cell of his
A-60
wife. Applications of cloning techniques could be used to assist in ART by the splitting of embryos, so increasing the number of embryos for later transfer, facilitating fertilisation in women over 40 (by cloning of the mitochondrial or gene set (cytoplasm replacement)), or replacing defective mitochondrial genes that cause disease.
4.19 If any of these procedures were to be undertaken in ART programs, statutory and/or ethical committee clearance would be required. Assisted reproductive technology is regulated by specific legislation in three States. There is a system of self-regulation and accreditation comprising the RTAC and its Code of Practice for units using IVF and related reproductive technologies, with RTAC setting professional and laboratory standards for clinical practice under this system of accreditation.
Status Of Children Legislation
4.20 The status of any child born in an ART program is addressed in State and Territory legislation. This legislation was introduced so that any person donating gametes to another person in an assisted reproductive process was not the parent at law of that child. In essence this legislation established the principle that the recipient social parent, rather than the biological parent, assumed all responsibilities at law for that child. In addition, the legislation also established that the person contributing the gametes did not assume any parenting responsibilities at law under such an arrangement.
4.21
This legislation rests on the donation of gametes rather than the contribution of genetic material. In a scenario where an infertile husband contributes his own genetic material by way of nuclear transfer, the genetic as well as legal relationship is to the husband. On the other hand, were the genetic material to be contributed by a person other than the husband, current legislation may not apply.
Replacing Human Tissue and Organs
4.22 In Chapter 2 there was discussion about early stage research into the development of cell lines from embryonic stem cells. This research may illuminate understanding of the programming and reprogramming of cell lines. Understanding of the process of differentiation and dedifferentiation could be the key to provide an unlimited source of therapeutic cells from which transplantable tissue and organs might result.
Human Tissue Legislation
4.23 All Australian States have enacted legislation regulating the donation and transplantation of human tissue. The definition of “tissue” is not identical, but in NSW includes “an organ, or part, of a human body and a substance extracted from, or from a part of, a human body.” In essence, this legislation requires the consent of the parties involved for the donation and for the acceptance of the human tissue in a transplantation procedure.
4.24 Current human tissue legislation may apply to some aspects of proposed cloning techniques. Where a cloning technique uses material from one body for transplantation to another or for research or other purposes, the consent provisions of the human tissue legislation would apply.
Cloning an Individual Human Being—Prohibitions in Australia
State Legislation
Victoria
4.25 The Victorian Infertility Treatment Act 1995 deals specifically with cloning and defines it as the formation “outside the human body” of “a human embryo that is genetically identical to another human embryo or person.” The Act prohibits a person from carrying out or attempting to carry out cloning. The Victorian Act contains prohibitions on destructive research on embryos. There are several clauses with a very direct bearing upon cloning.
A-61
Western Australia
4.26 In Western Australia, the Human Reproductive Technology Act 1991 establishes a regulatory structure and Code or Practice. The Act itself contains a list of offences including any procedure directed at human cloning or producing a chimaera.
South Australia
4.27 The South Australian Code of Ethical Research Practice also contains a list of prohibitions which include: cloning altering the genetic structure of a cell while that cell forms part of an embryo or an ovum in the process of fertilisation; replacing the nucleus of a cell of an embryo or of an ovum in the process of fertilisation with any other nucleus; and placing reproductive material in the body of an animal.
4.28 The procedure of nuclear transfer which does not involve human semen may not be regulated by the Act or the South Australian Code of Ethical Clinical Practice. The Code of Ethical Clinical Practice does not contain a definition of the term “cloning.”
NHMRC Ethical Guidelines on Assisted Reproductive Technology
4.29 The NHMRC Ethical Guidelines list a number of practices which are considered to be ethically unacceptable and to be prohibited. These include experimentation with the intent to produce two or more genetically identical individuals, including development of human embryonic stem cell lines with the aim of producing a clone of individuals.
4.30
Supplementary Note 7 to the NHMRC Statement on Human Experimentation clearly states that the introduction of pieces of DNA or RNA into germ (reproductive) cells or fertilised ova is not acceptable, because there is insufficient knowledge about the potential consequences, hazards, and effects on future generations.
4.31 Specific accreditation standards have been formulated by the RTAC and the Fertility Society of Australia has included in its Code of Practice a specific prohibition on nuclear transfer.
Comment
4.32 Embryo splitting and nuclear transfer for the specific purpose of cloning an identical human being is either prohibited or against the intention of the regulatory framework established in Victoria, Western Australia, South Australia and the NHMRC Ethical Guidelines. Production of embryonic stem cell (ES cell) lines is contravened by the Victorian and Western Australian Acts and NHMRC Ethical Guidelines.
Common Law
4.33 There is a general principle that contracts whose formation or performance is contrary to public policy are not enforceable in a court. In determining whether contracts are contrary to public policy, courts can have regard to relevant legislation. Thus, where statutes prohibit cloning, there would be grounds for concluding that a contract to provide tissue for the purpose of cloning an individual human being was contrary to public policy and thus unenforceable. Unenforceability alone does not, of course, provide a ground for prohibition of such contracts and does not mean that the parties by their contract have acted illegally.
Privately Funded Institutions
4.34 A concern at this stage is whether a private, rather than publicly funded, organisation in a State or Territory other than Victoria, Western Australia or South Australia might consider a venture in cloning of human being or cloning of human parts without the approval of an IEC under NHMRC guidelines.
Currently, the NHMRC guidelines are only enforceable against institutions receiving NHMRC funding. The possibility exists that a private institution could decide to undertake such work. Without legislation the NHMRC cannot stop private institutions conducting such work.
A-62
References
International Reports
President’s Commission Report. (1983) The Study of Ethical Problems in Medicine and Biomedical and Behavioural Research: Deciding to Forego Life Sustaining Treatment Washington.
The Belmont Report. (1979) Ethical Principles and Guidelines for the Protection of Human Subjects of Research The National Commission for the Protection of Human Subjects of Biomedical and Behavioural Research, Department of Health, Education and Welfare, pub. No. (OS) 78-0012 U.S. Government Printing Office Washington.
Council of Europe. (1996) Convention for the Protection of Human Rights and Dignity of the Human Being with Regard to the Application of Biology and Medicine Strasbourg (164).
CPMP/ICH. (1995) Code of Good Pharmaceutical Practice/ International Conference on Harmonisation Note for Guidance on Good Clinical Practice–135/95. See also ISO 14155 Clinical Investigation of Medical Devices.
CIOMS. (1993) Council for International Organizations of Medical Sciences (CIOMS) in collaboration with the World Health Organization (WHO), International Ethical Guidelines for Biomedical Research Involving Human Subjects Geneva Switzerland.
Canadian Code. (1997) Code of Ethical Conduct for Research Involving Humans, which is a tri-partite effort by the Medical Research Council, Natural Sciences and Engineering Research Council and the Social Sciences and Humanities Research Council of Canada.
Royal College of Physicians. (1996) Guidelines on the Practice of Ethics Committees in Medical Research Involving Humans London.
Health Research Council of New Zealand. (1997) Ministry of Health Review of the Ethical Review Structure in New Zealand Health Department New Zealand.
Nuffield Council on Bioethics. (1999) The Ethics of Clinical Research in Developing Countries United Kingdom.
National Bioethics Advisory Commission. (1997) Cloning Human Beings: Report and Recommendations of the National Bioethics Advisory Commission Washington.
Legislation
Poisons Act. 1966 New South Wales (see also the Poisons Act in ACT 1933; WA 1964; Tas 1971 and Vic Drugs Poisons and Controlled Substances Act 1991; Qld Health Act 1937; NT Poisons and Dangerous Drugs Act 1983).
Status of Children Act. 1978. (Qld) (and Artificial Conception Act 1984 (NSW); Status of Children Act 1974 (Vic); Family Relationships Act 1975 (SA); Status of Children Act 1974 (Tas); Artificial Conception Act 1985 (WA); Artificial Conception Act 1985 (ACT); Status of Children Act 1978 (NT); Family Law Act 1975 (Cth.).
Freedom of Information Act. 1982 Commonwealth Parliament Australia.
Human Tissue Act. 1983 ss. 6–9 NSW (see also Transplantation and Anatomy Act 1978 ss. 6–10 ACT; Human Tissue Transplant Act, ss. 6–10 NT; Transplantation and Anatomy Act 1979 ss. 8–12 Qld.; Transplantation and Anatomy Act 1983 s.7–10, SA; Human Tissue Act 1985 ss. 5–9 Tas; Human Tissue Act 1982 ss. 5–12 Vic.; Human Tissue and Transplantation Act 1982 ss. 6–9 WA).
Reproductive Technology Act. 1988 No 10 Parliament South Australia.
National Health and Medical Research Council Act. 1992 No 225 of 1992 Commonwealth Parliament Australia.
Code of Federal Regulations. (1992) 21 Food and Drugs Part 56.
Therapeutic Goods Act. 1993 Commonwealth Parliament Australia.
Infertility Treatment Act. 1995 Victoria Parliament Australia.
Human Reproductive Technology Act. 1993 Parliament Western Australian.
A-63
Books and Articles
Annas G. (1984) Ethics Committees in Neo-Natal: Substantive Protection of Procedural Diversion? Am J Pub Health 74; 843.
Beauchamp T. and Childress J. (1994) The Principles of Biomedical Ethics (4ed ) Oxford UP.
Beecher H. (1966) Ethics and Clinical Research New Eng J of Med 274:1354.
Beecher H (1968) Ethical Problems Created by the Hopelessly Unconscious Patient New Eng J of Med 278:1425.
Bennett B. (1997) Law and Medicine, LBC.
Brazier M. (1990) Liability of Ethics Committees and their Members Professional Negligence 186.
Breen K. (1997) Ethics Laws and Medical Practice Allen and Unwin Melbourne.
Brody H. (1981) Ethical Decisions in Medicine 2d ed Little, Brown and Co.
Bulletin of Medical Ethics. (1999a) Medical Research has its Downsides Bulletin of Medical Ethics, November 152: 7 (citing reports in The Times, 2 October 1999; The Guardian 8 November 1999; and 1999 Brit Med J 319274).
Bulletin of Medical Ethics. (1999b) Helsinki Declaration Revising Continues Bulletin of Medical Ethics 146:3.
Capron M.A. (1985) Legal Perspectives on Institutional Ethics Committees Journal of College and University Law 11:7.
Chalmers D. and Pettit P. (1998) Towards a Consensual Culture in the Ethical Review of Research Med J of Aust 168:79.
Clarke C. (1998) Should There Be an Accredited Ethics Committee System of Centralised Review of Multi-Centre Clinical Research Med J of Aust 169:283 (and Henman M. et al. Med J of Aust 169:283–4; E. O’Brien E. (1998) et al. Med J of Aust 169: 284–5; S. Gandevia S. et al. Med J of Aust 169:285.
Cohen M.(1998) Should There Be an Accredited Ethics Committee System for Centralised Review of Multi-Centre Clinical Research? Med J. of Aust 168:528.
Darvall L. (1993) Autonomy and Protectionism: Striking a Balance in Human Subject Research Policy and Regulation Law in Context 11:82.
Editorial. (1976) 7 Med J of Aust 7:179–80.
Engelhardt H. T. (1986) The Foundations of Bioethics Oxford UP.
Fletcher J. (1973) Realities of Patient Consent to Medical Research Hastings Center Studies 1:1.
Freckelton I. and Petersen K. (1999) Controversies in Health Law Federation Press Sydney.
Freedman B. and Glass K. (1990) Weiss v Solomon: A Case Study in Institutional Responsibility for Clinical Research Law, Medicine and Health Care 18:395.
Furrow B. et al. (1995) Health Law West Law Publishing.
Giesen D. (1995) Civil Liability of Physicians for New Methods of Treatment and Experimentation: A Comparative Examination
Med LR 3:22.
Gillespie R. (1988) Research and Human Subjects: an Historical Overview Conference Proceedings: Can Ethics Be Done by Committee? Monash University Centre for Bioethics Australia.
Healy D. (1999) Clinical Trials and Legal Jeopardy Bulletin of Medical Ethics 153:13–18.
Jonas H. (1969) Philosophical Reflections on Experimenting with Human Subjects (1969) Daedalus 98.
Kelly F. and Boyages S. (1999) Pilot Programme to Reform the Ethics Committee System in NSW Med J of Aust 171:52.
Kirby M. (1983) IVF - The Scope and Limitation of Law Conference on Bio-ethics and the Law of Human Conception IVF 29–30 September London UK.
The Lancet. (1999) 353:400–3 (Cited in Bulletin of Medical Ethics 148:5).
Laufer S. (1990) The Regulation of Medical/Scientific Research Practices Involving Experimentation on Human Beings Law in Context 8:78.
Levine R. (1986) Ethics and Regulation of Clinical Research Urban and Schwarzenburg Baltimore.
Mant D. (1999) Can Randomised Trials Inform Clinical Decisions About Individual Patients? The Lancet 353:743–746.
A-64
McNeill P. (1993) The Ethics and Politics of Human Experimentation Cambridge UP.
Magnusson R. (2000) The Use of Human Tissue in Medical Research: Legal Issues for Human Research Ethics Committees J of Law and Medicine 7:390.
Merritt A. (1987) The Tort Liability of Hospital Ethics Committees Southern Cal LR 60:1239.
NCBHR. (1995) Protecting and Promoting the Human Research Subject: A Review of the Function of Research Ethics Boards in Canadian Faculties of Medicine NCBHR Communiqué 6:1–33.
Nelson D. and Weiss R. (1999)Hasty Decisions in the Race to a Cure? Washington Post Sunday Nov 21 p A1.
Neuberger J. (1992) Ethics and Health Care: The Role of Research Ethics in the UK Kings Fund Institute Research Report 13 London.
Pellegrino E. and Thomasa D. (1996) Christian Virtues in Medical Practice Georgetown UP.
Rawbone R. (2000) Observation from Six Years Experience of a Health and Safety Research Ethics Committee Bulletin of Medical Ethics 155:13.
Scott R. (1984) Experimenting with Life: Must Law-Makers Experiment Too? 5th International Conference on Forensic Science, Sydney Australia.
Skene L. (1998) Law and Medical Practice Butterworths Melbourne.
Parliament Debate
Commonwealth of Australia Parliamentary Debates: Senate Volume S154 36th Parliament, 1st Session 5th period 1991.
National Health and Medical Research Council and AHEC Reports
NHMRC. (1984) Supplementary Note on Embryo Flushing AGPS Canberra Australia.
NHMRC. (1985) Report on Workshops on the Constitution and Functions of Institutional Ethics Committees in Australia 1984–85 NHMRC. AGPS Canberra Australia.
Statement on Human Experimentation. (1992) NHMRC Canberra Australia.
AHEC. (1992) CTN Guidelines for Institutional Ethics Committees Canberra Australia (see also Clinical Trials of Drugs in Australia DEB 1 The Clinical Trial Notification (CTN) Scheme; Guidelines for Good Clinical Research Practice Therapeutic Goods Administration; Australian Guidelines: Clinical Trials Exemption (CTX) Scheme for Drugs DBE 5).
AHEC. (1993a) Report of the 1993 Workshops for Institutional Ethics Committees: Consultation with Researchers and Forum on HRECs
NHMRC Canberra Australia.
NHMRC. (1993b) Report of the 1993 Survey of Institutional Ethic Committees NHMRC AGPS Canberra Australia.
NHMRC. (1994) Annual Report 1993 AGPS Canberra.
Bienenstock J. (1993) Report of an External Review of the National Health and Medical Research Council AGPS Canberra Australia.
NHMRC. (1994) Report on Compensation, Insurance and Indemnity Arrangements for Institutional Ethics Committees, AGPS Canberra Australia.
AHEC. (1996) Ethical Guidelines on Assisted Reproductive Technology AGPS Canberra Australia.
NHMRC. (1997) Annual Report 1996 AGPS Canberra.
NHMRC. (1998) Annual Report 1997 AGPS Canberra.
AHEC. (1998) Report on the Scientific, Ethical and Legal Considerations Relevant to Human Cloning Commonwealth Minister for Health and Aged Care NHMRC Canberra Australia.
NHMRC. (1999) Annual Report 1998 AGPS Canberra.
NHMRC. (2000) Annual Report 1999 AGPS Canberra.
NHMRC. (2000) Guidelines under Section 95 of the Privacy Act 1998 Canberra Australia.
National Statement on Ethical Conduct in Research Involving Humans. (1999) Commonwealth of Australia AGPS Canberra (www/nhmrc.gov.au/ethics/statemen.htm).
A-65
Australian Reports
Allars M. (1994) Report of the Inquiry into the Use of Pituitary Derived Hormones in Australia and Creutzfeldt-Jakob Disease (CJD) Commonwealth of Australia AGPS Canberra Australia.
Baume P. (1991) A Question of Balance: Report on the Future of Drug Evaluation in Australia Report to the Commonwealth Minister for Aged, Family and Health Services AGPS Canberra Australia.
Chalmers D. (1985) Interim and Final Report of the Committee to Investigate Artificial Conception and Related Matters Government Printer Tasmania Australia.
Chalmers D. (1996) Report of the Review of the Role and Functioning of Institutional Ethics Committees Report to the Commonwealth Minister for Health and Family Services AGPS Canberra Australia.
Cornwall J. (1984) Report of the Working Party on IVF and AID (and 1987 Select Committee of the SA Legislative Council, Report on Artificial Insemination by Donor, In Vitro Fertilisation and Embryo Transfer Procedures and Related Matters in South Australia)
Government Printer South Australia Australia.
Day R. (1993) Review of the Clinical Trials Notification (CTN) Scheme: Report to the National Manager of the Therapeutic Goods Administration, Therapeutic Goods Administration Canberra Australia.
Demack J. (1984) Report of the Special Committee Appointed by the Queensland Government to Enquire into the Laws Relating to AID, IVF and Related Matters Government Printer Queensland Australia.
Family Law Council of Australia. (1985) Creating Children: A Uniform Approach to the Law and Practice of Reproductive Technology in Australia Family Law Council AGPS Canberra Australia.
Finn P. (1990) Health Ethics: The NHMRC and the NBCC Report to the Federal Minister for Health Canberra 29 October 1990.
Michael C. (1984) Interim Report of the IVF Ethics Committee of W.A. and a final report in 1986; Report of the Committee Appointed by the Western Australian Government to Enquire into the Social, Legal and Ethical Issues Relating to In Vitro Fertilisation and Supervision
Government Printer Western Australia Australia.
Privacy Commissioner. (1996) The Privacy Implications of Genetic Testing AGPS Canberra Australia.
Senate Select Committee. (1986) Human Embryo Experimentation in Australia Commonwealth Parliament Australia.
TGA. (1990) Australian Code of Good Manufacturing Practice for Medicinal Products AGPS Canberra.
Waller L. (1982–1984) Interim Report of the Committee to Consider the Social Ethical and Legal Issues Arising from IVF; Report on Donor Gametes in IVF; Report on Disposition on Embryos Produced by IVF Government Printer Victoria Australia.
Wills P. (1999) Virtuous Cycle Report to Commonwealth Government AGPS Canberra July 1999 which reviewed the structure and financing of medical research in Australia.
Court Decisions
Bennetts v Board of Fire Commissioners of New South Wales (1967) 87 WN (NSW) 307.
Bouvia v Glenchur (1986) No C 583828 Cal. S Ct. Los Angeles County 7 10.
Canterbury v Spence (1992) 464 F2d 772.
Davis v Rodman (1921) 146 Ark. 385, 227 SW 612.
Halushka v University of Saskatchewan (1965) 53 DLR(2d) 436.
In Re Quinlan (1976) 70 NJ 10; 355 A 2d 647 Certificate denied, 429 US 922.
Nyali Ltd. v the Attorney-General (1956) 1 QB at 16–17.
R v Ethical Committee of St Marys Hospital ex-parte Harriott (1988) 1 FLR 512.
Reibl v Hughes (1980) 114 DLR (3d)1.
Rogers v Whitaker (1992) 67 ALJR 47; (1992) 175 CLR 479.
Tobacco Institute of Australia Ltd v National Health and Medical Research Council and Others (1996) 142 ALR 1.
Weiss v Solomon (1989) 48 CCLT 280.
A-66

LOCATION OF THE
OFFICE FOR PROTECTION FROM RESEARCH RISKS WITHIN THE NATIONAL INSTITUTES OF HEALTH: PROBLEMS OF STATUS AND INDEPENDENT AUTHORITY
Commissioned Paper John C. Fletcher University of Virginia
B-1
I. Introduction
Task and Methods. The task is to examine the location of the Office for Protection from Research Risks (OPRR) within the National Institutes of Health (NIH) and its effects on the mission of the Office. Recommendations will accompany the findings.
The issue of location is conceptually related to OPRR’s mandate, the institutional histories of OPRR and the NIH with regard to human subjects research (HSR), and the general performance of the U.S. system for protection of human subjects of research (HSoR).1 These themes will be addressed in the report, although the discussion will mainly address the location issue.
In addition to literature on the strengths and weaknesses of other federal regulatory agencies, the author reviewed the history and present mandate of two federal bodies with similar missions and past problems of conflicts of institutional interests: 1) the Office of Government Ethics (OGE) and 2) the Nuclear Regulatory Commission (NRC).
Interviews
| n |
September 4, 1997 (telephone) Charles R. McCarthy, former Director, OPRR |
| n |
September 11, 1997 (on-site, 10:00 A.M. – 3:00 P.M.) Gary B. Ellis, Director, OPRR J. Thomas Puglisi, Human Subject Protections, OPRR |
| n |
September 25, 1997 (telephone) Alexander M. Capron, Professor of Law, University of Southern California |
| n |
September 30, 1997 (telephone) James P. O’Sullivan, Associate General Counsel, U.S. Office of Government Ethics |
| n |
September 30, 1997 (telephone) J. Samuel Walker, Historian, Nuclear Regulatory Commission |
| n |
October 3, 1997 (telephone) Richard A. Merrill, Professor of Law, University of Virginia |
| n |
October 5, 1997 (telephone) Jay Katz, Professor Emeritus, Yale University |
| n |
October 17, 1997 (telephone) Robyn Y. Nishimi, Ph.D., Director, Presidential Advisory Committee on Gulf War Veterans’ Illnesses |
| n |
October 20, 1997 (telephone) Mary Ann Dufresne, Staff Aide to Sen. Glenn |
| n |
October 22, 1997 (on-site, 10:00 A.M. – 12:00 P.M.) Gary B. Ellis, Director, OPRR F. William Dommel, Director of Education, OPRR |
| n |
October 27, 1997 (telephone) Richard Riseberg, Chief Counsel, Public Health Service |
| n |
November 10, 1997 (telephone) James H. Jones, Professor of History, University of Houston |
B-3
Executive Summary and Major Findings
A. On the Location of OPRR in Government
1) OPRR’s location within the NIH is a structural conflict of missions and incompatibility of functions. This structural conflict gives rise to several troubling and persistent problems—including conflicts of interest—for the professional staff of OPRR and the NIH officials who administer OPRR.
The report’s arguments are based on these points and findings:
| n |
OPRR’s mission is to uphold the primacy of the rights and welfare of HSoR. This mission is enveloped within the NIH’s scientific mission and its powerful interests in funding and conducting research. This conflict of missions weakens OPRR’s authority and stature and engenders conflicts of interest. |
| n |
The most compelling evidence of conflict of interest is that OPRR is far more effective and authoritative in regulating grantee institutions than Department of Health and Human Service (DHHS) agencies. |
| n |
The NIH is in the implausible position of regulating itself. Internally, the NIH leadership suffers from institutional blindness to the structural problem and the issue of conflict of interest. Externally, the NIH suffers a credibility problem. Others, such as the General Accounting Office (GAO), the Human Research Ethics Group, and this observer, clearly see a conflict of missions that lead to conflicts of interest. The NIH leadership neither acknowledges nor moves to remedy the situation. In that the NIH is an agency of the DHHS and part of the Executive Branch of government, the White House and DHHS have the ultimate responsibility for the problems that weaken OPRR and its mission in HSR. |
| n |
An inappropriate location for OPRR imposes burdens that weaken the entire system, e.g., reduced status and lack of respect, political pressure from the NIH requiring problematic compromises, and inordinate time and effort to correct noncompliance and other significant problems. |
| n |
OPRR’s present location is entirely inappropriate for any future system of universal protection of human subjects as envisioned by Senator Glenn and other sponsors of federal legislation, the Advisory Committee on Human Radiation Experiments (ACHRE), the Human Research Ethics Group, or the National Bioethics Advisory Commission (NBAC) itself.2 |
| n |
The history of two other national agencies offers relevant analogies and remedies: the NRC and the OGE. B. The U.S. System of Protection of HSoR Has Significant but Remediable Problems |
1) Federal legal protections exist only for HSR that is a) conducted or supported by any of 17 Federal Departments or Agencies that adhere to the Common Rule or b) regulated by the Food and Drug Administration (FDA). A substantial volume of HSR occurs beyond the perimeter of those protections;
2) Sanctions are inadequate for violations of federal regulations to protect HSoR;
3) No permanent national forum exists for informed debate, continuing interpretation, and application of ethical principles and rules for HSR, consideration of problematic cases, or formulation of policies to meet new needs;
4) OPRR, the federal office for oversight of human subject Assurances representing approximately 5,000 domestic and foreign institutions and for consultation with 17 Federal Departments or Agencies that conduct or sponsor HSR, is now severely undersized and compromised in effectiveness, given the magnitude of its oversight of HSR activities within its current authority. If there were universal protection of HsoR, the current OPRR would be totally inadequate to the task.
B-4
Recommended Remedies:
For A.1, B.3, and 4: Elevated status, independent location, and adequate funding for a successor to OPRR: the National Office of Human Subjects Research (NOHSR) along with a National Advisory Committee for Human Subjects Research (NACHSR).
For B.1 and 2: Federal legislation that confers the protections of informed consent and Institutional Review Board (IRB) review for all HSoR, with appropriate sanctions for violators.
II. Moral and Political Reflection on the U.S. System to Protect
Human Subjects
| A. |
Moral Reflections |
| 1. |
How Vigorously Should Society Protect HSoR? |
Answers to this question depend on ethical perspectives on the status of research. Given society’s major goals and interests, is there a defensible moral imperative to conduct biomedical research and human experimentation? Is there a moral obligation—arising from the needs of society and the social contract with its members—for biomedical scientists to conduct research and for persons who are sick or well to participate in it? Does society have “rights” in human experimentation that it should claim to procure knowledge to save lives and reduce the incidence of disease? McDermott argued for a strong version of such a position in the 1960s.3 If his argument prevails, then the reasons for society to protect HSoR are weaker than reasons that flow from a different moral argument.
Jonas saw no moral duty to conduct research and especially HSR. Contrary to McDermott and other scientists who argued for the moral priority of society’s need for knowledge to struggle against death and sickness, Jonas defended the dignity of the individual over the advance of knowledge. He wrote that social progress through medical progress is an “optional goal, not an unconditional commitment….”4 His words capture the moral sense that, in my view, deserves the stronger loyalty in this debate. Jonas wrote: “Let us also remember that a slower progress in the conquest of disease would not threaten society, grievous as it is to those who have to deplore that their particular disease be not yet conquered, but that society would indeed be threatened by the erosion of those moral values whose loss, possibly caused by too ruthless a pursuit of scientific progress, would make its most dazzling triumphs not worth having.”5 Higher loyalty to the dignity and welfare of HSoR ought (almost always) to prevail over loyalty to the cause of science and the needs of society for knowledge, relief of suffering, and cure and prevention of disease. The origin of this loyalty is respect for persons and their capacity for expressions of altruism and sacrifice—the ideal (although rarely the actual) moral source of participation in research. As Jonas pointed out, society has no special claim or command over the altruism and sacrificial gifts of subjects of research, especially those who are sick. Conscription for research is unethical in any society. The “yes” to participate in research is one that only the individual or a legally authorized representative has the authentic moral capacity to give, despite all of the other real influences on subjects’ motivation, including financial inducements and physicians’ recommendations.
The caveat of “almost always” above recognizes those periods in social life when morally justified wars and national emergencies can lead to troubling degrees of relaxation of normal moral boundaries for the sake of survival. Even on these extraordinary occasions, however, there should be no involuntary experimentation on members of the armed services, prisoners of war, or otherwise incarcerated research subjects. At such times, some degree of secrecy about specific research projects may be required to protect the national interest. Even in this special context, all HSR in secret or protected projects should still have the twin protections of prior review and informed consent.
B-5
U.S. law and regulations on HSR fall far short of the moral ideal, in that legal protections are extended only to subjects who participate in certain federally funded or regulated projects. Universalizing the scope of legal protection, as has now been done by the 21 member countries of the Council of Europe,6 is now a moral imperative for the U.S. Congress. A large and unknown number of human subjects are at risk in research projects funded through the private sector. The nations belonging to the Council of Europe have implemented the first truly international legal protection of all human subjects.
Higher loyalty to the welfare of HSoR does not mean that no loyalty at all is owed to science’s quest for truth or to the needs of society to reduce and prevent disease. There is an important right of scientists to seek knowledge that can be infringed rarely and with a compelling public interest as the test. This right is constitutionally grounded in the right of “free speech.”7 There is at least a nonbinding civic obligation (but not a stringent moral duty) for members of modern and democratic societies to support scientific investigation and to participate if able in research conducted within prevailing ethical and legal norms. This civic duty arises from the value of science to democracy and from a shared commitment to resolve significant social and scientific disputes by evidence rather than ideology.
Rather than a sharp “either-or” division of loyalty that places all moral weight on protection of HSoR and none on any other related cause or claim, it is practical to recognize a hierarchy of loyalties in research activities. Loyalties are owed, in this order, to 1) protection of HSoR, 2) protection of scientific and academic freedom, 3) commitment to meeting society’s needs for biomedical knowledge, and 4) concern for the welfare of particular research institutions and investigators. Such a hierarchy of loyalties underlies the author’s views and recommendations of this report. The societal obligation to protect HSoR is higher than the other three, but it is also morally justifiable to be loyal to the other claimants when doing so does not override and unjustifiably infringe loyalty to protecting HSoR.
The guiding moral premise of this report is that Congress originally created the mandate that was delegated to OPRR out of fidelity to higher loyalty to the protection of HSoR. However belated this recognition by Congress in 1974, it is the moral core of OPRR’s mission. Further reasons to protect human subjects arise from three realities of HSR: 1) HSR is mainly for the benefit of society and the medical sciences, 2) HSoR are vulnerable—they frequently volunteer with motives driven by a “therapeutic misconception”8 that research will benefit them as well as trust in their physicians who refer or recruit them, and 3) the motivation of physicians who are also investigators studying their own patients is extremely complex and vulnerable to internal and external influences that can run counter to the welfare of the subjects—e.g., competition for scarce funding, career advancement, and financial inducements to enter patients into studies.9
| B. |
Political Reflections |
| 1. |
The Mandate of OPRR |
Congress amended the Public Health Service Act (July 12, 1974) with Public Law 93-438, the National Research Act. This law directed the Secretary, DHEW, to 1) promulgate regulations regarding IRB review and institutional Assurances, 2) establish a program of ethical guidance, and 3) establish a process for responding to violations of the rights of HSoR. The second item was handled by OPRR’s predecessor, the NIH Institutional Relations Branch, and was formally delegated by the Secretary to OPRR. OPRR is thus the DHHS-wide authoritative voice on clarification and guidance on ethical issues. The first and third items have always been done exclusively by OPRR.
2. The U.S. System of Protection of HSoR
Turning attention to the U.S. system of protection of HSoR and to OPRR’s place within it, a very mixed picture of strengths and weaknesses emerges. Justified pride is due in that the United States was the first nation to extend
B-6
legal protection for HSoR in federally funded research. A vast and very diverse network of IRBs, estimated at between 3,000 and 5,000, has evolved. These IRBs serve as the nation’s primary resource for the protection of HSoR by examining the ethical aspects of a project before it begins. A morally valid process of informed consent to the particular research project is the second major resource to protect HSoR.
IRBs and their authority have gradually been accepted by clinical investigators with rare exceptions. However, the nation’s IRBs have well-known problems, such as poor relationships to their local communities, inadequate education and training for members, inadequate scientific expertise, misallocation of effort to assure scrutiny of studies carrying greatest risk, poor quality control of reviewer performance, poor performance in continuing review, and little first-hand exposure to the context of clinical investigation and specific studies.10 These problems need attention within cooperative efforts between the local and federal partners in the enterprise. In my view, significant improvements will not occur without a national strategy, adequate funding incentives, and a strengthened successor to the OPRR, which is charged by Congress with the role of education and IRB welfare. Small staff and other pressures greatly limit OPRR’s role and effectiveness in IRB education and oversight as compared to its role with Assurances and compliance.
Nishimi’s testimony11 captures the history of the U.S. system of protection of HSoR. She explains that the approach that the federal government employs to protect HSoR is intentionally decentralized and diffused. The structure of the current system has changed very little from the approach set out by the 1966 Public Health Service (PHS) guidelines. Local review has been the centerpiece of protection, based on the belief that a local group of relatively disinterested individuals is most desirable because they are in the best position to know the prevailing values and ethics of the community and proposed subject population. At the NIH from 1966–1969, the author witnessed the earliest stage of the PHS regulation of HSR. The NIH leadership believed that local review coupled with a very modest NIH-based oversight mechanism would suffice. In 1982, the author interviewed Dr. James Shannon, former Director, NIH, and other NIH and PHS officials about the main features of the Surgeon General’s policy and their memories of the need for it.12 Dr. Shannon stated, “None of us wanted a bureau of ethics in Bethesda. Local prior group review was the linchpin of the policy.” Despite the wish of Dr. Shannon and others, the OPRR, if not a bureau of ethics, is the sole official voice and continuing presence within government with a priority of protecting HSoR. The OPRR is inadequate, for several reasons, to do this task within its current mandate. Problems arising from location contribute to this condition. The NIH exercises a dual role to promote and regulate HSR. Although the NIH’s problem is far less dangerous, there is a historical analogy in the Atomic Energy Commission’s (AECs) failure from 1951–1973 to hold together both the promotion of nuclear energy and regulation of its uses. DHHS and Congress should face and resolve a persistent conflict of missions and interests between the NIH and OPRR.
III. Location of OPRR: Impact on its Mission
A. Historical Background on HSR and the NIH
The argument in this report is that structural conflicts of mission between OPRR and the NIH engender conflicts of interest for OPRR’s staff and NIH officials. How does this report use the term “conflicts of interest?” In his discussion of this topic in the context of health care, Erde first describes an “artificially narrow account” of a conflict of interest, i.e., “conflicts of interest occur when and only [when] a [physician] strays or is tempted to stray from...role mandated duties for the sake of...economic benefit.”13 Erde goes on to discuss a much broader range of causes (e.g., motives, situations, and structures) that may or may not influence conflicts of interests. This report seeks an understanding of conflicts of interest informed by Erde’s broader discussion, e.g., in this situation—for regulators (at OPRR) and for funders and sponsors of HSR (at the NIH)—conflicts of interests are either “motives that …[regulators or funders/sponsors] have and/or situations in which we could reasonably
B-7
think...[their] responsibilities to observe, judge, and act according to the moral requirements of their role are or will be compromised to an unacceptable degree.”14 The next several parts of the report provide historical background and data to support the argument.
1. Historical Background
A brief historical background should preface a discussion of OPRR’s location. The history of NIH’s role in the protection of HSoR can be evaluated from different standpoints. Viewed from within the NIH, there is much in which to take pride. From 1953, a form of prior group review at the Clinical Center, NIH, was an early predecessor of IRBs. The NIH leadership responded in the early to mid-1960s to social and media criticism of a lack of protection of HSoR and to the legal risks to clinical researchers.15 As described below, the NIH’s intramural leaders continued to improve a very effective research review system from 1966 to the present. The NIH also helped to staff and support the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (1974–78), whose work developed consensus and a foundation for a systematic ethical perspective and body of ethical guidance on HSR. The work of the Commission, especially on research with children, had immediate effects within the intramural program. The NIH also funded and housed the OPRR to the present time.
From outside the NIH and the PHS, critical questions can be raised about the HSR record of the nation’s major funder and sponsor of biomedical research. One finds at different periods examples of “institutional blindness” to HSR issues,16 to congruence of public accountability between the NIH’s intramural and extramural programs, and to the OPRR’s legitimate authority. The first two examples are preludes to a condition of institutional blindness to the conflict of interests issue embedded in OPRR’s location within the NIH.
a. Early History of NIH-PHS and HSR: How Could the Tuskegee Study Have Endured So Long?
The founders of the NIH’s intramural program, which began when the Clinical Center opened in 1953, were very conscious of their moral responsibilities in HSR. Accordingly, they created and continued to improve forms of prior group review suited to the requirements of the intramural program. These efforts from 1953–1977 are described below. In this period, there was a greater degree of protection for normal volunteers and patients in research carrying higher risk than for patients in research with lower risks or who were being followed and studied in experimental conditions. The ethos of these years was also grounded in deep commitments to scientific freedom and flexibility for researchers to follow the implications of their discoveries with particular patients. It is important to remember that, in this period, there was no systematic body of ethical principles and guidance for HSR. As in the wider research community,17 the norms of the NIH culture permitted wide latitude with regard to informed consent and did not require prior group review of each research project with patients or of a single experiment involving one or a few patients.
In the 1950s and 1960s, the NIH was a relatively new agency where streams from two research cultures and one research bureaucracy met, but with apparently little creative or critical interaction. The first was an older pre-WWII research culture marked by a few general moral norms and an overriding degree of ethical relativism. It was this culture that created and supported the PHS-Centers for Disease Control (CDC) Tuskegee syphilis study from 1932–1972. The second was a post-WWII and post-Nuremberg research culture. It was marked by high commitment to the best science, to informed consent (tinctured heavily with flexibility and the therapeutic privilege), and to new forms of prior peer review of proposed research. The founders of the intramural program were largely members of this second culture. A third stream, a research bureaucracy with written ethical requirements on HSR, grew up around the NIH’s extramural grants and contracts program in the 1960s. The 1966 and 1971 PHS-NIH policies requiring local IRBs and prior group review were required of grantees and contractors in this program.
A question deserving of more historical research arises as to whether the principals in these three arenas seriously discussed ethical issues among themselves. If they did so, it was without much perspective on the
B-8
implications that strong commitments to post-Nuremberg research ethics within the intramural program had for the extramural program or for earlier research (e.g., Tuskegee syphilis study) being conducted by PHS and CDC physicians. If one hypothesizes great social distance between these three arenas, and such could be demonstrated, it would help greatly to explain subsequent events.
How else could the most dramatic example of institutional blindness to HSR issues in the history of the PHS-CDC be explained? Jones18 describes the mid-1960s confrontation of PHS and CDC officials about the Tuskegee study by Peter Buxton, a PHS venereal disease interviewer and investigator. These officials19 could find no ethical reasons to criticize or halt a longstanding (1932–1972) Tuskegee study of untreated syphilis, even after the discovery of penicillin. The depth of blindness and resistance to Buxton’s moral claims can also be measured by two factors. First, awareness of the civil rights movement should have focused PHS’s concern on the fact that all the subjects were black and totally uninformed.20 Second, it is also striking that the officials’ resistance to Buxton’s criticisms occurred at exactly the same time that the PHS-NIH was requiring prior group review of HSR in response to other famous cases, scandals, and Dr. Henry Beecher’s historic article.21 In fact, the PHS-NIH requirement of local prior review grew directly out of a decade of experience in the NIH intramural program. Did the right hand (PHS-CDC) know what the left hand (NIH-extramural/intramural) was doing? More historical research is needed to answer this question and to explain the reasons for such profound silence about the implications of post-Nuremberg ethics, as practiced at the intramural NIH, for evaluation of the Tuskegee study.
b. Applying Federal HSR Regulations to NIH’s Intramural Program
A second but less dramatic example of institutional blindness is a ten-year (1971–1981) period in which federal regulations incongruently applied to extramural grantees and contractors but not to the intramural research program. In government generally prior to this period, there was institutional blindness and a slow learning process as to the need for reforms in HSR ethics.22 The learning process within the PHS and the NIH was provoked by crises that sparked reforms and resulted in more NIH commitment to bioethics.
In 1966, PHS promulgated a Surgeon General’s policy requiring local prior group review of all grant applications to PHS to involve human subjects.23 The 1966 policy was revised in 1971 (“the Yellow Book”) to require IRBs to have outside members who were nonscientists. However, this policy did not apply to the NIH’s intramural research at the Clinical Center. The policy was translated into federal regulations in 1974. Notably, the 1974 federal regulations requiring IRBs24 stated:
46.1 Applicability
| (a) |
The regulations in this part are applicable to all Department of Health, Education, and Welfare grants and contracts supporting research, development, and related activities in which HSoR are involved. |
The regulations did not apply to NIH’s intramural program until the 1981 revised regulations25 were published, but with a loophole to provide flexibility:
46.101 To what do these regulations apply?
| (a) |
Except as provided in (b) of this section (i.e., categories of exempted research), this subpart applies to all HSR conducted by the Department of Health and Human Services and funded in whole or in part by a Department grant, contract, cooperative agreement or fellowship. |
(1)This includes research conducted by Department employees, except each Principal Operating
Component head may adopt such nonsubstantive, procedural modifications as may be appropriate from an administrative standpoint.
In 1991, Subpart A of the regulations was extended by the Common Rule to apply to all HSR conducted, supported, or otherwise subject to regulation by any Federal Department or Agency.26
B-9
In 1993, Congress finally closed the gap by specifically requiring all research conducted by the NIH be subject to IRB review:27
Section 492A (a) Review as Precondition to Research
A) … [requirement of prior IRB review of all applications to the Secretary for financial assistance to conduct research…]
B) In the case of research that is subject to review under procedures established by the Secretary for the protection of human subjects in clinical research conducted by the National Institutes of Health, the Secretary may not authorize the conduct of the research, unless the research has, pursuant to such procedures, been recommended for approval.
What explains this long period of incongruence and differences of public accountability to federal regulation? Three factors influenced this delay. The first factor was that the source of leadership for reform of research ethics in the mid-1960s as well as the substance of that reform arose from within the NIH and was promulgated outward for grantees and contractors. NIH officials, especially Dr. James Shannon, led the response to widespread evidence of abuses of HSoR and fashioned the requirement of local prior group review as U.S. public policy.28 Dr. Shannon and the Surgeon General, Dr. Luther Terry, presented the arguments for this policy to the National Advisory Health Council in September 1965.29 It did not occur to them to require prior group review intramurally because it was already being done. Later, directors of the NIH and leaders of the intramural program in the period 1971–1981 probably did not believe that the regulations should apply to them because they were already highly self-regulated and believed that they were doing what the regulations required. In truth, a great deal had been done.30
1) Protection of HSoR Within the NIH Intramural Program
When the Clinical Center opened in 1953, a document had been prepared, based on extensive discussion, requiring “group consideration” of clinical research procedures that “deviated from acceptable medical practice or involved unusual hazard.”31 A Clinical Research Committee (CRC) was organized as a subcommittee of the Medical Board of the Clinical Center. The CRC was designed as an “expert body” to deliberate scientific and ethical questions in research proposals that were referred to it. Between 1953 and 1966 three types of research were required to be referred to the CRC: research with patients involving unusual hazard (1953), research with normal volunteers (1954), and purely investigational (nontherapeutic) research with patients (1961). The director of the NIH exercised second-level review of normal volunteer studies. Also, from 1953, internal Clinical Center staff who volunteered for research had to meet written consent requirements.
Prior to 1966, NIH intramural leaders changed policy and procedures to ensure more protection of HSoR. In 1964, an ad hoc committee was appointed by Dr. Jack Masur, Director of the Clinical Center. The group was charged with the evaluation of practices in group review and informed consent since the 1953 document. Led by Dr. Nathaniel Berlin, the National Cancer Institute (NCI), the committee did a major study of the existing system and interviewed each clinical director and many senior investigators. Its recommendations were adopted in July 1966, and prevailed until further revisions were made in 1976 and 1977.
The specific change was to require review bodies (CRCs) within each institute. These bodies were charged to review patient research that fell outside the boundaries of accepted practice. The institute CRC or clinical director could refer a controversial project to the medical board’s CRC. Written informed consent was required only of normal volunteers. Patient consent could be given verbally with a note in the chart by the responsible physician. All normal volunteer studies remained under the aegis of the medical board’s CRC.
Federal regulations of 1974 led to a response from the intramural program and more changes in 1975–1977. All patient and normal volunteer studies were centralized in a two-level system of review.32 The official review bodies in each institute were renamed Institute Review Subpanels,33 and their membership enlarged to include
B-10
a richer mix of scientists and nonscientists from outside government. The author served as an outside member on a Subpanel at the NCI from 1975–1977. After 1977, I was responsible for helping NIH intramural officials to complete the process of shaping the Subpanels.
The drafters of the 1974 regulations were NIH officials whose attention was aimed at reducing research risks in the extramural program.34 Under congressional pressure, the 1974 regulations were hurriedly constructed. Little attention was devoted to bringing the intramural research programs under the regulations, because intramural research was not covered in the 1971 policy that served as a model for the regulations. These officials were also confident that the intramural program was reasonably well regulated.
Pressure for congruence of applicability of the regulations began to mount in the mid-1970s due to OPRR’s mandate and influences of the work of the National Commission on the intramural program. The revised 1981 regulations created congruence (with a loophole), and the intramural program’s Assurance was negotiated and approved by OPRR in 1981.
2) Protection of the NIH Intramural Program
A second factor influencing a degree of institutional blindness to the incongruence was the prominent and protected environment of the NIH intramural program in this period. One must assume efforts by NIH’s directors to protect scientific freedom and flexibility in the intramural program, as well as their belief that its internal practices of peer review were sound. Flexibility and freedom from restrictions on research were prized values. Many research ideas were born by experimentation and observation in a single patient. Regimentation of almost any kind was considered an anathema.
The first three years of the author’s service in the intramural program (1977–1987) were marked by challenges to a long tradition of freedom from external oversight and treasured flexibility in research practices.35 The areas of sharpest conflict were over a) complaints from patients and family members about lack of informed consent, b) the obligation to seek informed assent of children to research or major medical procedures, c) disclosure of psychologically sensitive information to patients, d) changing protocol strategy in midcourse without Subpanel permission, e) conflicts of interest in Subpanel review of protocols of Scientific and Clinical Directors of the Institutes, f) testing normal volunteers for psychopathology, and g) complaints of pressure on normal volunteers to complete studies.
At this time, there were internal struggles between advocates of NIH’s past and advocates for change. Many intramural officials felt strong pulls from both sides. The former argued for a type of “ethics exceptionalism” allied with the strong research culture. NIH scientists and officials with careers spanning the 1960s and 1970s tended to view their roles and mission as exceptional. They also viewed subjects’ participation in clinical research largely as beneficent, in part due to the quality of medical care received. Also contributing to this view was the fact that the costs of research and patient care were borne by the federal government, including patient and family travel costs and housing. Advocates for change appealed to the larger claims of social movements, of values that informed legal issues in medicine, and of bioethics as a discipline. The work of the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (1980–1983) made a strong case for these claims bearing on the practice of medicine. The work of the President’s Commission had effects in the intramural program. The same officials who wrote the 1974 regulations had been strongly influenced by the work of the National Commission and the President’s Commission. They saw the imperative for congruence of public accountability between the two programs and effected it in 1981.
3) Social Distance Between Extramural and Intramural
A third factor was the social distance between intramural and extramural programs described above. Each program had different leadership who rarely talked with one another. Neither wanted to be governed by the other. Failure of intramural leaders to communicate with extramural leaders was a significant reason, among others, why the protection of HSoR was not extended to the intramural program.36
B-11
| B. |
Problems and Conflicts Linked to OPRR’s Location |
| 1. |
OPRR’s Authority and (NIH’s) Institutional Blindness to Conflicts of Mission and Interests |
OPRR’s authority to require Assurances derives from the 1974 Act, which formalized the practice of obtaining written Assurances from DHHS-funded research institutions of their commitment to the ethical conduct of research. Before the 1974 Act, NIH had already developed such Assurance documents with many research universities, which were reviewed by OPRR. Even today, approval of an Assurance does not involve a site visit but reviews of paperwork and telephone discussions.
OPRR’s Assurances are of several types. MPAs pledge compliance for all federally funded projects as well as a voluntary pledge regarding compliance in the context of privately funded research. Renewals are for a five-year period. OPRR currently has 448 MPAs with 756 entities. At non-MPA institutions, a Single-Project Assurance agreement must be negotiated with OPRR for each individual study. OPRR must negotiate each of these agreements as well as approve the consent document. OPRR today is holding approximately 3,000 active Single-Project Assurances. There are also cooperative project Assurances for large multiple site studies. Today, OPRR has more than 1,500 active cooperative project Assurances.
The NIH is an MPA holder with the OPRR. OPRR is the authority for assessing the NIH’s compliance with federal regulations to protect human subjects. There have been longstanding concerns about the independence of OPRR and its ability to oversee the NIH itself, especially the NIH’s intramural program. The GAO report to Senator Glenn cited above pointed to “a potential weakness…because NIH is both the regulator of human subjects protection issues as well as an institution conducting its own human subjects research. The Director of NIH, therefore, has responsibility for both the success of NIH’s intramural research program and for the enforcement of human subjects protection regulations by OPRR.”37 The GAO report was also critical of the fact that it took the NIH five years to respond to compliance violations in the intramural program as noted by OPRR in 1991.
A recent report of the Human Ethics Research Group of the University of Pennsylvania recommended that “the placement and role of the (OPRR) in the regulatory system should be reassessed.” The report stated:
The primary mission of the federal regulations is to protect research subjects. One important obstacle to reform in this area is structural: The agency charged with enforcing and interpreting the regulations, the OPRR, is part of a larger bureaucracy that is also its major client and one of the nation’s leading sources of research funding, the NIH. As a matter of principle, the agency should not be located within the structure of any government funder, and its charter should specify that it is independent. Obviously, the agency would have to continue to be accountable to the professional and lay constituencies which its serves, and a suitable reporting structure would have to be devised.38
Dr. Harold Varmus, NIH Director, denied any conflict of missions or institutional interests. He wrote in response to the GAO report, “In fact, the OPRR oversees and interacts with the NIH just as with any extramural institution.”39 Dr. Varmus argued that there was no weakening of OPRR’s independent oversight and authority, because “the lines of authority of the NIH Deputy Director for Intramural Research and the OPRR Director do not cross within NIH.” He also attributed the five-year span to resolve the violations to “the complexity of fully implementing the corrective actions rather than a function of weakness in the OPRR’s ability to enforce human protection regulations within the NIH organizational structure.” Dr. Varmus did not discuss the nature of the “complexity” or address the proposition that the NIH was demonstrating by its behavior the basic conflict of institutional interests. His answer to GAO’s critique was essentially that it was resolved internally as a matter of lines of authority. The GAO report rightly reiterated before closing, “We disagree with NIH’s conclusion and believe that a potential weakness exists in OPRR’s ability to enforce human subject protection regulations within NIH.”40
B-12
Representative Shays (R-Conn) questioned Dr. Varmus at a recent (May 8, 1997) hearing of a House subcommittee on the conflict of interest issue in regard to the location of OPRR at the NIH. Dr. Varmus responded, as reported in “The Blue Sheet”:
It is important to remember that the office (OPRR) does not have any vested interest in seeing the research go forward….The research is being funded by CDC or the institutes, each of which has its own authorization and its own appropriation and it is the institutes that are responsible for funding the studies, so there really isn’t any conflict of interest.41
If Dr. Varmus was correctly quoted, this answer evades the basic question of conflict of missions and interests between OPRR and NIH by focusing on funding as the causative factor of conflicts of interest. The fundamental question is whether OPRR is less than effective—due to pressure from conflicts of interests—by being located at the NIH. In my view, the GAO’s term “potential weakness” as applied to OPRR’s ability to enforce the regulations within the NIH is more accurately termed a “past, present, and persistent weakness” due to location in a powerful parent organization that, in effect, looks down on OPRR, rather than respecting its authority and moving quickly to correct violations.
On behalf of human subjects, OPRR as the enforcer of federal regulations can use requirements for IRB review and informed consent to reduce excessive risks. However, when it comes to confronting powerful political and bureaucratic interests, OPRR’s power on behalf of human subjects is greatly limited by its location and identity as an office of the NIH. OPRR does not, as a matter of fact, have effective and independent oversight over NIH’s intramural or extramural programs, nor the research programs of other DHHS agencies, e.g., the CDC or the FDA, on the relatively rare occasions when it conducts or sponsors research. The records and documents that I have examined, while confidential in many details, strongly support this finding.
The tools that OPRR has developed in order to gain compliance from other institutions are: 1) fear of loss of funding, 2) respect for OPRR (the office/the authority), 3) respect for the primacy of human subjects protection, and 4) fear of bad publicity. The first tool is utterly useless in PHS agencies, since funding for the agencies is assured and self-administered. The second tool is greatly diminished in PHS agencies, because they perceive OPRR as a small and weak office within the NIH. Respect for the primacy of HSoR protection is missing to an often startling degree in PHS agencies, as evident in recent documents which I have examined. Taken all together, OPRR lacks the political capital to 1) impose serious measures and 2) to move an agency quickly towards correction of problems, especially when CDC or NIH performance regarding compliance is a subject of scrutiny.
Specific Examples:
The following are specific examples of problems posed by OPRR’s location:
1) Burdened policy and rule-making process. Proposed changes in rules or regulations must be vetted by officials at a minimum of 11 sign-off points within the NIH bureaucracy, even before moving out to PHS and DHHS levels.42 Each one of these levels of bureaucracy has its own vested interests in funding of science, in a scientific mission, or in an aspect of NIH-related activity. The process of consideration of rules and policy changes regarding protection of human subjects is subjected to multiple sets of vested interests in an institution that is supposed to be regulated by OPRR.
2) Resources. OPRR’s resources (i.e., funding and staff) have remained static for years, despite growth through the 1980s and 1990s in appropriations and a concomitant increase in volume of proposals for biomedical and behavioral research sponsored by the NIH. OPRR is currently funded at $2 million with 22 staff members who devote some or all of their time to HSoR protection and another eight staff members devoted to animal
B-13
welfare. That figure includes two volunteers and a consultant who have been recruited to the workforce. Congress itself—not the agency that would have to divert funds that it might wish to expend for other purposes—is the proper body to assess the funding and staffing needs of a national agency for oversight of human subjects.
3) Climate and morale. The performance of OPRR employees and promotions and awards are assessed by officials in an agency responsible for a scientific mission that houses OPRR. Although the performance of any OPRR director and his or her small staff may be outstanding, considered within the circumstances and pressures within which they work, the decisionmaking climate and morale are too dependent on concern about consequences within the NIH itself for the OPRR.
4) Lack of respect for OPRR’s authority. OPRR is specifically located within NIH’s Office of Extramural Research. In an interview43, the Office’s Deputy Director, Mr. Geoff Grant, described “various compliance requirements governing human subjects, animal welfare, and conflict of interest” as a “robbery” that is “distracting to research.” Dr. Ellis asked him if he had been quoted accurately in the article, and he verified that the quote was indeed accurate.
Another example of lack of respect emerges by comparing the time required for the NIH to make changes regarding compliance with the performance of other institutions. GAO identified 17 instances (including NIH itself) from 1990–1995 in which OPRR imposed a restriction on an institution’s authority to conduct HSR. GAO found those restrictions were lifted by OPRR in most cases after 12 to 18 months, when appropriate institutional corrective actions were taken. The NIH needed five years to implement corrective actions after being cited by OPRR in 1991 for compliance violations.
Analysis of time domains of OPRR’s governance of HSoR protections in another DHHS agency (documents are marked “confidential”) is similarly telling. The agency reported to OPRR, and OPRR independently identified a number of instances in which the agency failed to ensure that performance site institutions (in the dozens) conducting agency-supported research held an applicable OPRR-approved Assurance of compliance with the human subjects regulations. OPRR advised agency officials of these findings during the closing session of an August 1993 site visit. Twenty-one months later (September 25, 1995), OPRR reported that “...agency officials have informed OPRR that awards management procedures were recently modified to ensure that all institutions participating in human subjects research supported by—the agency—hold applicable OPRR-approved Assurances.” However, the truth is that the agency is still working to provide information and documentation to OPRR that will permit Assurance for all of the agency’s human subjects research. Four years have elapsed and the problems are still not solved. The numbers involved are very large.
Responses as sluggish as those seen in DHHS agencies are unknown among other institutions assured by OPRR. The protracted time periods consumed by DHHS research agencies to bring ongoing human subjects research into compliance with (what for these agencies are longstanding) regulations for protection of human subjects demonstrate that OPRR is not effecting proper HSoR protections from its position within the NIH. In the larger framework of government, DHHS and the Office of the President bear the ultimate responsibility for this problem and for initiatives regarding solutions.
5) Misunderstanding the scope of the Assurance. A final example is related to the OPRR-approved Assurance of Compliance held by the NIH. This example illustrates the NIH’s lack of understanding of, and/or lack of respect for, the authority of OPRR and, together with the comparatively sluggish response to citations, refutes Dr. Varmus’ assertion that “the OPRR oversees and interacts with the NIH just as with any extramural institution.”
B-14
The July 1, 1992, Assurance is “applicable to all research activities that, in whole or part, involve human subjects if…the research is conducted or supported by or under the direction of any employee of the NIH in connection with his/her institutional duties, regardless of the site of the activity....” On February 9, 1994, the NIH official signing the Assurance informed OPRR that “NIH has amended the Applicability section of its Multiple Project Assurance [MPA] with the following rewrite:”
applicable if ‘the research is conducted or supported by the Intramural Research Program (IRP) of the NIH by or under the direction of any employee of the NIH, regardless of the site of the activity….’
NIH stated the change reflected “a more precise statement of the fact that the NIH MPA does not apply to all NIH employees or research activities, but only to those individuals, either intramural or extramural, whose research is conducted or supported by the IRP in connection with their institutional duties.” In response (February 14, 1994), OPRR acknowledged receipt of the “proposed” (OPRR’s pointed characterization of NIH’s February 9, 1994, memorandum) amendment to NIH’s OPRR-approved MPA. OPRR reminded NIH that the terms of the NIH MPA approved by OPRR in July 1992 “remain in effect.” OPRR stated that it “looks forward...to negotiating any changes” in the MPA that NIH may elect to pursue. More to the point, OPRR stated: “Before OPRR can consider approving the proposed amendment, it will be necessary for NIH to clarify and define with as much specificity as possible the full dimensions of the ‘Intramural Research Program.’” NIH did not respond to OPRR. The revision pursued by the NIH signatory official would have, inexplicably, left the human subjects in research conducted by some number of NIH employees (i.e., those not supported by the IRP) without the institutional protections conferred by an Assurance.
Some three years later (April 21, 1997), OPRR found that the electronic text of the July 1992 NIH MPA existing on the NIH website differed from the OPRR-approved MPA in an important way. The “Applicability” had been altered to omit the language in effect (i.e., applicability to research undertaken by “...any employee of the NIH….”) and bore the new language sought by NIH in its February 9, 1994, correspondence to OPRR. Within two days after OPRR called this deviation to NIH’s attention, the actual “Applicability” language currently in force appeared on the NIH website.
In concluding this part, the report has provided examples of the effects of conflicts of interests that arise from a basic conflict of missions between the OPRR and the NIH. The latter’s mission is to promote, fund, and to conduct biomedical research. The NIH’s housing the OPRR is an arrangement that may have been acceptable in the past but does not fit the current scope and mission of OPRR in the 1990s and beyond. The basic mission of OPRR as regulator is organized around the primacy of the rights and welfare of human subjects. Like human subjects themselves, the OPRR’s mission is confronted by and too often subjugated to a powerful and complex set of countervailing interests: a) scientific and funding interests and b) political and bureaucratic interests. The best remedies for the aforementioned problems of conflicts of mission and conflicts of interests are independent oversight and unfettered lines of authority.
IV. Lessons from Other Regulatory Agencies
One does not need to look far to find similar histories in two other federal agencies. A clear parallel exists in the creation of the NRC from the AEC in 1974. The AEC came under massive public and congressional criticism for trying at once to promote nuclear power and regulate its uses. Similar incompatibility of functions led to an imperative to move the OGE out of the Office of Personnel Management in 1989. Some of the problems of adequate staffing and freedom of action that burden OPRR’s effectiveness were resolved by creating new
B-15
agencies. Both agencies today are independent and adequately funded for their tasks.44 There is a striking contrast between the OGE’s and the OPRR’s resources for education. OPRR has no staff dedicated solely to education of IRBs, although Congress mandated this role. In 1992, OGE had five staff dedicated to education of ethics practitioners and trainers.
Both agencies have capabilities that would strengthen OPRR or its successor. They can propose and finalize regulations in the Code of Federal Regulations; visit and/or audit their clientele; promulgate guidance and educational materials for consumption by their clientele; and independently govern pertinent activity within another Federal Department or Agency.
| V. |
Recommendations A. Elevation and Independent Location |
Despite a political climate that mitigates against the direction of these recommendations, the time has come to elevate the OPRR and create an adequate agency with an independent location. Initiatives from the DHHS and the Office of the President would greatly strengthen the plausibility of such solutions. An initiative from the White House is appropriate, inasmuch as OPRR’s successor should be separate from the DHHS agencies that it oversees (NIH, CDC, FDA, and others) and have authority in relation to the 17 other Federal Departments or Agencies that conduct HSR according to the Common Rule. OPRR is a consultant to these agencies, but has no direct authority over them. Also, if the direction of universal protection of human subjects is legally and ethically sound, all human subjects of research in privately funded projects and their sponsors will require representation and oversight. That there are many examples and complaints regarding exploitation of “most vulnerable” research subjects beyond the scope of existing legal protections has been documented by Dr. Ellis in a communication to NBAC.45 If Congress legislates to guarantee legal protection of all research subjects and impose sanctions for violations of federal policies and rules for HSR, broadening the authority of a successor to OPRR to regulate all HSR activities would be a logical step. An agency with such authority would quickly move from negotiating Assurances with research sponsors to a simple requirement for annual registration. Registration would involve research sponsors providing information on the twin protections of HSoR: informed consent and IRB review. Registration would also yield more data about the actual incidence and magnitude of HSR in the United States. This information is not currently available.
Recommendation 1: That the NBAC endorse the creation by Congress of a successor to OPRR: the National Office of Human Subjects Research (NOHSR). The NOHSR will have all of the present functions of OPRR with respect to DHHS and its Agencies. Additional authority should be given to NOHSR over all Federal Departments or Agencies conducting or funding HSR, as well as over privately funded HSR. The NOHSR should be headed by a single Director46 to be nominated by the President, subject to the advice and consent of the U.S. Senate. The NOHSR should be accountable to Congress and funded by congressional appropriation. A location within the Executive Branch is a logical step, similar to the OGE, but it should be an independent agency accountable to Congress and reporting to the President. The NOHSR’s initial resources would require a staff of 45 to 50 individuals and a funding level of $6 million to $7 million.47 The report strongly recommends moving OPRR outside the PHS as a permanent solution to the conflict of missions and conflict of interest problems. If creating a new independent agency may be problematic for Congress at this time, an interim solution would be to relocate OPRR alongside or within an existing and effective independent agency, e.g., the OGE. Other partial solutions would be intolerable. For example, some consider reinventing OPRR by investing its mission and mandate in the Human Subjects Research Subcommittee of the Committee on Health, Safety, and Food, National Science and Technology Council. The Subcommittee was originally chartered to write the Common Rule and continues to meet six times annually as a discussion
B-16
group of issues facing the 17 Departments covered by the Rule. This body has no staff and no funds. Locating OPRR within this weak entity makes no practical or political sense.
Part of this recommendation is to require that only Subpart A of DHHS regulation—the Common Rule—apply to new research sponsors and private sector institutions. The other subparts of DHHS regulations are dated and require scrutiny.
Recommendation 2: Congress should also create a National Advisory Committee for Human Subjects Research (NACHSR) through the Federal Advisory Committee Act. NACHSR’s role is to be the main source of advice and guidance on HSR policy and ethical issues to the NOHSR and to the nation. The NACHSR (11 to 13 members) will serve as a permanent forum for debate and resolution of issues referred to it by the nation’s IRBs, new ethical issues arising in HSR, problematic cases, and ongoing interpretation and application of ethical principles and rules governing HSR. The NACHSR would answer longstanding appeals by Katz and others48 for such a body. These appeals for such a permanent body extend back to the report of the Ad Hoc Advisory Panel that examined the Tuskegee Syphilis Study (1973).49 The NACHSR should have terms of office not to exceed three years, with one-third of members able to succeed themselves one time; it should meet quarterly and on special request of the Director, NOHSR, and its chairperson could succeed him or herself for one second term.
Twenty-seven other nations have established standing national bodies commissioned to work on bioethical issues.50 Seventeen nations have national bodies with specific missions to work on HSR policy and guidance to IRBs. These nations are listed in Attachment 2. The United States should not only create such a permanent advisory body alongside the NOHSR but should lead the rest of the world in strengthening the governmental voice of HSR protections, elevating its status, and providing an independent and less problematic location for it.
Attachment 1
Chart of Sign-Off Points Within NIH
B-17
Attachment 2
Other Nations with Standing National Commissions or Agencies with Oversight for HSR Policy and Practices
Argentina - National Bioethics Commission (1992) - secretarial.
Canada - National Council on Bioethics in Human Research (1989) - Established by the Medical Research Council, National Health and Welfare Canada, and Royal College of Physicians and Surgeons. Defines guidelines, advises IRBs, and promotes public and professional education in research ethics.
Denmark - Central Scientific-Ethical Committee (CSEC) (1978) - Given statutory authority in 1992. Acts on disputed proposals and in cases where a matter of principle needs to be decided.
Danish Council of Ethics - Broader mandate and disagrees with CSEC on issues of preserving brain tissue for research and teaching and on definition of death. Parliament told them to cooperate.
Finland - Finnish National Research Ethics Committee (1991) - A permanent advisory body of the government. Makes proposals, gives expert statements, promotes research ethics (has no teeth).
France - French National Consultative Ethics Committee on Life and Medical Sciences (1983) - Created by the President (Mitterand) to advise the government on issues of bioethics. French Parliament uses its work to make law. Has a small staff.
Hungary - Scientific and Research Ethics Committee (1987) - Established by the Hungarian Scientific Research Council. Parent forum overseeing HSR; coordinates regional research ethics committees, publishes guidance.
Israel - Supreme Helsinki Committee - Convened by the Director General of the Ministry of Health when research in sensitive areas is proposed.
Italy - National Committee on Bioethics (1990) - Created by the President of the Council of Ministers. Provides advice to Parliament (meets in closed sessions, no staff).
Mexico - National Bioethics Commission (1992) - Reports to the Ministry of Health.
Netherlands - Commission on Health Ethics and Health Law (1977) - Sponsored by the Health Council, this commission transmits findings to the government of the work of subcommittees organized by the Health Council. In 1989, Minister of Health created Dutch Interim Central Committee on Ethical Aspects of Medical Research. This national advisory commission on research ethics directly advises local medical ethics boards, not the government; recommendations are nonbinding.
New Zealand - Health Research Council Ethics Committee (1990) - Advises the Health Research Council on ethical issues in research.
Norway - Parliament created three bodies: 1) National Committee for Medical Research (already there but non-statutory), 2) for social sciences, and 3) for science and technology (1989).
Phillipines - National Ethics Committee and IRBs (1987) - Created by Phillipine Council for Health Research and Development.
Poland - Ethics Review Committee in Biomedical Research (1977) - Created by Ministry of Health;
Commission for Supervising Research on Human Subjects (1982) - Created by Ministry of Health and Social Welfare; and Commission for Research Ethics (1991).
Russia - Russian National Committee on Bioethics (1992).
B-18
Sweden - Medical Research Council houses a central committee that oversees local research ethics committees concerned with individual research projects. National Council on Medical Ethics - (1985) - Links science, public, and Parliament.
U.K. - Several bodies, including the Nuffield Council on Bioethics (1991) - A private group that acts as though it was government appointed. Establishes working groups and has an executive secretary and two staff members. No oversight of local research ethics committees.
Source: U.S. Congress, Office of Technology Assessment. Biomedical Ethics in U.S. Public Policy Background Paper, OTA-BP-BBS-105. Washington, DC: U.S. Government Printing Office, June 1993.
Notes
1 The abbreviations HSoR will be used for “human subjects of research” (focus on the human beings who are research subjects) and HSR for “human subjects research” (focus on the activities of research involving human subjects).
2 At its May 17, 1997, meeting, the NBAC voted unanimously for this statement: “No person in the United States should be enrolled in research without the twin protections of informed consent by an authorized person and independent review of the risks and benefits of the research.”
3 McDermott, W., Opening Comments. The Changing Mores of Biomedical Research. A Colloquium on Ethical Dilemmas from Medical Advances, Ann Int Med 67 (Supp.7, No. 3-Part II):39–42, 1967. “…the hard core of our moral dilemmas will not yield to the approaches of ‘Declarations’ (i.e., Helsinki) or ‘Regulations’ (i.e., the FDA’s 1967 human subjects regulations); for as things stand today such statements must completely ignore the fact that society, too, has rights in human experimentation” (p. 42).
4 Jonas, H., Philosophical Reflections on Human Experimentation, Daedalus 98:245, 1969.
5 Ibid.
6 Dommel, F.W., Alexander, D., The Convention on Human Rights and Biomedicine of the Council of Europe, Kennedy Institute of Ethics Journal 7(3):259–276, 1997.
7 Robertson, J.A., The Scientist’s Right to Research: A Constitutional Analysis, Southern California Law Review 51:1203–1279, 1977.
8 This term was coined by Paul Appelbaum, and the widespread power of its influence was ascertained in the Subject Interview Study of the Advisory Committee on Human Radiation Experiments, in which 1,882 patients receiving medical care in 16 outpatient facilities of private and public hospitals were surveyed.
9 “Drug manufacturers offer clinician-investigators financial inducements to enter patients into studies, typically $2000 to $5000 per patient. By contrast when a patient is entered into a NIH-sponsored study, the clinician-investigator receives capitation of approximately $1000 per patient to cover the costs of the physician-investigator’s time, the data manager’s salary, and additional expenses (secretarial, photocopying, etc.) incurred in participating in the study.” Shimm, D.S., Spece, R.G., DiGregario, M.B., Conflicts of Interest in Relationships Between Physicians and the Pharmaceutical Industry, in Spece, Shimm, and Buchanan (eds.),
Conflicts of Interest in Clinical Practice and Research, New York: Oxford University Press, 1996, 323.
10 These problems are described in three recent reports: U.S. General Accounting Office, Scientific Research, Continued Vigilance Critical to Protecting Human Subjects, 1996. GAO/EHS-96-72; Advisory Committee on Human Radiation Experiments, Research Ethics and the Medical Profession, JAMA 276:403–409, 1996; and Moreno, J.D., Caplan, A.L., Wolpe, P.R., and the Members of the Project on Informed Consent, Human Research Ethics Group, “Updating Protections for Human Subjects Involved in Research, JAMA, 1998 280(22):1951–1958.
11 Nishimi, R.Y., Testimony for the House Committee on Government Operations, The Federal Role in Protecting Human Research Subjects, 103rd Congress, 2nd Session, September 28, 1994: 158–160.
12 The occasion for the interviews was to prepare papers for presentation at the 125th anniversary of the Norwegian Academy of Sciences and for subsequent publication; i.e., Fletcher J.C., The Evolution of the Ethics of Informed Consent. In Research Ethics, Berg K., TranØy K.E. (eds.), Alan R. Liss, Inc., New York, 1983, 187–228; Boverman M., Fletcher J.C., The Evolution of the Role of an Applied Bioethicist in a Research Hospital. In Research Ethics, Berg K., TranØy K.E. (eds.), Alan R. Liss, Inc., New York, 1983, 131–158.
B-19
13 Erde, E.L., Conflicts of Interest: A Conceptual Overview, in Spece, Shimm, and Buchanan (eds.), Conflicts of Interest in Clinical Practice and Research, New York: Oxford University Press, 1996, 13.
14 Adapted from Erde, see note 13, p. 33.
15 The impact on the NIH of a case involving Dr. Chester Southam’s research at the Jewish Hospital for Chronic Diseases in Brooklyn, New York, had, in the author’s view, the most telling and persuasive influence leading to change. Dr. Southam’s license to practice medicine in New York was suspended for one year, and then he was placed on probation. For accounts of this case in historical context, see Langer E., Human Experimentations: New York Affirms Patients’ Rights. Science 151:663–665, 1966; Fletcher J.C., The Evolution of the Ethics of Informed Consent. In Research Ethics, Berg K., TranØy K.E. (eds.), Alan R. Liss, Inc., New York, 1983, 187–228.
16 The term “institutional blindness” refers to the end-state of excessive loyalty to the welfare of an institution and one’s role within it. The stronger and more uncritical the loyalty to an institution and role, the more impaired are independence of observation, judgment, and action with respect to prevention or moderation of conflicts of interest. Some professions are much better prepared and trained than others to detect and prevent conflicts of interests. Physicians and biomedical researchers do not receive the same degree of education and training about such issues as attorneys and behavioral scientists. For example, “because physicians are not trained to look for conflicts of interest, they often find themselves enmeshed in them without recognizing the problem.” Spece R.G., Shimm D.S., Buchanan A.E., Conflicts of Interest in Clinical Practice and Research, New York: Oxford University Press, 1996, preface.
17 See the ACHRE report cited in note 10 for description of the norms of the wider research community, at 404–405.
18 Jones, J.H., Bad Blood, 2nd ed., New York: Free Press, 1993, 191–196.
19 What was the involvement of the NIH, if any, in the Tuskegee study? The pre-1950s NIH was involved in analyzing spinal fluid and autopsy tissues from the subjects. Jones, see note 18, 124, 136. It is likely that no NIH physician-investigator or official was directly involved in the study itself or in its defense against Buxton’s challenges. (James Jones, personal communication, November 10, 1997). Dr. John Heller was a junior officer in the PHS Division of Venereal Diseases who was actively involved in the study. Following his retirement as President of Sloan Kettering Hospital, he was in residence at the National Library of Medicine. In an interview with James Jones in 1977, Dr. Heller described his experience in meetings led by Dr. Raymond Vonderlehr, with the medical societies and boards of health of four Alabama counties in 1933: “...no one questioned whether the experiment was ethical; no one even came close to doing so. ‘I don’t recall any philosophical discussions at all,’ declared Dr. Heller. What emerged from his comments was the image of a profession whose members had closed ranks behind a study they were told had real merit. The experiment obviously had struck their sense of scientific curiosity, and it did not occur to anyone to suggest that it should not be conducted.” Jones, see note 18, p. 144.
20 Although Peter Buxtun, a PHS employee, challenged the ethics of the Tuskegee study from within DHEW as early as November, 1966, PHS officials did little to heed his criticism. The Tuskegee story was broken by the Associated Press on July 25, 1972, in a report by Jean Heller. Cf. Jones, J.H., Bad Blood, 2nd ed., New York: Free Press, 1993, 188–205. The author conducted numerous interviews and 10 focus groups with scientists and clinical investigators at the NIH from 1966 to 1968 in preparation for a Ph.D. dissertation on the ethics of medical research. No one brought up the Tuskegee study. The author was unaware of it until the news story.
21 Beecher, H.K., Ethics and Clinical Research, N Engl J Med 74:1354–60, 1966. The occasion for the interviews was to prepare papers for presentation at the 125th anniversary of the Norwegian Academy of Sciences and for subsequent publication, i.e., Fletcher, J.C., The Evolution of the Ethics of Informed Consent. In Research Ethics, Berg K., Tranfy K.E. (eds.), Alan R. Liss, Inc., New York, 1983.
22 In an earlier interview, Charles R. McCarthy, former director of the OPRR, commented: “It seems to me that…for the most part government was passive, a few farsighted individuals such as Shannon and Stewart in the Executive Branch, and Ted Kennedy in the Congress, initiated procedures that have matured into a remarkable system. These few individuals were both learners and teachers, but the government as a whole was at best a sleepy, distracted pupil, awakened periodically by a scandal, but otherwise content to ‘get by’ without having to recite” (personal communication, May 14, 1993).
23 Surgeon General, PHS, DHEW, Investigations Involving Human Subjects, Including Clinical Research: Requirements for Review to Ensure the Rights and Welfare of Individuals, PPO 129, Revised Policy, July 1, 1966.
24 DHEW, 45 Protection of Human Subjects 46, Federal Register, Vol. 39, No. 105, Part II, 46.1(a) (b), 1974.
25 Federal Register, Vol. 46, No. 16, January 26, 1981.
26 45 CFR 46.101(a), 56, Federal Register 28003, June 18, 1991.
27 The National Institutes of Health Revitalization Act of 1993, Public Law 103-43, June 10, 1993, Section 492A.
B-20
28 This story is well told in Faden, R.R., and Beauchamp, T. L., A History and Theory of Informed Consent, New York: Oxford University Press, 1986, 206–215.
29 Ibid., 208.
30. A fuller history of HSR protection and the evolution of prior group review in the NIH intramural program is found in Boverman M., Fletcher, J.C., the Evolution of the Role of an Applied Bioethicist in a Research Hospital. In Research Ethics, Berg K., TranØy K.E. (eds.), Alan R. Liss, Inc., New York 1983, 131–158.
31 NIH. 1958. Group Consideration of Clinical Research Procedures Deviating from Accepted Medical Practice or Involving Unusual Hazard. (Memorandum, approved by the Director, NIH, 1953); Sessions, S.M., What Hospitals Should Know About Investigational Drugs—Guiding Principles in Medical Research Involving Humans, Hospitals 32:44–64.
32 Lipsett, M.B., Fletcher, J.C., Secundy, M., Research Review at NIH, Hastings Center Report 9:18–27, 1979.
33 These programs were called “Subpanels” to overcome the difficulty of having each chartered under the Federal Advisory Committee Act, because each had one or more outside members.
34 The members of the drafting committee were Charles Lowe, Jane Fullerton, and Charles McCarthy (Charles McCarthy, personal communication, November 11, 1997).
35 See note 30.
36 McCarthy, C.R. (personal communication, November 11, 1997).
37 U.S. General Accounting Office, Scientific Research, Continued Vigilance Critical to Protecting Human Subjects, 1996. GAO/EHS-96-72, 20.
38 Project on Informed Consent of the Human Research Ethics Group. Updating Protections for Human Research Subjects, submitted for publication, 1997.
39 Letter, Harold Varmus to Sarah F. Jaggar, February 15, 1996 (see GAO report, 33).
40 Note 37, at 25.
41 Research Administration, OPRR Location Questioned by Rep. Shays at Hearing, The Blue Sheet 40(20):2, May 14, 1997.
42 See Attachment 1 for a chart showing sign-off points within the NIH bureaucracy. Proposals for changing federal regulations that arise from NBAC’s deliberation on HSR, e.g., regarding studies involving cognitively impaired subjects, would in the near future necessarily be introduced through OPRR and be subject to the same vetting and sign-off process depicted in Attachment 1. Many of the entities in Attachment 1 have strong vested interests in the subject matter.
43 The NIH Record, June 18, 1996, 4.
44 U.S. Office of Government Ethics, Second Biennial Report to Congress, March, 1992; Walker, J.S., A Short History of Nuclear Regulation, January 1993 (NUREG/BR-1075).
45 Letter, Gary B. Ellis to James F. Childress, April 10, 1997.
46 The preference for agencies headed by a single administrator over a commission form of agency has been generally favored for some time by scholars in the administration sciences and based on research sponsored by the Committee on Government Operations. See 95th Congress, 1st Session. Study on Federal Regulation. Vol. 1. The Regulatory appointment Process, January, 1977.
47 Ellis, Gary B. (personal communication, October 18, 1997).
48 Katz, J., Do We Need Another Advisory Commission on Human Experimentation? Hastings Center Report 25(1):29–31, 1995.
49 U.S. Department of Health, Education and Welfare, Final Report of the Tuskegee Syphilis Study Ad Hoc Advisory Panel, 1973, U.S. GPO: 1973-747-022/5334, Region No. 4.
50 See Nishimi testimony, note 11.
B-21
PRIVACY AND
CONFIDENTIALITY IN HEALTH RESEARCH
Commissioned Paper
Janlori Goldman and Angela Choy Georgetown University

C-1
The Health Privacy Project is dedicated to raising public awareness of the importance of ensuring health privacy in order to improve health care access and quality, both on an individual and a community level.
Abstract
Health research can offer many benefits, such as the improvement of clinical practices, public health programs, and health products; the reduction of public health threats; the advancement of basic biomedical science; and the development and improvement of pharmaceuticals and medical devices.1 All of this research, however, requires access to a great deal of individuals’ data. This need for data often runs counter to the public’s desire to keep health information confidential. The public may have some reason to be concerned about the confidentiality of their health information. At present, there is no comprehensive federal law protecting the confidentiality of health information. The patchwork of state and federal laws varies in scope and tends to protect specific types of information collected and maintained by particular entities. A significant amount of research is conducted without federal oversight or review. Ultimately, the public’s fear and anxiety over the loss of privacy and confidentiality can threaten the research initiatives meant to benefit them. The federal government, researchers, Institutional Review Boards (IRBs), and research institutions will need to work together to provide strong privacy and confidentiality protections to build public trust and encourage continued participation in research.
I. Introduction
Individuals share a great deal of sensitive, personal data with their physicians.2 Full disclosure to health care providers is necessary for accurate diagnosis and treatment of the patient. While patients may expect—or desire—to have all of their health data kept confidential, it is not possible to protect confidentiality absolutely. In seeking health care, patients will likely experience some loss of privacy and confidentiality. Health data may be shared with pharmacies, employers, researchers, and even marketers for reasons not related to diagnosis and treatment. In fact, it is estimated that when a person goes to the hospital, approximately 150 different people will look at his or her records.3 But since patients are often not involved in decisions about the disclosure of their health data, they may be taken by surprise when they learn of disclosures—including disclosures to researchers. A recent Department of Health and Human Services (DHHS) Inspector General report found that “patients are often unaware that their records are being reviewed by persons other than their physicians and these records may be used to contact them about participating in research.”4 Historically, there has been tension between privacy advocates and researchers over how to address privacy and confidentiality issues. Consumer advocates often view research initiatives as threats to individual privacy, while researchers may treat privacy as a barrier to improving health. There is a fear that protecting confidentiality will prevent the free flow of health data for research, public health initiatives, and other health-related activities.5 Protecting privacy and confidentiality and promoting health, however, are values that go hand-in-hand. Without trust that the personal, sensitive data that they share with researchers will be handled with some degree of confidentiality, subjects will not participate in research projects.6 If people continue to withdraw from full participation in their own care, the personal health data from medical files and patient databases that researchers may rely on to recruit subjects or conduct records-based studies will be inaccurate and incomplete.
Researchers therefore need to be aware of potential privacy and confidentiality issues throughout the course of the research to incorporate privacy protections and minimize potential breaches of confidentiality. Public policies should also incorporate privacy standards so individuals will have greater trust in research enterprises and to ensure that there is accountability for breaches of confidentiality. Researchers may becoming more attentive to issues of security and use physical and technological measures, such as locked filed cabinets and
C-3
passwords to help protect against unauthorized access to data. But these security requirements do not answer the larger policy questions about how data should be used, shared, and exchanged.7 The key issue here is to determine which disclosures in health research are acceptable invasions of privacy and which limits are acceptable on confidentiality.
Currently, there is no comprehensive federal law that protects the confidentiality of all personal health data. Third-party access to medical records and other data—including researcher access to this data—is governed by a loose configuration of state and federal law, common law, and professional ethics. There are federal regulations that apply to some research involving human subjects. These rules, however, may be applied unevenly and may not be relevant for different kinds of research. Furthermore, it is generally believed that a significant amount of research falls outside the scope of these regulations. Reform efforts that seek to bolster existing rules and to expand the kinds of research subject to the rules, however, are met with a common critique: that the existing system of research review is already over-extended and that new requirements could place undue burdens on the system.
This paper addresses 1) the definitions of privacy and confidentiality; 2) the potential threats to privacy and confidentiality in research with a focus on the use of medical records and databases in health research;8 3) public concerns and potential consequences or harm from violations; 4) the existing statutory and regulatory requirements with regards to privacy and confidentiality in health research; 5) the potential impact of DHHS proposed federal health privacy regulations on health research; 6) what data exist on current research review policies and practices regarding privacy and confidentiality when health research is subject to IRB review and when it is not; and 7) what data exist regarding enforcement of the privacy and confidentiality requirements in the Common Rule. It concludes with a set of recommendations for addressing some of the weaknesses in the current system of research review.
II. Defining Privacy and Confidentiality
The terms privacy and confidentiality are often used interchangeably, although they are distinct concepts. Privacy is a state or condition of physical or informational accessibility.9 Many sources attempt to define and distinguish privacy and confidentiality. One frequently cited source is Privacy and Freedom, by Alan Westin, who defines privacy as “the claim of individuals, groups or institutions to determine for themselves when, how and to what extent information about them is communicated to others.”10 Professor Anita Allen, Professor of Law and Philosophy at the University of Pennsylvania, breaks down the concept of privacy into four types: physical privacy, informational privacy, proprietary privacy, and decisional privacy. Physical privacy is “spatial seclusion and solitude.” Informational privacy is “confidentiality, secrecy, data protection and control over personal information.” Proprietary privacy is “control over names, likenesses and repositories of personal identity.” Decisional privacy is “allowing individuals, families and other nongovernmental entities to make many of the most important decisions concerning friendship, sex, marriage, reproduction, religion, and political association.”11 A common justification for protecting privacy is the principle of respect for personal autonomy—“personal rule of the self that is free from both controlling interferences by others and from personal limitations that prevent meaningful choice.”12 The right to privacy should not be confused with the right to act autonomously. As Tom Beauchamp and James Childress explain in Principles of Biomedical Ethics, rights of privacy are valid claims against unauthorized access based in the right to authorize or decline access.13 In an 1890 law review article, Louis Brandeis and Samuel Warren argued that the right to privacy is “the right to be let alone,” the right to live without unwarranted interference by the public in matters with which the public is not necessarily concerned.14 Today, the right to privacy is not only a right to “retreat from the
C-4
world” but also a right to “step forward and participate in society,” sharing information about oneself with others while still maintaining some control over the data.15 Rules of confidentiality protect an individual’s privacy interests in the data collected about him or her. In cases involving the collection, use, and disclosure of health data, it becomes even easier to confuse the terms privacy and confidentiality. A person, however, can surrender some privacy and still maintain some control over the information generated about him or her. Alan Westin distinguishes confidentiality from privacy by defining confidentiality as “how personal data collected for approved social purposes shall be held and used by the organization that originally collected it, what other secondary or further uses may be made of it, and when consent by the individual will be required for such uses,” whereas information privacy is “the question of what personal information should be collected or stored at all for a given function.”16
III. Issues Confronting Researchers and IRBs: Threats to Privacy and Confidentiality
Again, there is no comprehensive federal law that protects the confidentiality of personal health data. However, there are federal regulations that apply to most research receiving federal funds, commonly referred to as the Common Rule, or research conducted in anticipation of approval by the Food and Drug Administration (FDA). Most federally funded research involving human subjects falls under the Common Rule,17 a federal policy adopted by 17 federal agencies in 1991 to protect “the rights and welfare of human research subjects,” including their personal health information.18 The FDA has established similar regulations for research involving the development of a product regulated by the FDA.19 The Common Rule requires research organizations to establish and operate IRBs, administrative bodies, to protect the rights and welfare of human research subjects. However, privately funded research that does not involve a federally regulated product is not subject to federal requirements. Some institutions that are not required to follow the Common Rule may choose to subject all research at their institutions to the Common Rule, while others apply the federal rules only where required. For example, an institution that conducts a large number of federally funded studies may enter into multiple project assurances (MPAs), which require all research at that institution to comply with the Common Rule.
Given the limited applicability of the federal regulations, it is generally believed that a significant amount of human subjects research is conducted in the absence of federal regulation, such as some privately funded research conducted by pharmaceutical companies, health plans, and universities not in anticipation of product approval by the FDA. An IRB chair commented at a U.S. House Commerce Committee hearing in May 1999 that “Today, if I want to study the medical history of Congressional representatives, and I don’t use federal funds, I may be able to get access to your medical records without going through any meaningful review process.”20 A recent Institute of Medicine (IOM) workshop found that much health services research using large databases falls outside the scope of federal regulations because the research is privately funded by organizations without federal MPAs.21 In addition, even where organizations submit research to an IRB for review, certain activities that involve identifiable health data and other human subjects research may not be defined by the organization as research, and therefore are left without any oversight and accountability.22 For example, the IOM found that IRBs vary in how they interpret federal guidelines regarding the definition of research, specifically whether or not a project is intended to yield “generalizable knowledge.”23 Some institutions may differ in how they interpret activities that might be considered quality assurance or quality improvement, taking the view that as long as the findings will be disseminated outside the division or department conducting the project, the project is research and thus subject to IRB review.24 While IRB review does not necessarily ensure that issues of privacy and confidentiality are adequately addressed, it does provide some level of accountability and oversight.
C-5
Health researchers encounter privacy and confidentiality issues at various stages of research, from recruitment of participants and data gathering, to data processing and analysis, to data storage, data dissemination, and the publication of research results. Researchers and IRBs need to be aware of and understand the range of privacy and confidentiality concerns in health research to adequately protect the privacy interests of their subjects and the confidentiality of personal health data.
A. Recruitment and Follow-Up
Where there is a lack of direct contact in research with subjects, individuals may have little or no knowledge that data collected from them in a clinical setting are being used for purposes other than for their treatment and payment. For research involving interaction with individuals, such as clinical trials, prior to contact with potential research participants, the researcher has to determine where and how to recruit participants. Most people are not concerned about researchers who are also physicians searching their own patient database to identify eligible subjects; they are concerned about someone other than their physician accessing their medical records to screen for potential subjects and contacting them about participation.25 A physician may have patients who would meet the criteria for subjects in a research project, but the potential participants may consider direct recruitment by a researcher a violation of privacy, whereas recruitment by the physician may be considered acceptable. Patients expect a certain level of confidentiality when they share sensitive information with their physicians. Therefore, when individuals are contacted by someone whom they were not aware had access to their medical information, they may consider the contact an invasion of privacy.
A recent DHHS Inspector General report on recruitment of subjects for industry-sponsored clinical research found that in a rush to recruit subjects, investigators might compromise privacy and confidentiality. The Inspector General found that patients were often unaware that someone other than their physician may be reviewing their records and using them to contact them about participating in research. Some IRBs have received complaints of harassment from potential participants.26 However, nothing in the federal regulations specifically prohibits access to these records by researchers, and there is little guidance from DHHS on acceptable recruitment practices.
After a research project is completed, a researcher also may decide to conduct follow-up studies or a different project. However, the subjects of the first study may not have been asked whether they would want to be contacted for other studies, and some of them may find subsequent contact from the researcher an invasion of privacy, particularly if contact occurs many years after completion of the first project.
B. Access to Health Records and Databases
Even if a research protocol does not call for direct contact with individual subjects, the researcher still must determine whether or not he or she will require access to personally identifiable health data. There are confidentiality concerns when researchers want access to personally identifiable data from health care providers, insurers, state registries, and any other entity that collects data from individuals in the course of treatment and payment. For example, many states maintain a cancer registry of which many patients are not even aware. Researchers may have access to the registry to conduct epidemiological studies and examine trends among cancer cases on behalf of a state’s health department. In a few states, researchers can obtain access to data from the cancer registry without first obtaining permission from the patient.27
C. Redisclosure
After a researcher receives or collects health data, there are confidentiality concerns regarding redisclosure of those data to third parties. Latanya Sweeney, Assistant Professor of Public Policy and of Computer Science at Carnegie Mellon University, stated at a recent Senate briefing that even if the original data holder imposes
C-6
privacy and confidentiality requirements on a third party requesting access to the data, once the data are disclosed to the third party, the third party may redisclose the data to others without restrictions.28 Similarly, Dr. Carolin Frey, Chair of the Geisinger Medical Center IRB, stated at a July 1999 House Commerce Committee hearing that when identifiable data travel between institutions, “it is possible for only [a] portion of an individual’s record to be within the purview of an IRB.”29 As an example, she noted that medical records are protected by the hospital IRB when the records are used in research but are not protected when the data travel to a third party payer.
Some researchers, however, are restricted from redisclosing data. For example, for data requests from other DHHS employees and contractors, the Health Care Financing Administration (HCFA) requires data use agreements that indicate the requestor’s understanding of the confidentiality requirements of the Privacy Act and HCFA’s data release policies and procedures. These agreements include a requirement that those receiving information from HCFA use it only for its approved purpose. Subsequent use for a different purpose is prohibited without further approval.
Without uniform rules for all research that limit redisclosure of personal health data, data collected for one purpose will continue to be disclosed and used for another purpose without the knowledge or consent of the subjects of the data. For example, for 52 years, research has been conducted using data from medical examinations, food diaries, X-rays, and blood samples of 10,000 Massachusetts residents in a long-term study known as the Framingham Heart Study. Originally, the participants signed on to a National Institutes of Health (NIH)-funded heart disease project.30 Now, Framingham Genomics Medicine (FGM) proposes to correlate the genetic information from blood samples with the study’s clinical data to create a huge database and sell the data to biotechnology and pharmaceutical companies. The major concern here is whether or not FGM will contact all the living study participants and relatives of the deceased for informed consent to use the information for this new project. Will strong and effective measures be implemented to protect the privacy of the subjects and the confidentiality of the genetic information? How meaningful is informed consent if sensitive health information is used for different purposes years later?
In another example, in December 1998, Iceland’s parliament authorized a license to deCODE genetics, a for-profit U.S. corporation, to use data already collected by the government to create a database (Icelandic Healthcare Database) of the medical records of all Icelandic citizens. This privatization plan raised a number of ethical questions, including the role of individual informed consent. The primary purpose of deCODE is to collect and analyze DNA samples for commercial purposes. Individual consent was not obtained prior to the transfer of medical data to the database, although individuals have the right to withhold their records by filing paperwork to opt out of the program.31 Those who do not opt out are presumed to give consent.
D. Conflicting Requirements and Policies
In a research study, it also may be technically difficult for an IRB and investigators to determine how it is required to protect privacy and confidentiality. Inconsistencies or conflicts may exist among legal requirements and institutional policies and practices. Some IRBs, for example, believe that unless a study impacts ongoing care, the consent forms for the study should not be included in a subject’s medical record.32 There is a fear that the consent form itself may reveal information about a patient that the patient wants to keep confidential. In one project, a medical resident discovered that his consent form for participation in research was placed in his medical record, even though the research had nothing to do with treatment. In fact, he was participating as a control subject for a study on coping behavior involving HIV. While the resident was not HIV-positive, the consent form in his medical record indicated he was participating in a study involving HIV. The Joint Commission on the Accreditation of Healthcare Organizations (JCAHO) requires consent forms to be included in a patient’s medical record, so in compliance with JCAHO requirements, the medical records department at this hospital
C-7
placed the consent form in the resident’s medical record. There is limited guidance for IRBs on how to reconcile conflicting policies and requirements.
E. Other Potential Violations of Privacy and Confidentiality
Researchers and IRBs also face other potential privacy and confidentiality issues. The method of contact, such as a postcard notice or e-mail regarding participation in a research project, may be considered a breach of confidentiality, because information on the postcard or e-mail may suggest information that the potential subject considers confidential. For example, a recruitment postcard for a study that is sent to an individual’s home may suggest that the recipient of the postcard has a specific disease. Even if the individual does have the disease, he may have kept it a secret from the rest of the household, and the postcard would be considered a breach of confidentiality.
If subjects get paid for participation in a project, parties providing compensation also need to be sensitive to concerns that the form of payment may contain information that would indicate to a third party a subject’s participation in a research project. For example, there may be information on a check that could constitute a confidentiality breach, not only because it is apparent to the bank that the recipient of the check is a research subject, but because the information can presumably be transferred to an affiliate of the bank, such as an insurer.
Another potential breach of confidentiality can occur with projects that involve periodic tests or visits with a physician. Reminders are often sent out to subjects at their home addresses, which may have information suggestive of the addressee’s health status or participation in research. There are also special considerations for research involving minority groups. A research study may focus on a particular group because of specific physical, social, or cultural attributes, possibly threatening the privacy of a small community. Dr. William Freeman, IRB chair at the Indian Health Service, stated at an IOM workshop that with certain minority groups, such as the American Indian and Alaska Native, the communities are small and isolated and the members are well known to each other, making it difficult to ensure individual privacy.33 If a minority group, however, perceives a research study as a threat to the privacy of the individual members or the group, they will be less likely to cooperate with the researchers.
F. New Technology
Individuals usually expect that the information they provide to their physicians will be kept confidential. Today, a growing number of disclosures occur without the express consent of the individual, stimulated in part by technological and scientific advances. The growth of information technologies for the delivery and payment of health care may offer significant opportunities for improved access to quality care at reduced costs. However, growing demands for access to health data and easier and cheaper storage and access to such data pose greater threats to privacy and confidentiality.
1. Health Databases
Organizational and structural changes in the delivery of health care call for the use of information technology to coordinate care and to integrate and disseminate information among providers, institutions, and managed care organizations. The demand for better quality care and the desire for reduced health care costs have also contributed to the rising need for patient data. The management of care in this environment requires data about what, where, and when health care services are provided, by whom for whom, and at what cost to determine the value and appropriateness of care. Such changes have led to the creation of large databases of health information, data linkage within and across data sets, and the ability for more people to access medical records and other personal health data from remote locations.
C-8
In fact, most data that move through health information systems end up in databases.34 While many of the databases are not organized optimally for research, researchers can avoid the costs of original data collection by using the available data. For example, one of the largest databases in the world is the U.S. Medicare database system, which process over 600 million reimbursement claims records yearly.35 Researchers have access to this database provided that they meet HCFA’s criteria for release of the data.36 The database includes data on enrollment, eligibility, and utilization. The data may not be of the highest quality or fully standardized, but they provide a great deal of information about the health status and health care of millions of patients. With the recent release of the final rule on national standards for electronic transactions by DHHS, however, there will be greater standardization of data transmitted for health care transactions.37 Standardization creates the potential for data linkage within and between data sets. Data linkage provides greater opportunities for research. It allows researchers to make associations between data on subjects from one source or multiple sources. For example, researchers can link workplace exposures with suspected illnesses. Such research may not require identifiable data, but the existence of large databases—especially those that are public databases—raise particular concerns. Chief among these concerns is that the more data are linked from different sources, the more likely it is that individual people or particular groups of people can be identified. Data may be aggregated from several sources without individual knowledge or consent and accessed by parties outside the health care treatment environment.
As Latanya Sweeney demonstrated at a policy briefing on medical and genetic privacy on July 14, 2000, “nonidentifiable” data can be combined with publicly available data to easily identify people.38 For example, most cities sell locally collected census data or voter registration lists, which include the date of birth, name, and address of the residents. These data may be linked to de-identified medical data, containing dates of birth and zip codes, to re-identify individuals, particularly in smaller communities.39 With an increasing focus on the health of a population rather than an individual comes the greater need for comparable data across health care organizations. Some of the sources of the data come from hospital databases, but a growing number of databases exist outside the health care environment. If personally identifiable data are used, the question is whether or not the subjects of the data need to be asked consent for the new use of their information. Locating and contacting subjects may be more difficult and prohibitively expensive. Where consent is waived, however, it is particularly important that there is objective review of the research protocol to ensure that safeguards are in place to respect the privacy of the subjects and protect the confidentiality of the data.
2. Internet
Increasingly, Internet sites are created to help consumers, patients, and health care professionals find information about their health and health care. Internet sites include peer support sites, sites that provide information on the latest research, and sites that provide a means for providers and patients to communicate outside the office.40 Researchers are using Internet chat rooms to conduct studies with and without the knowledge of chat room participants. According to clinical psychologist Storm King, there are “easily hundreds of researchers” conducting research on the Internet.41 Conducting research on the Internet presents new concerns because of the ability of both the participants and the researchers to assume anonymous or pseudonymous identities. In addition, there are new challenges, such as how to obtain informed consent, how to determine the expectations of privacy, and how to determine what data provided online would be treated as confidential. According to Jeffrey Cohen, Associate Director for Education, former Office for Protection from Research Risks (OPRR), breach of confidentiality is the primary source of harm in most Internet research.42 While health-related sites are generally more attentive to the need for privacy policies, some Web sites have yet to post privacy policies.43 For example, Pharmaceutical Research Plus, Inc., helps researchers recruit
C-9
participants via the Internet by offering a Web site at www.clinicaltrials.com that allows individuals to sign up for participation in clinical trials. On the patient registry page, an individual is asked to provide various identifiable data about himself or herself, including name, address, phone number, e-mail address, date of birth, and illness of interest. The site, however, is not secure, and there is no privacy policy that informs individuals what data are being collected, for what purpose, and who will have access to the data.
The lack of confidentiality protections is particularly troubling because Internet users may consider themselves anonymous or their activities as private. Chat room participants, especially those participating in support groups, often perceive these chat rooms as private sites when they exchange sensitive information about themselves.44 However, researchers are often not asking for consent to quote the participants, and a review board is not reviewing the research to ensure that the research is conducted ethically.45
3. Genetic Research and Testing
Scientific developments in genetics have given society a greater understanding of alterations in genes that are associated with human diseases, providing opportunities for better diagnosis, treatment, and prevention of disease. On June 26, 2000, two groups of scientists announced that they had completed a rough draft of the human genome, a breakthrough that may revolutionize the practice of medicine.46 With a rough draft complete, biomedical researchers can begin their search for disease-causing genetic mutations and develop therapies to treat disorders at the molecular level. Scientists may eventually be able to identify from birth the diseases a person may develop and tailor treatment to that individual.
However, with the ability to better detect genetic aberrations comes the questions of how genetic information should be protected and used and who should have access to that information. Genetic research on stored samples, such as blood samples, biopsy specimens, and organs and tissues, raises questions about privacy, consent, and confidentiality. Unlike most other biomedical research, genetic studies involve families. Research findings about individual subjects have direct implications for biological relations of the research participants because they may reveal information about the likelihood that members of the family are carriers or will be affected by a disease. The ethical question here is whether or not the research findings become part of the study without consent from the subjects of the findings.
Genetic research involving groups of people with specific genetic attributes also raise concerns about privacy. The Iceland example mentioned earlier concerns not only individual privacy but also group privacy. Like the American Indians, the Amish, and Ashkenazi Jews, Icelanders have a relatively homogenous gene pool, which improves the likelihood that researchers will find the genetic mutations associated with a disease. However, population-based genetic studies can lead to stigmatization. Specific groups of people may become identified with certain diseases, even if these diseases do not affect them disproportionately.
There is also public concern that access to genetic information by others, such as insurers and employers, will increase the potential for discrimination based on such information. Many people shy away from genetic testing because they fear that too many people have access to their information and that it can be used against them. Such fears may be justified: A 1992–1993 pilot study documented 206 instances of discrimination (loss of employment and insurance coverage or ineligibility for benefits) as a result of access to genetic information.47 The primary risks of genetic research are social and psychological rather than physical harm. Confidentiality concerns are a significant barrier to genetic research. According to a 1997 national survey conducted by the U.S. Department of Labor, 63 percent of people reported that they would not take genetic tests for diseases if insurers or employers could access the tests.48 One in three women invited to participate in a breast cancer study using genetic information refused because they feared discrimination or loss of privacy.49 More recently, a CNN-Time magazine poll found that 46 percent of the respondents expect harmful results from the Human Genome Project. Only about 20 percent said the genetic information should be available to insurance companies, and only 14 percent said it should be available to the government.50 While a number of states have passed
C-10
laws to provide greater confidentiality protections and to prohibit genetic discrimination to encourage more people to seek genetic testing and counseling, protections are still piecemeal.
G. Identifying Research
Needless to say, research is only subject to IRB review if it is indeed research as defined in the federal regulations. It is not, however, always easy to determine which activities are regulated research and thus subject to IRB review.
It is particularly hard to distinguish between health services research and health care operations and quality assurance activities, for example. Many aspects of health services research are similar to quality assurance and improvement activities. Research is defined in the Common Rule as “a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge.”51 While quality improvement activities at an institution are intended to affect the population of participants, the data may or may not be generalizable to others within and even outside the institution.52 Government Accounting Office (GAO) investigators found that several managed care organizations did not define records-based quality improvement activities as research, so these activities do not undergo IRB review, while some organizations do define these studies as research and thus submit them for IRB review.53 Alternatively, what begins as an internal review of quality of care may evolve into an activity that could be classified as health research. Even after an institution discovers that it may be engaging in research, however, it may choose to publish its results without seeking IRB review.54
IV. Public Concerns and Consequences of Violations of Privacy and Confidentiality
In general, research involving human subjects does not directly benefit the subject. Some health research can even pose potential harm to the subject physically and emotionally. Health research, however, can offer many societal benefits. To justify placing individuals at risk for the greater good, therefore, requires that research be conducted with respect for the rights and welfare of the individual subjects. Whether research involves collecting information or samples from individuals or getting access to medical records and databases, respect for the individual requires that researchers strive to protect the privacy of their research subjects by obtaining voluntary informed consent and ensuring that data are safeguarded against unauthorized access.
A 1993 survey conducted by Louis Harris & Associates found that 64 percent of the public wanted to be asked their permission before medical records are used for research.55 Furthermore, a 1996 Louis Harris & Associates survey found that only 18 percent of the public considers the use of patient records for medical research without prior permission to be very acceptable. The public’s comfort level increased if the information released did not identify individual patients, but one-third found it not at all acceptable for researchers to use health information without patient consent, even if their identities were kept confidential.56 The public is right to be apprehensive about invasions of privacy and lack of protections for their personal health data. While there are few widely publicized cases of violations of privacy and confidentiality in the research environment, in a recent GAO report, investigators noted that “during a research presentation at a national meeting, notes on a patient suffering from extreme depression and suicidal impulses stemming from a history of childhood sexual abuse were distributed. The notes included the patient’s identity, medical history, mental status and diagnosis, as well as extensive intimate details about the patient’s experience.”57 Because the study did not receive federal funding, there was no legal recourse for the research subjects. In a separate investigation, the former OPRR found that a university inadvertently released the names of study participants testing positive for HIV to parties outside the research project, including a local television station.58
C-11
Such breaches of confidentiality raise concerns not only about individuals being exposed or embarrassed, but also concerns that access to personal health data would allow others to use the information against the individuals to deny insurance, employment, and housing or to expose them to unwanted judgments and scrutiny. According to a California HealthCare Foundation survey, one in five U.S. adults believes that a health care provider, insurance plan, government agency, or employer has improperly disclosed personal medical information. Half of these people say it resulted in personal embarrassment or harm.59 Today, people engage in a variety of “privacy-protective” behaviors to protect themselves from what they consider harmful and intrusive uses of their health information. Privacy-protective behavior includes paying out of pocket for health care, seeing multiple providers, providing inaccurate or incomplete information, or avoiding care altogether. One in six adults in the United States engage in some form of privacy-protective behavior when seeking, receiving, or paying for health care.60 Engaging in such behavior not only puts the patient at risk, but affects the accuracy and integrity of health data for downstream users, such as individuals engaged in public health initiatives and health services research.61 Lack of privacy protections erodes public confidence and trust in the health care and research community, potentially resulting in the reluctance and unwillingness of individuals to participate in important research.
V. U.S. Regulation of Human Subjects Research
While there is not yet any comprehensive federal legislation that protects the confidentiality of health information, there is a patchwork of federal and state legislation, constitutional law, case law, and rules of civil procedure that provide limited protection. These laws address specific aspects of patient privacy and confidentiality of personal health data: 1) researcher access to data; 2) disclosure of data by the researcher; and 3) safeguards for research participants. Some of the laws provide substantial protections for the confidentiality of sensitive medical information, such as drug and alcohol abuse data, but without a comprehensive federal law protecting the confidentiality of all health information, most health information will continue to be subject to inconsistent legal standards and requirements.62
A. Common Rule
Currently, most research that receives federal funding is subject to the Common Rule. The Common Rule requires research institutions and federal agencies conducting research with human subjects, which includes the use of “identifiable private information,” to establish IRBs to review research proposals. The role of the IRB is to determine if the rights and welfare of the subjects will be safeguarded. While IRBs can help to ensure that a study’s procedures observe sound research design and that there is adequate informed consent, they do not directly observe the research study or the process in which consent is obtained. IRBs periodically review previously approved research to determine whether the study should be allowed to continue.
IRBs review the risks and benefits of the research and also make sure that adequate plans are made by the researcher to protect the privacy of subjects and maintain the confidentiality of the data. Among the criteria for IRB approval of research are requirements that 1) the risks to subjects are minimized; 2) the risks to subjects are reasonable; and 3) when appropriate, there are adequate provisions to protect the privacy of subjects and to maintain the confidentiality of the data. There is no further guidance in the Common Rule, however, for evaluating privacy and confidentiality issues when reviewing a research protocol.
Although most federally funded health research involving human subjects generally requires IRB review, there are exceptions to full IRB review and consent requirements. Records-based research, for example, is often subject to an expedited review process.63 Under the Common Rule, research activities that involve only minimal risk or “research involving materials that have been collected, or will be collected solely for nonresearch purposes” may be eligible for expedited review, which is carried out by the IRB chair or one or more of the IRB members.64 The IRB member or members conducting expedited review must follow the same standard of
C-12
review; however, the protocol may lack the evaluation that a full board review can offer. The level and adequacy of IRB review depend on the expertise and capabilities of the IRB members.
In particular, it appears that records-based research that does not involve any direct contact with patients may be reviewed differently by IRBs. According to Elizabeth Andrews at Glaxo Wellcome, “a fairly small proportion of research that is currently being reviewed by IRBs is [research for which there is no medical risk to the patient and relies purely on existing medical records] so IRBs typically have less experience reviewing this kind of research.”65 The typical procedure is to automatically assume that research using existing records is “minimal risk” and allow the study to undergo expedited review.66 Furthermore, the current regulations were largely written for interventional research studies, such as clinical trials, so there is less guidance for research that uses personally identifiable data without physically involving the individual in the research.67 Under the Common Rule, some research may be exempt from IRB review. The Common Rule lists many kinds of research that are not subject to IRB review, such as research that only involves “the collection or study of existing data, documents, records, pathological specimens, or diagnostic specimens, if these sources are publicly available or if the information is recorded by the investigator in such a manner that subjects cannot be identified, directly or through identifiers linked to the subjects.”68 However, what is “identifiable” or “nonidentifiable” is subject to interpretation. IRBs may find projects eligible for exemption because of how they interpret the definition of nonidentiable data, so they may come to different conclusions regarding subject consent for the same kinds of research. Not everyone grasps the distinction between identifiable and nonidentifiable data, so exemptions may be misapplied. According to Daniel Nelson, director of Human Research Studies at the University of North Carolina-Chapel Hill, some investigators and IRBs consider data stripped of the common identifiers, such as name, address, and Social Security number, as nonidentifiable and therefore not subject to IRB review.69 Professor Latanya Sweeney has often shown in her published work and presentations how difficult it is to produce nonidentifiable data in today’s society. As she puts it, “anonymity is in the eye of the beholder.”70 Data that appear anonymous can be linked or matched to other databases (public or private) to re-identify individuals; a person can also look at unique characteristics in the fields and records of the database to identify individuals.71 DHHS-proposed health privacy regulations do not cover information that has been de-identified. To be considered de-identified under the proposed regulations, a covered entity must remove, code, or encrypt specified identifiers outlined in the proposed regulation and have no reason to believe that the information can be used by recipients to identify an individual. Some of the identifiers may be retained if the covered entity has appropriate statistical experience and expertise and determines that the probability of identifying the individuals with these identifiers is very low. The new definition of de-identified information may help researchers and IRBs better distinguish between identifiable and nonidentifiable information; however, some comments from the public on the proposed definition indicates that further clarification and guidance will be needed to ensure proper compliance with the regulations. The National Bioethics Advisory Commission (NBAC) report on human biological materials also provides a breakdown of unidentified, unlinked, coded, and identified samples, which may be helpful to IRBs considering these terms in research protocols.72 For human subjects research not exempt from review, informed consent of the research participants is required, unless an IRB waives the informed consent requirements, including the requirement to inform participants of the extent to which their information will be kept confidential. If an IRB finds that the research is not likely to cause harm to the subjects and the research could not otherwise be carried out without waiving consent, the IRB may waive consent.73 For example, an IRB may decide to waive informed consent for a project involving access to the medical records of 10,000 patients because it may consider the researcher’s access to these records minimal risk. Furthermore, the IRB may find that such research could not practicably be conducted if consent was required from all 10,000 patients. Consent waivers, however, raise concerns about adequate considerations for privacy and confidentiality.
C-13
B. Health Insurance Portability and Accountability Act of 1996
Congress recognized the importance of medical privacy when it passed the Health Insurance Portability and Accountability Act of 1996 (HIPAA).74 In response to growing public pressure for a comprehensive federal health privacy law, Congress imposed a deadline on itself in HIPAA to enact a privacy law by August 21, 1999. Congress’ failure to meet that deadline triggered a requirement in HIPAA for the Secretary of DHHS to issue final health privacy regulations. The Secretary published proposed regulations on November 3, 1999, and the public comment period closed on February 17, 2000. The final regulations are expected by fall 2000, with a 24-month implementation period to follow before the law takes effect.
The proposed regulation would directly cover only three entities: health care providers who transmit claims in electronic format; health insurers; and health care clearinghouses. As such, the regulation does not directly cover most researchers. Only researchers who provide care are considered providers and are thus subject to the regulations. The regulation will, however, have a large impact on researchers because it establishes rules for when a covered entity may disclose “protected health information”75 to researchers without the informed consent of the subject of the information. The regulation outlines specific criteria that must be met to disclose “protected health information” to a researcher without informed consent:
| 1. |
The research protocol must be approved by a review committee: an IRB or “privacy board” and |
| 2. |
The review committee must determine that the research meets certain criteria. The proposed regulations also include additional confidentiality criteria for IRBs and privacy boards beyond what is currently required under the Common Rule. If informed consent is waived, information can only be released to researchers if they meet the following criteria: |
Common Rule provisions for the waiver of informed consent:
| 1. |
The use or disclosure of protected health information involves no more than minimal risk to the subjects; |
| 2. |
The waiver will not adversely affect the rights and welfare of the subjects; |
| 3. |
The research could not practicably be conducted without the waiver; |
| 4. |
Whenever appropriate, the subjects will be provided with additional pertinent information after participation; New criteria required by the proposed federal health privacy regulations: |
| 1. |
The research could not practicably be conducted without access to and use of the protected health information; |
| 2. |
The research is of sufficient importance so as to outweigh the intrusion of the privacy of the individual whose information is subject to the disclosure; |
| 3. |
There is an adequate plan to protect the identifiers from improper use and disclosure; and |
| 4. |
There is an adequate plan to destroy the identifiers at the earliest opportunity consistent with conduct of the research, unless there is a health or research justification for retaining the identifiers. |
If a researcher is also providing health care to the subjects of the research and processes claims electronically, then the researcher is considered a provider and must abide by additional rules outlined in the proposed regulations. These include:
C-14
Research data that are unrelated to treatment may not be disclosed without specific voluntary patient authorization for purposes of treatment, payment, or health care operations. The proposed regulations, however, do not cover all researchers. For example, the regulation does not address use and disclosure of health data generated by researchers, if they are not based within a covered entity and do not provide health care.
In effect, the proposed regulations would change research requirements in two significant ways: 1) extend application of the Common Rule provisions for waiver of informed consent by requiring all research involving individually identifiable electronic health information regardless of the source of funding to undergo some form of review (IRB or privacy board) and 2) add additional criteria for review of such research.
It should be emphasized that the regulation will not apply to all researchers or all research. The proposed regulations do not cover researchers who generate their own data or who receive data from any entity not covered by the regulation. Much research conducted by pharmaceutical companies, for example, will not be covered by the regulations.
C. The Privacy Act
In 1974, concern about computerized data systems led to the passage of the Privacy Act,77 which covers all personally identifiable data held by the federal government. The Privacy Act limits the ability of federal agencies to disclose personally identifiable data. It also provides people the right to access and amend their records. The act, however, only applies to federal government agencies and their contractors. While it may prevent most nonconsensual access to government-held health records by insurers or the general public, the records are accessible to researchers and other federal and state agencies. The “routine use” exception in the act gives broad discretion to disclose information when compatible with the purpose for which the information was obtained. Over time, the volume of routine use exceptions has increased and government officials have interpreted the exception to allow disclosure that is compatible with any original purpose for which records were collected.78 For example, government officials have interpreted the routine use exemption to allow the computerized matching of separate agency records, even though a literal reading of the act does not appear to permit matching.79 On May 14, 1998, President Clinton issued a memorandum directing each federal agency to review its information practices to ensure compliance with the Privacy Act.80 As a result of this memorandum, in January 1999, the Office of Management and Budget (OMB) issued guidance stating that agencies can protect privacy by limiting the amount of data they maintain about individuals and ensuring that such data are relevant and necessary to accomplish an agency purpose, which would include research purposes. The OMB instructs the agencies to 1) designate a Senior Official for Privacy Policy; 2) review and improve the management of Privacy Act systems of records; 3) ensure notices describing systems of records are up-to-date, accurate, and complete; 4) identify any unpublished systems of records; and 5) review information sharing practices with state, local, and tribal governments.
C-15
D. Other Federal Laws
At the federal level, there are strict laws limiting access to data about individuals with certain sensitive conditions. However, these laws apply only to specific types of data collected and maintained by particular entities.
The Alcohol, Drug Abuse, and Mental Health Administration Reorganization Act amended the Comprehensive Alcohol Abuse and Alcoholism Prevention, Treatment and Rehabilitation Act of 1970 to make records of the identity, diagnosis, prognosis, or treatment of substance abuse patients confidential and require express authorization for disclosure.81 The Controlled Substances Act allows the Attorney General to authorize persons engaged in drug abuse research to withhold the names and other identifying characteristics of research subjects. Researchers with this authorization cannot be compelled in any federal, state, or local civil, criminal, administrative, legislative, or other proceeding to identify the research subjects for which the authorization was obtained.82 The Public Health Service Act also prohibits personally identifiable information from research, demonstration projects, and evaluation conducted or supported by the Agency for Health Care Policy and Research (now known as the Agency for Healthcare Research and Quality) from use, publication, or release for any purpose other than the purpose for which it was supplied.83 Under the Public Health Service Act § 301(d), the Secretary of DHHS may authorize persons engaged in biomedical, behavioral, clinical, or other research to protect the privacy of research subjects by withholding the subjects’ names or other identifying characteristics from persons not connected with the research in any federal, state, or local civil, criminal, administrative, legislative, or other proceedings. Persons so authorized would receive a Certificate of Confidentiality.84 Individually identifiable information obtained in the course of activities supported or undertaken by the Agency for Healthcare Research and Quality or the National Center for Health Statistics (NCHS), Centers for Disease Control and Prevention (CDC), cannot be used for any purpose other than the purpose for which it was obtained, unless the establishment or person providing the information gives consent for its use. Furthermore, individually identifiable information obtained in the course of health statistical or epidemiological activities may not be published or released if the person or establishment providing the information has not given consent.85 Data collected by NCHS may be used only for the purpose of health statistical reporting and analysis. The Director of CDC can issue an Assurance of Confidentiality, which protects both individuals and institutions from court-ordered release of identifiable information. This assurance is used for studies conducted by CDC staff and/or contractors.86 In addition, under the Justice System Improvement provisions, no officer or employee of the federal government or any recipient of assistance under Title 42, which covers various public health and welfare programs such as the Public Health Service, Family Violence Prevention Services, Civil Rights, and the National Space Program, can use or reveal individually identifiable research or statistical information provided by any person under title 42 for any purpose other than the purpose for which the information was obtained.87 The Department of Education (DOE) also offers additional safeguards for children under the Protection of Pupil Rights Amendment.88 No student will be required to submit to a DOE-funded survey, analysis, or evaluation that reveals information concerning the student’s attitudes, beliefs, or habits in seven areas—including mental and psychological problems potentially embarrassing to the student or family, sexual behavior and attitudes, and legally recognized privileged or analogous relationships, such as those with lawyers, physicians, and ministers—without the prior consent of the student (if the student is an adult or emancipated minor) or the parent.
While the above mentioned laws attempt to provide some protection for personally identifiable health data, a recent provision in OMB’s appropriation for FY1999 provides public access under some circumstances to research data through the Freedom of Information Act (FOIA). The provision directed OMB to amend its Circular A-110 to require “federal awarding agencies to ensure that all data produced under an award be made available to the public through the procedures established under FOIA.”89 Circular A-110 applies only to grants,
C-16
not to contracts and to data produced with federal support that are cited publicly and officially by a federal agency in support of an action that has the force and effect of law. It covers data collected by institutions of higher education, hospitals, and nonprofit institutions receiving grants from federal agencies, but not data collected by commercial organizations or most data collected by state and local governments.90 The new law was widely criticized by the scientific community, and OMB tried to narrow the scope of the law by applying it only to published research and to research that is used as a basis for making federal policy or rules. OMB has defined research data as “the recorded factual material commonly accepted in the scientific community as necessary to validate research findings,” but the research community still has concerns about what data would fall under this definition.
Finally, under the Financial Services Modernization Act (more commonly referred to as Gramm-Leach-Bliley),91 banks can share with their affiliates (which include insurers and others) a consumer’s personal data, including health data, without the consumer’s knowledge or consent. For example, if a researcher pays a subject with a check and the check has information on it that is suggestive of the subject’s health status or participation in a study, the bank that cashes that check could presumably pass the information along to its affiliates. The law also allows the sharing of this information with others not affiliated with the bank if the bank or insurer gives the consumer notice that it intends to share the information and the opportunity to opt out of the disclosure.
In cases where insurance companies may cover treatment administered in the course of a clinical trial, the health insurer would be covered by the HIPAA regulations governing individually identifiable health information. While Gramm-Leach-Bliley itself is silent on whether or not it supersedes or limits the provisions of HIPAA, the regulations promulgated by the Department of the Treasury (Office of the Comptroller of the Currency and Office of Thrift Supervision),92 Federal Reserve System,93 Federal Trade Commission,94 Federal Deposit Insurance Corporation,95 Securities and Exchange Commission,96 and the National Credit Union Administration97 specifically state in their final regulations on the Privacy of Consumer Financial Information that they do not modify, limit, or supersede the HIPAA standards.
E. Case Law
Information privacy is not constitutionally protected as a fundamental right. While there is some judicial protection of privacy interests, application of federal or state law is often limited to specific factual situations. Most federal and state courts have recognized a right to informational privacy; however, the scope of privacy protection varies. Furthermore, courts often balance an individual’s privacy interest against the compelling interests of the state or other individuals, and few cases, if any, adequately explain how such interests should be weighted.98 The lack of uniform protection through the judicial system leaves individuals vulnerable to potential intrusions on their privacy.
In Griswold v. Connecticut, the Supreme Court found that the First, Third, Fourth, Fifth, and Ninth Amendments “have penumbras, formed by emanations from those guarantees that help give them life and substance” and create zones of privacy. While the Griswold Court limited the zones of privacy to the marriage relationship when it overturned state law that prohibited the use or dissemination of contraceptives, it did recognize that a constitutional interest in privacy exists.
Over a decade later, in Whalen v. Roe, the Supreme Court examined whether there was a right to privacy with regard to the collection, storage, and dissemination of information in government databanks. The Whalen Court upheld the requirement that names of individuals obtaining abusable prescription drugs be reported, but it observed that the “right to collect and use such data for public purposes is typically accompanied by a concomitant statutory or regulatory duty to avoid unwarranted disclosures.” The Court found that the safeguards implemented by the New York Health Department had sufficiently shown “a proper concern with, and protection of, the individual’s interest in privacy.”
C-17
In United States v. Westinghouse Electric Corp., a Third Circuit court held that the invasion of privacy was justified when the director of the National Institute for Occupational Safety obtained a federal subpoena ordering an employer to disclose information from employee medical records. The court established a five-part test for determining whether the government’s “right to know” justifies invasions of privacy. The test requires a balancing of the following factors:
| 1. |
the type of health record and type of health information required; |
| 2. |
the potential for harm in any subsequent nonconsensual disclosure; |
| 3. |
the injury from disclosure to the relationship in which the record was generated; |
| 4. |
the adequacy of safeguards to prevent unauthorized disclosure; and |
| 5. |
the degree of need for access.99 |
F. Rules of Civil Procedure
In civil and criminal cases and when the government conducts an investigation, the courts have the authority to compel disclosure of relevant information, including scientific data and health information, by judicial subpoenas. In addition to Griswold and Whalen, the Federal Rules of Civil Procedure provide some level of protection against subpoenas or other court orders in federal courts. Section 26(a) of the Federal Rules limits discovery, but, generally, if a court finds that certain information is relevant to the requesting party’s case, it will order disclosure of that information. If the information is of questionable importance or relevance, the court will examine the requesting party’s need for the information before granting or denying a motion to quash the subpoena. For example, in one case, a plaintiff put her medical condition at issue by seeking damages for pain and suffering, so her gynecological records were held relevant to possible alternative causes of her medical problems and her claim of emotional distress.100 In a suit against Procter & Gamble to recover damages for toxic shock syndrome allegedly caused by a tampon manufactured by P & G, Farnsworth v. Procter & Gamble Co.,101 the court of appeals held that the CDC’s interests in keeping confidential the names and addresses of its participants in research on toxic shock syndrome outweighed the discovery interests of Procter & Gamble. The Farnsworth court emphasized the compelling social interest in promoting research and the potential harm to the CDC’s public health mission if the information were released.
Even when research data are discoverable, Rule 45(c)(3)(B) of the Federal Rules of Civil Procedure allows the court to quash or modify a subpoena, if the subpoena 1) requires disclosure of a trade secret or other confidential research, development, or commercial information102 or 2) requires disclosure of a) an unrelated expert’s opinion or information that does not describe specific events or occurrences in dispute and b) information from an expert’s study which was not made at the request of any party to the lawsuit.103 For example, in Bluitt v. R.J. Reynolds Tobacco Co., the court upheld a U.S. Magistrate Judge’s order to quash a subpoena, based on Rule 45(c)(3)(B), for data and supporting documentation from the Louisiana State University Medical Center for research involving environmental tobacco smoke and cancer in women.104
G. Certificates of Confidentiality
Health researchers, federally and privately funded, can also apply for Certificates of Confidentiality, so they “may not be compelled in any federal, state, or local civil, criminal, administrative, legislative, or other proceeding to identify [subjects of research].”105 Certificates of Confidentiality were originally enacted in 1970 as part of the “War on Drugs” to allow studies of drug addiction and abuse. Because potential research subjects were involved in illegal activity, they needed to be assured that the information they shared with researchers would remain completely confidential. Of particular concern was disclosure to law enforcement. In 1988, biomedical
C-18
or behavioral research information that an investigator deems to be “sensitive” was incorporated into the Public Health Service Act.
The Public Health Service has the authority to issue Certificates of Confidentiality to researchers to protect the identities of the research participants; however, the research must be of a “sensitive nature where the protection is judged necessary to achieve the research objective.”106 The Certificates legally free the researcher from obligations to comply with a subpoena, court order, or mandatory reporting, but the researcher can still voluntarily disclose the information to other interested parties. The Certificate allows the holder to use it to resist compulsory disclosure. No court decisions challenging Certificates of Confidentiality have been found.
It is important to recognize that the protections of the Certificate of Confidentiality are exclusively for identifiable research data and do not extend to clinical information or medical records. In addition, according to Olga Boikess from the National Institute of Mental Health at NIH, the Certificates are issued sparingly and are only intended to provide additional confidentiality protections.
Certificates are issued on a project by project basis, and they are administered out of multiple agencies. Therefore, there may be inconsistent administrative guidance. According to Moira A. Keane, Director of the Research Subjects’ Protection Program IRB/IACUC at the University of Minnesota Health Center, it also can be very time-consuming, taking several months to get a Certificate of Confidentiality.107 Furthermore, even in cases where IRBs find a protocol that seems to fit all the requirements for a Certificate, applications for Certificates have been denied. For example, the IRB at UNC asked some researchers to apply for a Certificate of Confidentiality for a project on illegal activity, HIV, and drug use, but the application was rejected.108 Authorizations of confidentiality are also available for research requiring an Investigational New Drug exemption under section 505(i) of the Federal Food, Drug, and Cosmetic Act109 or to approved new drugs that require long-term studies, records, and reports. For research directly related to law enforcement activities concerning drugs or other substances that may be subject to control under the Controlled Substances Act, the Attorney General has the authority to issue grants of confidentiality.110
H. State Law
For privately funded research that does not involve approval of an FDA-regulated product, the researcher need only comply with state law. There is little uniformity in how state statutes regulate researcher access to people’s health information. Virtually every state has some law aimed at the confidentiality of patient health information in the health care environment, but very few states have anything approaching a comprehensive health privacy law, and so the requirements for researchers are scattered or nonexistent.111 Most state health privacy laws were never intended to be comprehensive.112 They were enacted at different points in time, over many years, to address a wide variety of uses and public health concerns. The statutes are generally entity specific or condition specific because they are often crafted to speak to the unique needs of the patient population and the information needs of particular entities in the state. Many states, for example, have privacy laws governing hospitals and clinics, but not health plans and HMOs. Finally, many of the heightened privacy protections at the state level also were enacted hand-in-hand with mandatory reporting laws.113 Many states require patient authorization prior to disclosure. Researcher access, however, is almost always built-in as an exception to these statutes. The vast majority of laws, therefore, allow researchers broad access to patient records. Minnesota, for example, however, is an exception. For records generated after January 1, 1997, the health care provider must first advise the patient in writing that his records may be released to researchers. If the patient objects, the records may not be released, but they still may be used by researchers within the entity holding the data.114 Some states place restrictions on researcher access by requiring IRB approval, patient authorization, or justification of the need for the individually identifiable information. There also may be specific requirements for
C-19
information such as HIV/AIDS or genetic information. While researchers are generally given broad access to patient data, some states place limits on researchers once they obtain the data. For example, in Michigan, information, records of interviews, written reports, or records that came in the possession of the department of health through a medical research project may not be admissible as evidence in a legal proceeding against an individual.115 In South Dakota, information may be released for the purpose of research into the causes and treatment of alcohol and drug abuse, but the researchers are prohibited from publishing the data in such a manner that identifies individuals.116 Researcher access to patient data held by state government entities is also often subject to different rules.117 (For a more comprehensive review of the role of states in the oversight of human subjects research, see, in this volume, the commissioned paper by Jack Schwartz from the Office of the Maryland Attorney General entitled Oversight of Human Subjects Research: The Role of the States.)
VI. International Principles for Ethical Research
Historically, privacy and confidentiality in research received little attention until the early twentieth century. The first set of principles for protection of human subjects was codified in 1946 as part of the verdict of the Nuremberg War Crime Trials after World War II. In 1964, the World Medical Association adopted the Declaration of Helsinki, which includes among its principles the following: “Every precaution should be taken to respect the privacy of the subject” and “Concern for the interests of the subject must prevail over the interests of science and society.” More recently, the European Union (EU) passed a Data Protection Directive that took effect in October 1998.118 The World Medical Association also announced that it will draft international guidelines on the use of centralized health databases to address issues of informed consent, privacy, confidentiality, individual access, and accountability.119 The EU Directive protects the privacy rights of its citizens, setting conditions on the international transfer of personal information from the EU to nonmember countries, such as the United States. The Directive prohibits the transfer of data to any country that fails to ensure an “adequate” level of protection. Such a prohibition can potentially impede the flow of personal health data from the EU to the United States, since the United States lacks a comprehensive health privacy law or nationally enforceable regulations or policies.
In an attempt to avoid punitive measures, the United States has been negotiating a safe harbor agreement with the EU this past year, which would make U.S. businesses responsible for safeguarding the confidentiality of personal data they collect or receive about European consumers. EU members have approved the U.S. proposal in principle; however, the European Parliament rejected the proposal on July 5, 2000, saying “key provisions needed to be renegotiated to strengthen data privacy and protection rights.”120 Nevertheless, the Internal Market Commissioner, Frits Bolkestein, is expected to recommend that the European Commission approve the agreement—a recommendation that likely will be accepted by the Commission.121
VII. Enforcement of Common Rule and Institutional Policies and Practices
There are an estimated 3,000 to 5,000 IRBs in the United States associated with a hospital, university, or other research organization. IRBs also exist in managed care organizations, government agencies, and as independent entities that review protocols for a fee. There is no accurate count, since IRBs are not required to register with any entity. Each of the 17 federal Common Rule agencies has independent responsibility for oversight of IRBs reviewing the research that it supports.122 Some researchers or research facilities conducting research that falls outside the scope of the Common Rule or FDA regulations use external research ethics or advisory boards. There are no data on the number of such review boards in the United States. At a July 1999 House Commerce Committee hearing, Greg Koski, the recently appointed director of the Office for Human Research Protections
C-20
(OHRP), stated that only about 1,200 of the 5,000 or so IRBs that currently review research in the United States come under the Common Rule.123
A. Office for Protection from Research Risks/Office for Human Research Protections
Within DHHS, until recently, OPRR oversaw implementation of the Common Rule in all DHHS facilities and any institutions or sites receiving DHHS funds to conduct research involving human subjects. OPRR required these facilities and institutions to submit an “assurance” of compliance, a policy statement that sets forth the procedures they will use to protect human subjects. The assurance is a formal commitment to implement 1) widely held ethical principles; 2) 45 CFR 46 (the Common Rule and additional protections pertaining to research involving children, prisoners, fetuses, pregnant women, and human in vitro fertilization); and 3) institutional procedures adequate to safeguard the rights and welfare of human subjects. If a problem arises, OPRR uses the assurance to gauge an institution’s compliance with human subject protections.124 The former OPRR investigated allegations of noncompliance and had the authority to restrict an institution’s authority to conduct DHHS-funded human subjects research if there were a breach of confidentiality. OPRR handled most inquiries and investigations by telephone and correspondence. OPRR sometimes restricts further research until the researcher takes corrective action. For example, in one investigation, OPRR found that a university inadvertently released the names of study participants testing positive for HIV to parties outside the research project, including a local television station.125 The OPRR worked with the university to evaluate the extent of the breach of confidentiality. The university revised its internal systems to prevent a similar violation from occurring in the future.
In June 2000, the new Office for Human Research Protections in DHHS officially replaced OPRR. In 1999, the Advisory Committee to the Director of NIH had recommended that the role of OPRR be expanded and that the office be elevated in stature and effectiveness. There was growing recognition of the need for enhanced federal oversight of human clinical studies. As such, OHRP was established in the Office of the Secretary at DHHS with the responsibility for ensuring the safety and welfare of research participants in DHHS-sponsored research. An independent National Human Research Protection Advisory Committee has also been established to provide scientific and ethical guidance to OHRP in its oversight role.
In its regulatory role, OHRP monitors and evaluates an institution’s compliance with the rules governing human subjects research. OHRP has the authority to investigate complaints and require corrective action or suspend research at an institution until the problem is resolved. For example, OHRP recently shut down all government-funded human medical experiments at the University of Oklahoma Health Sciences Center in Tulsa because the researchers broke multiple rules designed to protect subjects and then tried to cover up their lapses by withholding information from the university’s IRB and subjects.126 In its educational role, OHRP provides guidance to IRBs, scientists, and research administrators on ethical issues related to medical or behavioral research involving human subjects. The office conducts national educational workshops and on-site technical assistance to institutions conducting DHHS-sponsored research.127 The former OPRR Institutional Review Board Guidebook provides some guidance for addressing privacy and confidentiality. The guidebook provides points IRBs should consider in reviewing research protocols.128 The OPRR does note, however, that even research in which there are privacy concerns, these concerns may not come to the attention of an IRB. For example, under the federal regulations, IRBs do not have to review proposed research involving observation unless someone, such as the investigator or department head, determines that it falls in the category of research requiring IRB review.
C-21
B. The Food and Drug Administration
The FDA also monitors and enforces human subject protections. The agency requires a promise from researchers that they will abide by FDA requirements for conducting drug, medical devices, and biologics research and conducts on-site inspections of IRBs that oversee such research. If there are serious violations, FDA may terminate the IRB’s authority to approve new studies or recruit new participants for ongoing studies until FDA is assured of corrective action. Both OHRP and FDA have oversight responsibilities for research involving an FDA-regulated product supported by DHHS.
However, a review of FDA’s inspection process for clinical investigators conducted by the DHHS Office of Inspector General shows that FDA’s main focus is procedural compliance with FDA regulations affecting IRBs rather than the content of IRB reviews. Furthermore, while its objectives for inspections are “ensuring the quality and integrity of data and information submitted to FDA as well as the protection of human research subjects,” the FDA has focused mainly on ensuring the integrity of the data submitted to the agency.129 The FDA monitors human subjects protection by conducting on-site inspections of the IRBs that oversee drug research. Its inspections have demonstrated that compliance with federal oversight rules are uneven. To enforce its regulations, the FDA uses four types of actions: 1) obtain a promise from the researcher to abide by FDA requirements; 2) impose restrictions on researcher use of investigational drugs; 3) disqualify researcher from use of investigational drugs; and 4) criminally prosecute the researcher.130
C. Research Institutions and IRBs
At the institution level, the institutions conducting or supporting the research are responsible for ensuring that the Common Rule requirements are met and for addressing violations of privacy and confidentiality. The IRBs and investigators are responsible for implementation of and compliance with the Common Rule. The IRB assists researchers in identifying possible threats to privacy and confidentiality. According to the 1999 GAO report on medical records privacy, IRBs rely on their organization’s policies for determining the appropriate actions for protecting the confidentiality of personally identifiable health data used in the projects at the organization. However, according to Moira Keane at the University of Minnesota Health Center, while IRB members have an appreciation of the need for privacy and confidentiality, unless members themselves are actively involved in research, the level of expertise of IRBs to adequately identify and address privacy and confidentiality varies.131 In addition, IRB and institutional oversight is generally limited to review of progress reports, such as a review of outcomes, implementation of research design, and adverse physical effects. The IRB does not audit the researchers to ensure compliance. A GAO report found that “while reasonable safeguards may be in place in these companies [organizations surveyed by GAO], external oversight of their research is limited, and even in those cases where IRBs are involved, they are not required to give substantial attention to privacy protection.”132 Even where there is subsequent and periodic review of the research approved by the IRB, privacy and confidentiality issues may be ignored once a project has been approved. The frequency of review may also depend on the level of risk the study poses to the subjects, but the focus is on physical or psychological risk, not threats to privacy and confidentiality.133 There is an expectation that the investigators will put in place the necessary privacy and confidentiality protections as specified in their research protocol. The principal investigators are ultimately responsible for ensuring that adequate safeguards are in place to protect privacy and confidentiality. As such, they may not follow all of the IRB’s instructions. For example, researchers may retain identifying fields as a matter of convenience or when there is no need for that information, even after an IRB has informed the researchers that retaining the identifiers may pose a confidentiality threat that can easily be eliminated without jeopardizing the study.134
C-22
D. Research Outside the Scope of the Common Rule and FDA Regulations
For research not subject to the Common Rule or FDA regulations, there are few data about criteria for addressing privacy and confidentiality. Some organizations choose only to apply the federal rules when they are required. They may also rely on their collaborating universities or institutions for informed consent procedures and IRB review.
HCFA imposes additional requirements on researchers who are not funded by a DHHS agency and want access to HCFA databases. The agency conducts a review to determine whether disclosure would be permitted under the Privacy Act and determines if the purpose of the research 1) requires identifiable data; 2) is of sufficient importance to warrant risk to the individual; and 3) is likely to be accomplished because the project is soundly designed and properly financed.
However, HCFA does not routinely monitor these researchers to prevent unauthorized disclosures or uses and to provide corrective action for violations of the agreement.135 The agency does not have a system for monitoring whether organizations outside of HCFA have established safeguards for personal health information received from the agency. Instead, HCFA relies on each organization to monitor its own compliance with the data use agreements.
A February 1999 GAO report shows that most of the organizations the agency surveyed have steps to limit access to personal health data, such as security safeguards to limit internal and external access to paper records and electronic databases.136 The agency, however, found that 2 of the 12 organizations contacted lacked written confidentiality policies restricting employee use and access to health information.137 Furthermore, while there may be some sanctions in place, there is little information on how violations are addressed. In addition, there are no guarantees that the institution’s own penalties will be imposed for violations of privacy or confidentiality. Without remedies or sanctions, the current framework of enforcement will be lacking.
E. Impact of Federal Health Privacy Regulations
Once the federal health privacy regulations are finalized, penalties may be imposed on researchers who are also health care providers and transmit or maintain health information in electronic form, if they wrongfully obtain or disclose individually identifiable health information. Penalties include fines and/or imprisonment. There are also penalties for noncompliance with the regulations. However, there is no individual right to sue, so if an individual finds that his or her rights under HIPAA have been violated, all he or she can do is file a complaint with DHHS.
VIII. Evaluation of the Current System of Research Review
There has been recent and growing concern about the adequacy of the current system of IRB review and oversight, particularly as it relates to the confidentiality of personal health information. A report commissioned by DHHS Secretary Donna E. Shalala concluded, “It is less clear that IRBs have been attending as vigorously to privacy risks as they have to physical and emotional risks.”138 Recent studies conducted by the Office of the Inspector General at DHHS and NIH have found that IRBs review too many studies too quickly and with insufficient expertise.139 There is little training for researchers and IRB members and minimal oversight of approved studies.140 The level of expertise across IRBs varies. For example, according to the DHHS Inspector General report, in June 2000, 25 percent of the IRB survey respondents did not even ask researchers to explain their recruitment practices in the application for review.141 Most studies on human subjects research and protection focus on specific topics, such as informed consent issues and injuries to subjects. There are smaller data gathering efforts, such as the GAO report on Medical Records Privacy142 and the IOM Workshop on data privacy in health services research,143 which provide a glimpse into the current system of review for research protocols.
C-23
Experts in the research community comment that the current IRB system works well with respect to most interventional protocols but not necessarily for observational research, that is, research involving only existing medical data. Among the weaknesses of the existing system:
There is also concern that the extension of the federal regulations to privately funded research under the proposed federal health privacy regulations will place further burdens on the IRB system.147
A. NIH Study on IRBs
In 1995, NIH conducted an evaluation of the implementation of the human subjects protection program, surveying IRB members and chairs from institutions that operated with MPAs.148 The main conclusion of this study was that IRBs are providing an adequate level of protection at a reasonable cost. However, there were only limited references to privacy and confidentiality issues. The emphasis of the survey was on broader issues of IRB workload, IRB personnel and policy practices, and the adequacy of protections for the rights and welfare of research subjects.
B. IOM Study on Health Data Privacy
Little is known about IRB practices and how IRBs function, particularly in health services research, which is largely research using databases of health information. The IOM convened a committee to gather information on the current practices and principles followed by IRBs to safeguard confidentiality of identifiable health data used for federally and privately supported health services research purposes. On August 14, 2000, the IOM released its recommendations regarding best practices for IRB review of health services research subject to federal regulations and IRB or other review board review of research outside the scope of federal regulations. Highlights of the IOM recommendations include the following:
C-24
C. GAO Report on Medical Records Privacy
In 1999 a GAO report on medical records privacy identified research that is and is not subject to federal oversight and examined how IRBs ensure the confidentiality of health data used in research. While the basis of its findings was limited to the information provided by federal agencies and organizations interviewed, the GAO concluded that external oversight of privately funded research is limited. Not all research is subject to outside review, and even when IRBs are involved, they are not required to give substantial attention to privacy protection.149 In addition, the agency found that “privacy protection is not a major thrust of the Common Rule and IRBs tend to give it less attention than other research risks because they have the flexibility to decide when it is appropriate to focus on privacy protection issues for review.”150 There are even fewer data on the research review policies and practices regarding privacy and confidentiality in institutions conducting privately supported research. GAO found that some of the organizations the agency contacted conform to the FDA regulations because the organizations conduct both FDA regulated and privately funded research. Some organizations have adopted internal policies that require all studies that meet their definition of research to follow the Common Rule requirements. However, not all organizations necessarily define the same type of activity as research. Hence, application of the Common Rule varies within and across organizations.151 The GAO also found that in some organizations no research receives IRB review. One pharmacy benefits manager used external advisory boards rather than IRBs to review research proposals.152
IX. Recommendations
Currently, there are only federal requirements for federally funded human subjects research or research involving an FDA-regulated product, leaving a significant amount of research outside the scope of federal regulation. NBAC itself has stated in its preliminary findings on the adequacy of federal protections for human subjects research that “the absence of federal jurisdiction over much privately funded research means that the U.S. government cannot know how many Americans currently are subjects in experiments, cannot influence how they have been recruited, cannot ensure that research subjects know and understand the risks they are undertaking, and cannot ascertain whether they have been harmed.”153 At the same time, the public has demonstrated a concern about the lack of protections for their sensitive personal health data, withholding information or providing incomplete information to prevent intrusive uses of their information and to avoid discrimination, stigma, or embarrassment. Ultimately, such actions not only hurt individuals, but also compromise important research initiatives. Public trust in the research community is the key to ensuring continued access to personally identifiable health data for health research.
To ensure adequate protections for research participants’ privacy and health data confidentiality and to improve implementation of existing federal requirements for human subjects research, we offer the following recommendations. We hope that NBAC will consider these recommendations in its review and evaluation of the current system of review for human subjects research.
Uniform Standards and Process
1. All research should undergo IRB review.
Today, research is subject to any number of review procedures—or subject to no review at all—depending on a fairly arbitrary set of circumstances, such as funding or the site of the research. Even recent attempts to create greater uniformity have fallen short. For example, the intent of the HIPAA regulations is to establish uniform rules and process for research regarding privacy and confidentiality issues regardless of the source of funding. However, the proposed regulations would allow the creation of privacy boards, which would only address the confidentiality concerns of a research project. Much of privately funded research will continue to be less
C-25
accountable if it is subject only to privacy board review. The benefits of the IRB system are not reflected in privacy boards. In the proposed regulations, privacy boards exist only to grant a waiver for patient authorization, whereas IRBs review every step of a research project. All health research involving human subjects should receive comprehensive review.
Establishing a truly uniform system of review would ensure oversight and accountability of all research. As Dr. Greg Koski, the recently appointed first director of OHRP, testified on July 15, 1999, before the Subcommittee on Health and Environment of the U.S. House Committee on Commerce, “having a separate process that causes segregation in the whole process for review and approval of research would not only undermine the process that is there, it would tend to dilute the process for protection of human subjects.”154 The most effective way to achieve uniformity is to subject all research to IRB review. Critics of this suggestion have argued that subjecting more research to IRB review will overburden a system that is already beyond capacity. Those concerns, however, can and should be addressed separately. In fact, adequate reform of the system can only take place when there is a single uniform system.
| 2. |
Uniform and objective standards should be established for all health research, regardless of the source of funding. |
Research projects should be held to the same standards to ensure equity, fairness, and accountability to bolster public trust and confidence in research.155 On June 8, 2000, Representative Diana DeGette introduced H.R. 4605, the Human Research Subject Protection Act of 2000, which would extend the Common Rule to human subjects participating in private sector research.
In the absence of a uniform review system—such as an IRB—all research should be held to the same standard. Therefore, private IRBs, internal review systems, or even newly created “privacy boards” should all be following the same set of rules and standards. In particular, there should be uniformity in decisions about when and under what circumstances a waiver of informed consent can be granted.
The privacy and confidentiality standards established for federally funded research should be the standard for all research. As these standards are revised, they should be incorporated into the policies of the bodies reviewing research proposals.
Oversight and Accountability
3. All IRBs should register with a federal agency.
Today, it is impossible to determine how many IRBs are in existence, so it is impossible to even accurately study IRBs, let alone ensure compliance with federal standards. Registration is a basic easy step to allow for greater oversight of IRBs.
Registration could be coordinated through the OHRP or with an office in each of the federal departments that provides funding for health research. According to Daniel Nelson, Director of Human Research Studies at the University of North Carolina-Chapel Hill, there is currently a national effort to require certification and accreditation of all institutions conducting research.156
| 4. |
There should be periodic review after a research project has been approved that includes continued consideration of privacy and confidentiality issues. |
Several recent reports have identified problems in the current IRB system, which could impact an IRB’s ability to address human subjects concerns, including privacy and confidentiality. Not only have these reports found that IRBs are understaffed and overburdened, but also there is little oversight once a project has received IRB approval. A DHHS Inspector General report found that continuing review has become a low priority at many IRBs.157 Review is largely paper based, and IRBs often rely on the investigators to provide timely and accurate reports.158 The system of review is generally based on trust and confidence that once a protocol is approved, the
C-26
investigators will implement appropriate privacy and confidentiality safeguards as specified in the protocol.159 Furthermore, the focus of subsequent review tends to be physical and psychological harm to the subjects.160 Continued periodic review, which includes an examination of privacy and confidentiality issues, would better ensure that IRBs and researchers address unanticipated privacy and confidentiality issues that may arise during the course of a study.
| 5. |
Researchers should be required to sign confidentiality agreements that prohibit a) the use of personally identifiable health data for purposes other than for the original study and b) redisclosure of such data, without specific voluntary consent from the individual. |
To maintain public trust and encourage individuals to participate in research, recipients of personally identifiable health data should be bound by the same requirements and obligations as the original data holder to protect the privacy of the subjects and the confidentiality of the data.
Training and Education
6. There should be more resources allocated to support and reform the IRB system.
DHHS Secretary Shalala announced on May 23, 2000, that DHHS will be undertaking an aggressive effort to improve education and training of clinical investigators, IRB members, and associated IRB and institutional staff on bioethics and human subjects research.161 However, there are other federal departments that engage in and sponsor health research, and they should also expand their educational efforts. Specifically, more education and training is required for researchers, IRBs, and institutions on 1) particular privacy and confidentiality issues arising from various types of health research and 2) the best policies and practices for safeguarding privacy and confidentiality More training and education of investigators and IRBs will be required as new opportunities for and types of health research arise, especially with the mapping of the human genome.162 Expanding the scope of IRB-reviewed research will also require more resources to ensure that adequate review is conducted.
Further Study and Guidance to IRBs and Researchers
The OHRP at DHHS and other federal departments all need to play a greater role in providing guidance and support to IRBs and researchers as they confront issues of privacy and confidentiality in their research. A recommendation for uniform and objective rules and standards would be meaningless without adequate guidance for investigators, IRBs, and research institutions to effectively implement these rules. Specifically:
7. A comprehensive privacy survey of all IRBs should be commissioned.
Today, there are few data on how IRBs function; how they currently identify and address privacy and confidentiality; and how research is reviewed (if at all) outside the IRB system. Furthermore, there is little information on how many IRBs exist and how many people are research subjects. A study on IRBs would provide data on the strengths and weaknesses of the current system with regards to the protection of privacy and confidentiality. A study can also help identify policies and best practices for safeguarding privacy and confidentiality that can be adopted by all IRBs and other review boards.
8. Model privacy and confidentiality policies and practices should be developed.
The IOM recently released a report with findings and recommendations, which include specific recommendations for ensuring health data privacy and confidentiality in health services research. Any entity collecting or receiving personal health data should do so under comprehensive policies.
C-27
9. Specific guidance is needed on the distinction between identifiable and nonidentifiable data.
Generally, there is broad agreement that the use of anonymous data in noninterventional research should not require informed consent of the subjects of the data. It is becoming increasingly difficult, however, to differentiate between identifiable and nonidentifiable (or anonymous) data. Data exist on a continuum of identifiability. The increasing amount of publicly available data means that seemingly anonymous data can now be used to identify individuals.
More guidance is needed for institutions, IRBs, and researchers to make determinations about whether data is truly anonymous. Such guidance should specifically comment on the amount, quality, and type of data that is publicly available. The guidance should also include commentary on the feasibility of using privacy-enhancing technologies in research, such as encryption.
10. Clearer definitions of health research are needed.
One of the major issues in health research is distinguishing activities that will require IRB review from activities that do not fall under the definition of research for purposes of federal regulation. Guidance to researchers, IRBs, and research institutions is needed on what activities must undergo IRB review, especially when an activity begins as quality assurance but evolves into health research.163
11. Additional guidance may be needed to clarify the new requirements specified in the HIPAA regulations.
New federal health privacy regulations are expected to be finalized by the fall of 2000. We have found that some IRBs and researchers are not aware of HIPAA and the impact that the new regulations will have on their research activities. Researchers, IRBs, and data holders will need guidance on implementation of the new rules and information about the possible penalties for noncompliance with the new regulations.
Enforcement
| 12. |
Research institutions should establish strong enforceable remedies and sanctions for violations of privacy and confidentiality protections. |
For rules and policies to be truly effective, strong, and enforceable sanctions need to be established for violations of privacy and confidentiality, inside and outside an institution. HIPAA penalties are limited in application, since they would apply only to researchers who fit the definition of a covered entity, such as researchers who are also health care providers who transmit or maintain health information in an electronic format.
Notes
1 William W. Lowrance, Privacy and Health Research: A Report to the U.S. Secretary of Health and Human Services 21–29 (May 1997).
2 Data are discrete pieces of information. Health information, as used in this paper, is the knowledge obtained from investigation or study of health data.
3 Associated Press, Medical data up for grabs, Nov. 9, 1998.
4 Office of Inspector General, Department of Health and Human Services, Recruiting Human Subjects: Pressures in Industry-Sponsored Clinical Research 24, OEI-01-97-00195 (June 2000) [hereinafter Office of Inspector General, Recruiting Human Subjects].
5 Janlori Goldman, Protecting Privacy to Improve Public Health, 17 Health Affairs 47, 48 (Nov.–Dec. 1998).
6 Ibid.
7 Health Privacy Project, Best Principles for Health Privacy: A Report of the Health Privacy Working Group 10 (July 1999), available at www.healthprivacy.org/resources/index.shtml.
C-28
8 We broadly define health research to include basic research, clinical trials, epidemiological studies, and health services research. Health services research is a multidisciplinary field of inquiry, both basic and applied, that examines the use, costs, quality, accessibility, delivery, organization, financing, and outcomes of health care services to increase knowledge and understanding of the structure, processes, and effects of health services for individuals and populations (Committee on Health Services Research: Training and Work Force Issues, Institute of Medicine, Health Services Research: Work Force and Educational Issues, 1995).
9 Tom L. Beauchamp and James F. Childress, Principles of Biomedical Ethics 407 (4th ed., 1994).
10 Alan F. Westin, Privacy and Freedom 7 (1967).
11 Anita L. Allen, Coercing Privacy, 40 Wm and Mary L. Rev. 723, 723–724 (1999).
12 Beauchamp and Childress, supra note 9, at 121.
13 Ibid., at 410.
14 Louis D. Brandeis and Samuel D. Warren, The Right to Privacy, 4 Harv. L. Rev. 193–197 (1890).
15 Janlori Goldman, Privacy and Individual Empowerment in the Interactive Age, in Visions of Privacy: Policy Choices for the Digital Age 97–115, 101 (Colin J. Bennett and Rebecca Grant, eds., University of Toronto Press 1999).
16 Alan F. Westin, Computers, Health Records, and Citizen Rights 6 (U.S. Government Printing Office, 1976).
17 Federal Policy for the Protection of Human Subjects, 56 Fed. Reg. 28002-28032 (1991); 45 CFR 46, subpt. A.
18 Department of Agriculture, Energy, Commerce, Health and Human Services, Housing and Urban Development, Justice, Defense, Education, Veterans Affairs and the Transportation; the National Aeronautics and Space Administration, The Social Security Administration; the Consumer Product Safety Commission; the Agency for International Development; the Environmental Protection Agency; the National Science Foundation; and the Central Intelligence Agency. The Common Rule provisions are codified in regulation by the individual agencies. The Food and Drug Administration issued its own regulations for research involving FDA-regulated products.
19 21 CFR Parts 50 and 56.
20 Medical Records Confidentiality in the Modern Delivery of Health Care: Hearing Before the Subcomm. on Health and
Environment of the House Comm. on Commerce, 106th Cong. 34 (1999) (Statement of Robert Amdur, Former Associate Professor of Medicine and Chairperson, Dartmouth Committee for the Protection of Human Subjects, Dartmouth Medical School).
21 Committee on the Role of Institutional Review Boards in Health Services Research Data Privacy Protection, Division of Health Services, Institute of Medicine, Institutional Review Boards and Health Services Research Data Privacy: A Workshop Summary 2 (National Academy Press, 2000) [hereinafter Workshop Summary].
22 Personally identifiable health data are data concerning a person’s health or treatment that are or may readily be associated with an individual. Synonyms include individually identifiable health data and personal health data.
23 Committee on the Role of Institutional Review Boards in Health Services Research Data Privacy Protection, Division of Health Services, Institute of Medicine, Protecting Data Privacy in Health Services Research 45 (National Academy Press, 2000) [hereinafter Institute of Medicine, Protecting Data Privacy in Health Services Research].
24 Ibid.
25 Office of Inspector General, Recruiting Human Subjects, supra note 4, at 24.
26 Ibid. at 25.
27 In Indiana (Ind. Code § 16-38-2-5), Nebraska (Neb. Rev. Stat. § 81-666), and Ohio (Oh. Rev. Code § 3701.263), for example, a researcher may get access to individually identifiable data from the cancer registry if they meet certain conditions specified by the state health departments, such as providing to the department information about the purpose of the project, the nature of the data to be collected, the records the researcher wishes to review, and the safeguards the researcher will put in place to protect the identity of the patients. See also, Office of Inspector General, Recruiting Human Subjects, supra note 4, at 24.
28 Alliance for Health Reform and The Forum on Technology and Innovation, Policy Briefing: Medical and Genetic Privacy (Washington, D.C., July 14, 2000).
29 H.R. 2470 Medical Information Protection and Research Enhancement Act of 1999: Hearing Before the Subcomm. On Health and Environment of the House Comm. On Commerce, 106th Cong. (1999) [hereinafter House Hearing] (Statement of Carolin M. Frey, Chair, Institutional Research Review Board, Pennsylvania State Geisinger Medical Center).
30 Editorial, Whose Heart Data? The Boston Globe, June 21, 2000; Ronald Rosenberg and Liz Kowalczyk, Heart Study Will Sell Patient Data for Profit, The Boston Globe, June 16, 2000, at A1.
C-29
31 George J. Annas, Rules for Research on Human Genetic Variation—Lessons from Iceland, 342 The New England Journal of Medicine (2000).
32 Ibid.
33 Workshop Summary, supra note 21, at 4.
34 William W. Lowrance, Privacy and Secondary Use of Data in Health Research, Proceedings of the Inaugural Robert H. Levi Leadership Symposium 13, 14 (April 2000).
35 Lowrance, Privacy and Health Research, supra note 1, at 19.
36 The criteria include the purpose of the research project 1) requires the use of identifiable data; 2) is of sufficient importance to warrant risk to the individual that additional exposure of the record might bring; and 3) is likely to be accomplished because the project is soundly designed and properly financed. U.S. General Accounting Office, Medicare: Improvements Needed to Enhance Protection of Confidential Health Information 39, GAO/HEHS-99-140 (July 20, 1999).
37 45 CFR Parts 160 and 162; The Health Insurance Portability and Accountability Act of 1996 (HIPAA) required the Secretary of DHHS to adopt national standards for electronic health care transactions. Today, health care providers and health plans that conduct business electronically use different formats for electronic transactions. The purpose of these standards is to improve the efficiency and effectiveness of the health care system. For more information, visit DHHS’ Administrative Simplification website http://aspe.hhs.gov/admnsimp/index.htm>.
38 Alliance for Health Reform and The Forum on Technology and Innovation, supra note 29; See also, Latanya Sweeney, Controlling Inference and Protecting Privacy by Constructing an Anonymous Data System, Carnegie Mellon University, unpublished paper, November 1998.
39 Latanya Sweeney, Weaving Technology and Policy Together to Maintain Confidentiality, 25 J.L. Med. and Ethics 98, 100 (1997).
40 Robert Mittman and Mary Cain, The Future of the Internet in Health Care 1 (January 1999), available on the web at http://ehealth.chcf.org/forecast4/index_show.cfm?doc_id=17.
41 Online Privacy: Researchers Use Internet Chat Rooms for Studies, California Healthline (May 1, 2000).
42 Jeffrey M. Cohen, Human Subjects Issues in Internet Research, 13 Health L. News 5 (2000).
43 A recent report sponsored by the California HealthCare Foundation profiled the policies and practices of 21 health-related web-sites and found that most of the privacy policies do not meet the minimum fair information practices, such as adequate notice and giving users some control over their information. Furthermore, the report shows inconsistencies between the privacy policies and the actual practices of the health websites. There were instances where personally identified data were transferred to third parties in direct violation of stated privacy policies. (Janlori Goldman et al., Report on the Privacy Policies and Practices of Health Web Sites [February 2000], available on the web at http://ehealth.chcf.org/priv_pol3/ index_show.cfm?doc_id=33).
44 Online Privacy: Researchers Use Internet Chat Rooms for Studies, supra note 41.
45 Ibid.
46 Associated Press, Scientists announce DNA mapping, June 26, 2000.
47 Lisa N. Geller et al., Individual, Family, and Societal Dimensions of Genetic Discrimination: A Case Study Analysis, 2 Science and Engineering Ethics 71 (1996).
48 U.S. Department of Labor, U.S. Department of Health and Human Services, Equal Employment Opportunity Commission, and U.S. Department of Justice, Genetic Information and the Workplace (Jan. 20, 1998), available at www.dol.gov/dol/_sec/public/media/reports/genetics.htm.
49 Ibid.
50 Reuters and The Associated Press, Genome announcement “technological triumph:” Milestone in genetics ushers in new era of discovery, responsibility (June 26, 2000), available at www.cnn.com/2000/HEALTH/06/26/ human.genome.04/ index.html.
51 45 CFR § 46.102(d).
52 David Casarett et al., Determining When Quality Improvement Initiatives Should Be Considered Research, 283 JAMA 2275, 2276 (2000).
53 U.S. General Accounting Office, Medical Records Privacy: Access Needed for Health Research, but Oversight of Privacy Protections Is Limited 11–12, GAO/HEHS-99-55 (February 1999).
C-30
54 Telephone interview with Daniel K. Nelson, Director, Human Research Studies, and Associate Professor of Social Medicine and Pediatrics, School of Medicine, University of North Carolina-Chapel Hill (July 14, 2000).
55 Louis Harris and Associates, Inc., Health Information Privacy Survey: A Survey of the Public and Leaders (1993).
56 Louis Harris and Associates, Inc., The 1996 Equifax-Harris Consumer Privacy Survey (1996).
57 U.S. General Accounting Office, Medical Records Privacy, supra note 53, at 17.
58 Ibid. at 16.
59 California HealthCare Foundation, National Survey: Confidentiality of Medical Records (January 1999), available on the Web at http://ehealth.chcf.org/cons_att2/index_show.cfm?doc_id=155>.
60 Ibid.
61 Goldman, Protecting Privacy to Improve Public Health, supra note 5, at 49.
62 Numerous federal reports in the past 20 years have recommended that comprehensive federal medial records confidentiality legislation be passed to protect patient privacy and the confidentiality of the health information. See National Research Council, For the Record: Protecting Electronic Health Information (1997); National Academy of Sciences, Institute of Medicine, Health Data in the Information Age: Use, Disclosure and Privacy (1994); Office of Technology Assessment, Protecting Privacy in Computerized Medical Information (1993); Advisory Committee on Automated Personal Data Systems, U.S. Department of Health, Education, and Welfare,
Records, Computers, and the Rights of Citizens (1973).
63 U.S. General Accounting Office, Medical Records Privacy, supra note 53, at 3.
64 45 FR § 46.110.
65 House Hearing, supra note 29 (Statement of Elizabeth Andrews, Director of Worldwide Epidemiology, Glaxo Wellcome).
66 Ibid.
67 Health Privacy Project, supra note 7, at 39.
68 Federal Policy for the Protection of Human Subjects, 56 Fed. Reg. 28003 (1991); 45 CFR § 46.101(b).
69 Telephone interview with Daniel K. Nelson, supra note 54.
70 Latanya Sweeney, supra note 39, at 100.
71 Ibid. at 98.
72 National Bioethics Advisory Commission, Executive Summary, Research Involving Human Biological Materials: Ethical Issues and Policy Guidance (August 1999).
73 45 CFR § 116(d).
74 42 USC §§ 1320d–1320d-8.
75 Under the regulations, “protected health information” is information that relate to a person’s physical or mental health, the provision of health care, or the payment of health care; identify, or could be used to identify, the person who is the subject of the information; be created by or received from a covered entity; and have been electronically maintained or transmitted by a covered entity at some point (Standards for Privacy of Individually Identifiable Health, 64 Fed. Reg. 59918, 60053 [1999]).
76 Exceptions are (1) inspection could be reasonably likely to endanger the life or physical safety of the patient or another person; 2) information identifies another individual and inspection is reasonably likely to cause substantial harm to that other individual; 3) disclosure is likely to reveal the source of information provided under a promise of confidentiality; 4) while the research study is in progress, and an IRB/privacy board has approved the denial of access and the participant has agreed to the denial when consenting to participation in the study; or 5) disclosure compiled for a legal proceeding (Standards for Privacy of Individually Identifiable Health, 64 Fed. Reg. at 60059–60060).
77 5 USC § 552a.
78 Jerry Berman and Janlori Goldman, A Federal Right of Information Privacy: The Need for Reform 14 (Washington, DC: Benton Foundation 1989); See also William W. Lowrance, Privacy and Health Research, supra note 1, at 59–60.
79 Berman and Janlori, supra note 78, at 15.
80 Memorandum from President William J. Clinton to the Heads of Executive Departments and Agencies, Privacy and Personal Information in Federal Records (May 14, 1998), available at www.pub.whitehouse.gov/uri-res/I2R?urn:pdi://oma.eop.gov.us/1998/5/14/8.text.1.
C-31
81 42 USC § 290dd-2. 82 21 USC § 872. 83 42 USC § 299a-1(c). 84 42 USC § 241(d). 85 42 USC § 242m(d).
86 42 USC §§ 242k and 242m(d).
87 42 USC § 3789g.
88 20 USC § 1232h.
89 Omnibus Consolidated and Emergency Supplemental Appropriations Act, Pub. L. No. 105-277.
90 Many states have Public Records statutes that provide access to information compiled by agencies of the state government. Some researchers have expressed concern that these state statutes may be used by individuals or corporations opposed to certain research to get access to research data that may identify subjects, threatening the privacy of the subjects and the confidentiality of their data. For example, in 1998, a law firm subpoenaed an environment scientist conducting research on pollutants, requesting records of private conversations and the scientist’s personal finances under the state’s open-records statute and FOIA. The scientist was forced to comply because her lawyers could not find recourse under state or federal law (Daniel K. Nelson, Vision 2030 Task Force for Social and Ethical Issues—Health and Biological Information).
91 Gramm-Leach-Bliley Act, Pub. L. No. 106-102, 113 Stat. 1338.
92 12 CFR Part 40.
93 12 CFR Part 216. 94 16 CFR Part 313. 95 12 CFR Part 573. 96 17 CFR Part 248. 97 12 CFR Part 716.
98 Joni Gray et al., Ethical and Legal Issues in AIDS Research 137 (1995).
99 638 F.2d 570 (3d Cir. 1980).
100 Isaacson v. Keck, 875 F. Supp. 478 (N.D. Ill. 1994).
101 Farnsworth v. Procter & Gamble Co, 758 F.2d 1545 (11th Cir. 1985).
102 See e.g., United States Environmental Protection Agency v. General Electric Co., 197 F.3d 592 (2d Cir. 1999).
103 Fed. R. Civ. P. 45(c)(3)(B)(i) and (ii).
104 1994 U.S. Dist. LEXIS 16933 (1994).
105 42 USC § 241(d).
106 Office for Protection from Research Risks, Office of Extramural Research, National Institutes of Health, U.S. Department of Health and Human Services, Certificates of Confidentiality: Privacy Protection for Research Subjects, available at http://ohrp.osophs.dhhs.gov/humansubjects/guidance/certconpriv.htm (last updated June 23, 2000).
107 Telephone interview with Moira A. Keane, Director, Research Subjects’ Protection Program IRB/IACUC, University of Minnesota Health Center (August 1, 2000).
108 Telephone interview with Daniel K. Nelson, supra note 54.
109 21 USC § 355(i).
110 21 CFR § 1316.21.
111 Joy Pritts et al., The State of Health Privacy: An Uneven Terrain (A Comprehensive Survey of State Health Privacy Statutes) (August 1999), available at www.healthprivacy.org/resources/index.shtml.
C-32
112 Hawaii and California are notable exceptions. Both states passed comprehensive health privacy laws in 1999. A few states are considering comprehensive health privacy legislation but are waiting for the release of the HIPAA regulations before passing any laws.
113 For example, HIV/AIDS statutes requiring physicians to report to the state health department the names and addresses of individuals suffering from HIV/AIDS also include restrictions on disclosure of such information to others. Such restrictions were passed in response to public fear that certain health information would be widely disclosed and used to deny benefits or cause other harm.
114 Minn. Stat. § 144.335(3a)(d). 115 Mich. Comp. Laws § 333.2632. 116 S.D. Codified Laws § 26-8-13. 117 Joy Pritts et al., supra note 111.
118 European Parliament and the Council of the European Union, Directive on the Protection of Individuals with Regard to the Processing of Personal Data and on the Free Movement of Such Data (95/46/EC), Official Journal of the European Communities No. L281, 31-50 (Nov. 23, 1995), available at www.privacy.org/ pi/intl_orgs/ec/final_EU_Data_Protection.html.
119 Kamran Abbassi, WMA to Produce Guidelines on Health Databases 320 BMJ 1295 (2000).
120 Associated Press, EU to Let U.S. Data Deal Stand, July 13, 2000.
121 Ibid.
122 U.S. General Accounting Office, Medical Records Privacy, supra note 53, at 5.
123 House Hearing, supra note 29 (Statement of Greg Koski, former Director, Human Research Affairs, Partners Health Care System).
124 Office for Protection from Research Risks, Office of Extramural Research, National Institutes of Health, U.S. Department of Health and Human Services, Protecting Human Research Subjects: Institutional Review Board Guidebook (1993) [hereinafter Institutional Review Board Guidebook]; 45 CFR § 46.103.
125 U.S. General Accounting Office, Medical Records Privacy, supra note 53, at 16.
126 Rick Weiss and Deborah Nelson, U.S. Halts Cancer Tests in Oklahoma, Wash. Post, July 11, 2000, at A1.
127 Information on the Office for Human Research Protections is available at http://ohrp.osophs.dhhs.gov.
128 1) Does the research involve observation or intrusion in situations where the subjects have a reasonable expectation of privacy? Would reasonable people be offended by such an intrusion? Can the research be redesigned to avoid the intrusion?
2) If privacy is to be invaded, does the importance of the research objective justify the intrusion? What if anything, will the subject be told later?
3) If the investigators want to review existing records to select subjects for further study, whose permission should be sought for access to those records? How should the subjects be approached?
4) Will the investigator(s) be collecting sensitive information about individuals? If so, have they made adequate provisions for protecting the confidentiality of the data through coding, destruction of identifying information, limiting access to the data, or whatever methods that may be appropriate to the study? If the information obtained about subjects might interest law enforcement or other government agencies to the extent that they might demand personally identifiable information, can a grant of confidentiality be sought from a federal or state agency to protect the research data and the identity of the subjects from subpoena or other legal process?
5) Are the investigator’s disclosures to subjects about confidentiality adequate? Should documentation of consent be waived in order to protect confidentiality? Institutional Review Board Guidebook, supra note 124, at 3–36 and 3–37.
129 Office of Inspector General, Recruiting Human Subjects, supra note 4, at 30.
130 U.S. General Accounting Office, Scientific Research: Continued Vigilance Critical to Protecting Human Subjects 5–6, GAO/T-HEHS-96-102 (March 12, 1996).
131 Telephone interview with Moira A. Keane, supra note 107.
132 U.S. General Accounting Office, Medical Records Privacy, supra note 53, at 21.
133 Telephone interview with Moira A. Keane, supra note 107.
134 Workshop Summary, supra note 21, at 19.
135 U.S. General Accounting Office, Medicare: Improvements Needed to Enhance Protection of Confidential Health Information, supra note 36, at 3.
C-33
136 U.S. General Accounting Office, Medical Records Privacy, supra note 53, at 17–18.
137 Ibid.
138 Lowrance, Privacy and Health Research, supra note 1, at 42.
139 Office of Inspector General, Department of Health and Human Services, Institutional Review Boards: A Time for Reform 5–6, OEI-01-97-00193 (June 1998); See also James Bell et al., Final Report: Evaluation of NIH Implementation of Section 491 of the Public Health Service Act, Mandating a Program of Protection for Research Subjects, Prepared for the Office of Extramural Research, National Institutes of Health 83–86 (June 15, 1998).
140 Office of Inspector General, Institutional Review Boards: A Time for Reform, supra note 139, at 6–8.
141 Office of Inspector General, Recruiting Human Subjects, supra note 4, at 26.
142 U.S. General Accounting Office, Medical Records Privacy, supra note 53, at 1–22.
143 Institute of Medicine, Protecting Data Privacy in Health Services Research, supra note 23, at 1–152.
144 House Hearing, supra note 29 (Statement of Carolin M. Frey, Chair, Institutional Research Review Board, Pennsylvania State Geisinger Medical Center).
145 Ibid.
146 Telephone interview with Daniel K. Nelson, supra note 54.
147 Health Privacy Project, supra note 7, at 37.
148 Bell et al., supra note 139, at 1–86.
149 U.S. General Accounting Office, Medical Records Privacy, supra note 53, at 21.
150 Ibid. at 13.
151 Ibid. at 10.
152 Ibid. at 12.
153 National Bioethics Advisory Commission, Summary of Preliminary Findings: Adequacy of Federal Protections for Human Subjects in Research, at bioethics.gov/finalmay3.pdf. (See Memorandum attached to Letter from Dr. Harold T. Shapiro, Chair of the National Bioethics Advisory Commission, to President William J. Clinton on the National Bioethics Advisory Commission Summary of Preliminary Findings: Adequacy of Federal Protections for Human Subjects in Research, May 4, 1999).
154 House Hearing, supra note 29 (Statement of Greg Koski, former Director, Human Research Affairs, Partners Health Care System).
155 Health Privacy Project, supra note 7, at 36.
156 Telephone interview with Daniel K. Nelson, supra note 54.
157 Office of Inspector General, Institutional Review Boards: A Time for Reform, supra note 139, at 6.
158 Ibid.
159 Telephone interview with Daniel K. Nelson, supra note 54.
160 Ibid.
161 U.S. Department of Health and Human Services, Fact Sheet, Protecting Research Subjects (May 23, 2000).
162 Telephone interview with Moira A. Keane, supra note 107.
163 In a recent article in the Journal of the American Medical Association, the authors suggest criteria to distinguish quality improvement activities from health research, proposing that an activity should be regulated as research if 1) the majority of participants involved are not expected to benefit directly from the knowledge to be gained or 2) additional risks or burdens are imposed to make the results generalizable. The authors acknowledge that such criteria may create greater burdens on health care institutions and IRBs by categorizing more initiatives as research but argue that “it makes little sense to reject these criteria, if they are otherwise sound, simply because they would create additional burdens for institutions (Casarett et al., supra note 52, at 2276–2279).
C-34

AN EXAMINATION OF ISSUES PRESENTED BY PROPOSALS TO UNIFY AND EXPAND FEDERAL OVERSIGHT OF HUMAN SUBJECT RESEARCH
Commissioned Paper C.K. Gunsalus
University of Illinois at Urbana-Champaign
D-1
Executive Summary
The National Bioethics Advisory Commission (NBAC) seeks to determine whether to improve the federal regulatory system for the protection of human subjects, and if needed, in what ways. This paper was commissioned to examine whether NBAC should recommend unifying federal oversight of federal and private human subjects research under a single government office such as the Office for Protection from Research Risks (OPRR).
The question posed by NBAC encompasses two related but distinct groups of issues: 1) those pertaining to unification of federal human subject protection oversight in a single agency or office and 2) those raised by expansion of the scope of federal oversight to cover not just federally funded, but also privately conducted human subjects research.
NBAC seeks to protect all human subjects of research against abuse or exploitation. But to get to that goal, NBAC must grapple with several fundamental questions: should citizenship or residency in the United States ensure a minimum level of protection against the risks inherent in research involving human subjects? If so, how is that level of protection defined? Is it possible to provide that level of protection efficiently, cost-effectively, and without burdening research that presents little or no risk to human subjects?
Our current system for protecting human subjects of research has many acknowledged strengths, and it balances effectively the competing interests always present in a regulatory system. It has served remarkably well for decades, and achieved many of the goals it was originally designed to meet. On the other hand, aspects of the system have known deficiencies that require correction and improvement. The recommendations in this paper are not designed to detract from the strengths of a good system, but to improve upon it in ways that will be beneficial without undue regulatory burden.
This paper recommends four elements for an improved regulatory system:
| 1. |
Correcting structural/organizational deficiencies in the present regulatory system, |
| 2. |
Unifying federal oversight of human subject research in one federal office or agency, but leaving in place the current jurisdiction of FDA over the approval of drugs, medical devices, and biologics, |
| 3. |
Using existing federal offices as structural models for unified oversight of human subjects research, and |
| 4. |
Expanding the scope of regulation incrementally rather than globally. This recommendation envisions an expansion of federal jurisdiction only to identified categories of research that meet the criterion of presenting known risks to human subjects of research. |
Correcting Deficiencies. A series of studies over recent years, culminating in the June 1998 Department of Health and Human Services (DHHS) Office of the Inspector General (OIG) report on Institutional Review Boards (IRBs) and the NBAC-commissioned papers by Drs. John C. Fletcher and Charles R. McCarthy, have identified deficiencies in our present system for protecting human subjects. These must be corrected in tandem with any expansion of federal oversight. Of particular concern are the conflicts of interest inherent in OPRR’s location within an agency for which it has a monitoring responsibility. Other key issues include the inadequate (and evidently declining) governmental resources allocated for the protection of human subjects; inconsistency of human subject protection across the government; and minimizing bureaucratic procedures in favor of educational efforts and true accountability.
Unification of Oversight Responsibilities. Responsibility for oversight of federally conducted or sponsored research should be consolidated into one federal agency or office. Responsibility for drug, device, and biologic approvals should remain with FDA, but the two agencies should develop a memorandum of understanding to codify their cooperation and coordination. Information is presented on existing governmental agencies that might serve as models for a reorganized and strengthened human subject protection office.
D-3
Recommended Strategy for Expanding Regulatory Scope. This paper proposes adopting a strategy of including all research posing “known risks” to human subjects of research under federal jurisdiction regardless of the source of funding or nature of the organization conducting the research. This approach is sensitive to current societal concerns about unchecked governmental regulation and should fare well under cost/benefit analyses. If NBAC adopts this proposed strategy, further work will be necessary, first to devise a mechanism for defining known risks, and then to develop a procedure for bringing relevant categories of research under federal jurisdiction.
I. Introduction
NBAC unanimously adopted a resolution on May 17, 1997, that “No person in the United States should be enrolled in research without the twin protections of informed consent by an authorized person and independent review of the risks and benefits of the research.”1 This position was reinforced when President Clinton asserted in a commencement address that same month that “[w]e must never allow our citizens to be unwitting guinea pigs in scientific experiments that put them at risk without their consent and full knowledge.”2 While the NBAC resolution and presidential declaration seem to be straightforward expressions of fundamental American beliefs about human rights and dignity, translating them into practice will be far from straightforward.
First, whether or not it is immediately apparent, these statements imply a sweeping expansion of federal regulation of research involving human subjects. Paradoxically, cats, dogs, rabbits, hamsters, guinea pigs, and nonhuman primates have more federal protection from the risks of participation in research than do humans.3 The federal government has regulated all research on these animals—regardless of the source of funding—since the Animal Welfare Act was first enacted in 1966. In contrast, the only research involving human subjects that is regulated by our government is that which a) is funded by one of seventeen federal agencies, b) is conducted without federal funds at an institution voluntarily extending federal oversight to the research, or c) involves drugs, devices, or biologics falling within the jurisdiction of the FDA. Absent these conditions, individuals with concerns or complaints about their treatment have no recourse except through civil litigation or criminal statutes. Thus at present, the minimum protections NBAC and the President seek are not even provided in all research conducted or paid for by the federal government, let alone that performed in the private sector.
While we cannot know how much unregulated research on human subjects takes place in the United States—precisely because it is not regulated—indications are that it is significant. Information about problematic practices in such research surfaces with sufficient regularity that expanded government oversight must be seriously considered.
Second, our system for the protection of human subjects of research is more than 30 years old, and, while the basic system is sound, we know that it has shortcomings. Beyond our knowledge of the existence of problematic unregulated research, we know that even regulated research may be exposing human subjects of research to inappropriate risks. Some of the deficiencies in the current regulatory structure and implementation are described in the Report of the DHHS Inspector General, Institutional Review Boards: A Time for Reform (June 1998), the Report of the Human Radiation Interagency Working Group (March 1997), the General Accounting Office (GAO) Report, Scientific Research: Continued Vigilance Critical to Protecting Human Subjects (1996), and the findings of the Advisory Committee on Human Radiation Experiments (ACHRE) (October 1995).4 To implement fully the NBAC resolution and give meaning to the President’s declaration, some of the identified problems in the current system must be corrected.
A pivotal issue is how federal oversight in our purposefully decentralized system of oversight for human subjects is fractionated, with 17 separate federal agencies holding responsibility. The decision to place primary responsibility for human subject protections with local IRBs at institutions conducting research is well suited in
D-4
many respects to our thriving research system. But federal oversight and protections are unevenly implemented and variably enforced, leading to serious gaps in human subject protections.
Another issue that NBAC must confront directly is the federal commitment to human subject protection as revealed through the resources devoted to the task. There is evidence that funding in this area has declined despite significant increases in research.5 Unless accompanied by adequate resources, neither reforms of our existing system nor expansion of federal protection will produce meaningful or long-lasting change.
Any proposal for change should be grounded in a clear statement of principles and goals: What is to be accomplished? The NBAC resolution already contains two goals: informed consent and independent ethical review for all persons “enrolled in research.” But the resolution does not define the “research” it intends to encompass or the level of risk at which these twin protections should attach.
Comprehensive application of the present federal definition of research—purposefully designed to be broad in its application and reach—could sweep myriad low-risk activities into a regulatory structure with unknown costs and implications. Activities that have never before been labeled as “research” could become subject to regulation, commanding resources for their review and oversight, ultimately to the detriment of human subjects in higher risk situations.
Many of this nation’s 3,000-plus6 IRBs are already overloaded by their current workloads. As the GAO report observes:
IRB reviews are labor intensive and time consuming, forcing boards to balance the need to make reviews thorough against the need to get them done. IRB members…are not paid for their IRB service. Board members themselves…face a heavy workload and others in the research community have raised concerns that heavy workload impairs IRB review.7
Research institutions would complain—and with some merit—if their workload is increased by a broad expansion of types of research requiring IRB review. One result could be a dramatic increase in the number of for-profit IRBs, or an incentive for IRBs to provide superficial reviews, or both. Careful design and implementation will be required to avoid a system that substitutes mechanical review for substantive ethical considerations.
Expanding federal jurisdiction to assure that “no person” is enrolled in research without the twin protections specified by NBAC requires care and focus—and will require changes in federal law and the commitment of additional federal resources to assure compliance with that law. To explore the issues raised by a unification of oversight into one federal agency and by a proposed expansion of federal oversight of research involving human subjects, we must examine 1) the present structure of federal regulatory protection, including its functioning, shortcomings, and the gaps in its coverage and 2) practical problems inherent in expanding the scope of federal oversight. These two issues are intertwined to a considerable degree.
II. The Present Federal System for Human Subject Protection
Government regulation frequently arises as a reaction to revelations that disturb the public conscience. The federal oversight of research involving human subjects is no exception. As recounted in David J. Rothman’s
Strangers at the Bedside: A History of How Law and Bioethics Transformed Medical Decision-Making,8 the entry of the federal government into this realm was driven by a combination of dramatic scientific/medical advances and scandals concerning abuses of human subjects of research. Medical advances in genetic engineering and heart transplantation gave rise to questions about the beginning, end, and quality of life. At the same time, disclosure of the now infamous Tuskegee experiment in 1972 and the abuses of human subjects detailed in Dr. Henry Beecher’s 1966 paper in the New England Journal of Medicine drew attention from the media and Congress.9 These in turn opened new areas of ethical debate including whether certain procedures should be governed outside the physician-patient relationship. More sophisticated versions of these questions are still with us today.
D-5
The reaction of the biomedical research establishment to these questions and to the prospect of government intrusion into the historical preserve of physicians and researchers was negative and strong—but not sufficient to convince Congress that patients and human subjects of research would be adequately protected without government intervention. Nonetheless, the strength of the reaction helped to shape the system of protection that resulted; similarly strong reactions can be expected to new proposals for change.
A. Background and Overview
Before moving to expand federal protections to subjects of currently unregulated research, we should examine the present system, which has grown incrementally over a period of years. The first federal policies covering research funded by the Department of Health, Education and Welfare (now DHHS) were issued in 1966. The first congressionally mandated commission, the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission), started its work in 1974.10 It produced ten reports over four years that provide the ethical foundation for the system of protections in place today. Even so, it took until 1991 for a subset of federal agencies to agree upon the Federal Policy for the Protection of Human Subjects11 as the core regulation governing research conducted by or under the auspices of the government. This policy is often referred to as “the Common Rule.” The Common Rule is not followed by all federal agencies, and it is unevenly enforced by those that do.
In 1994, Dr. Robyn Y. Nishimi of the Office of Technology Assessment (OTA), testified before Congress that:
No statute…governs the general oversight of research involving Americans. Moreover, the current system, while changing incrementally, has fallen short of implementing, or did not implement at all, recommendations made between 1973 and 1982 by an ad hoc committee of DHEW, a congressional report and two congressionally mandated commissions.12
Research involving human subjects may be regulated by the federal government through three separate mechanisms: a) because it is sponsored by a federal office or agency subscribing to the Common Rule; b) because an institution conducting research not sponsored by the federal government has voluntarily granted jurisdiction over the research to OPRR through a negotiated assurance; or c) because the research involves regulated drugs or medical devices over which the FDA has jurisdiction. An unknown quantity of research is not regulated either because the sponsoring/conducting agency does not subscribe to the Common Rule or has not negotiated an assurance extending federal jurisdiction or because the research is privately sponsored/conducted and not subject to FDA approval.
1. The Common Rule
Summary of Common Rule Provisions. The approach of the Common Rule to regulation of human subject research is decentralized, involving negotiation of assurances by the institutions where research is conducted with federal agencies certifying that certain procedural and substantive protections will be provided. While these assurances are received and overseen by the various federal agencies, review of specific proposed experimental protocols and informed consent forms occurs at the local level through IRBs. Federal requirements govern the composition and activities of IRBs, but as we shall see, true oversight and accountability for the rigor and consistency of IRBs has not been attained.
Six categories of research are exempt from full IRB review under the Common Rule.13 These review procedures permit research meeting specific, narrow criteria to proceed without any formal review. The six exemption categories, developed with public comment and through negotiation and policy formulation involving an interagency committee over a period of ten years, offer important insight into one mechanism that might be employed to address the practical problems that could arise from broadening the scope of federal regulation. (See below.) There are additional categories of research for which IRBs may use expedited review procedures,
D-6
on the theory that the types of research involved, like voice recordings or collection of fingernail clippings, are less intrusive and pose a low level of risk to the subject.14 Application of the Common Rule. Seventeen federal agencies that fund or conduct research subscribe to the Common Rule and thus use an approach similar to that of DHHS, the lead federal agency in this area, with the important exception that most do not have an active program for assuring compliance with applicable regulations. While there is no definitive assessment of how many federal agencies conduct or fund research on human subjects, the 1981 report of the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research, Protecting Human Subjects, documented that 23 federal entities funded research involving human subjects.15 Dr. Nishimi of OTA testified to Congress in 1994 that:
…a definitive picture of current federal implementation and oversight of existing regulations to protect human research subjects is not available….Currently, information from all agencies on the total number of all research grants or contracts, total funding for research and grants involving human subjects, and number of full time equivalent personnel devoted to assurance and compliance has not been collected in a coordinated or centralized fashion.…
For some agencies, information even limited to the number of, funding levels for, and types of research involved for current grants or contracts using human subjects could not be reported as recently as March 1994, although the common rule has been effective since June 1991. Without such information, ensuring that proper institutional assurances are in place and then overseeing compliance would appear to be problematic.16
Within DHHS, OPRR assumes oversight responsibility for both human and animal subjects of research. The FDA also has responsibility for protecting the rights and welfare of human subjects of research, in the context of its required approvals for drugs and medical devices. While both OPRR and FDA have mechanisms for reviewing cases of alleged noncompliance with federal regulations and responding to them,17 most other agencies do not. As Dr. Nishimi noted in 1994:
…agencies will not be aware of violations of existing regulations unless a rigorous system is in place to monitor compliance. Put another way, those Departments and agencies that are not looking for problems will not find any problems.18
The ACHRE inventoried federal experiments on human subjects and found that:
In most federal agencies, current mechanisms of oversight of research involving human subjects are limited to audits for cause and a review of paperwork requirements. These strategies do not provide a sufficient basis for assuring that the present system is working properly.19
2. OPRR Oversight System
OPRR relies heavily upon the assurances it negotiates with institutions conducting research. These assurances contain the institutions’ provisions for protecting the welfare of human subjects and generally follow common patterns. In addition to the promises institutions provide in their negotiated assurances, OPRR provides educational support and information to IRBs and queries institutions about reports of noncompliance. OPRR conducts a number of record reviews through paper correspondence and a much smaller number of on-site, for-cause reviews of IRB effectiveness. Both the GAO and the DHHS OIG compliment the effectiveness of OPRR’s compliance reviews, but both also comment upon the extraordinarily limited extent of on-site visits, due to staffing and budgetary constraints.
D-7
Currently, OPRR negotiates an assurance with each institution that receives research support from DHHS.20 Each assurance requires significant amounts of time and review by OPRR. According to the GAO, in 1996 OPRR had about 14 full-time equivalent staff devoted to human subject protection, with a budget for those activities of under $1 million. OPRR augments its professional staff with three physician volunteers.21 Most major institutions accepting federal research funding negotiate Multiple Project Assurances (MPAs) with OPRR through which they agree to provide the same protections to all subjects of research conducted at the institution that they do for research funded by DHHS. There are almost 450 MPAs covering more than 750 entities operating around 700 IRBs; they are virtually all in the United States.22 Two to four times as many institutions negotiate only Single Project Assurances (SPAs) for each individual project funded in whole or in part by DHHS (covering around 3,000 IRBs), or Cooperative Project Assurances (for multisite clinical trials), with another 1,250 associated IRBs.
There are about 3,000 active SPAs—locations where we know that some DHHS-regulated research is conducted but no MPA is in place to cover other research that may be performed at that institution.23 At these institutions, other research involving human subjects may occur without any governmentally provided protections for the subjects of that research. This does not necessarily mean that the research is not reviewed by an IRB, as institutions may choose voluntarily to extend those protections to all subjects of research—or they may not. It does mean that there is no federal jurisdiction to investigate if a subject of research files a complaint.
OPRR reports that negotiation of assurances for SPAs requires more time than other negotiations, because they usually involve OPRR scrutiny of protocols and informed consent documents from institutions with little or no history of review of research involving human subjects. Because DHHS funds research in 80 countries around the world, institutions negotiating SPAs are not all in the United States.
These and other recent reviews of the IRB system emphasize the changes that time and resource constraints have brought to their oversight by OPRR. While all contribute to a conclusion that OPRR does a good job of protecting human subjects of research, they also illustrate that its resources are inadequate for its present responsibilities and indicate areas where changes could strengthen its performance.24 The assurance negotiation process, for example, has by most accounts become routinized.25 The NBAC-commissioned paper by Dr. McCarthy provides background on the educational nature of the assurance negotiation process in its early phases: He implies that these negotiations were usually conducted on-site at institutions and describes how mutually beneficial these exchanges were, both for institutions with little background in these issues and for OPRR officials in gaining insight into the institution’s culture. By now, the negotiation process has lost much of this educational flavor; perhaps its time has just passed.
The McCarthy paper also describes an OPRR that was able to sustain a much larger educational program than is now the case. As an ongoing constant educational program is essential if consistency is to be achieved in a decentralized system, this is a serious matter. It is not an overstatement to suggest that, in a large distributed oversight system, high-quality educational programs are the cornerstone of true accountability. The report of the ACHRE went so far as to recommend that:
…efforts be undertaken on a national scale to ensure the centrality of ethics in the conduct of scientists whose research involves human subjects.…The necessary changes are unlikely to occur solely through the strengthening of federal rules and regulations or the development of harsher penalties.…The federal government must work in concert with the biomedical research community to exert leadership that alters the way in which research with human subjects is conceived and conducted so that no one in the scientific community should be able to say “I didn’t know” or “nobody told me” about the substance or importance of research ethics.26
D-8
Much of OPRR’s ability to conduct such programs has since been curtailed by budgetary reductions and limitations, although an ongoing set of programs is offered annually through co-sponsorship arrangements. Dr. McCarthy raises cautions against the conflicts of interest that can arise when regulated institutions are assuming responsibility for part of the cost of educational programs in this way.
OPRR’s reliance upon a paper-based and time-intensive assurance negotiation system is no longer desirable. OPRR agrees with the calls from external observers that it is time to make changes in the negotiation of assurances.27 Replacing the assurance system with a streamlined registration system seems a sound alternative. If change of this nature were adopted expeditiously, it would free some resources for activities more conducive to true accountability. OPRR should be able to make this change without regulatory modification, but should be encouraged to do so by NBAC.
Other recommendations of the OIG—several of which mirror changes OPRR staff have indicated they would like to adopt—will require more resources than are presently available to OPRR. This is a central issue with which NBAC must grapple as it formulates it recommendations.
3. FDA Oversight System
The FDA is responsible for the safety and effectiveness of medicines and medical devices. As part of its regulatory responsibilities, FDA requires that studies involving investigational new drugs, devices, and biologics receive review and approval by an approved IRB and that researchers submit statements that they will uphold ethical standards. FDA has “concurred” with the Common Rule, but has not adopted it in its entirety; while its regulations are largely congruent with those that OPRR enforces, there are differences in its IRB and informed consent regulations.
A major difference is that FDA does not require or negotiate assurances with institutions. It oversees IRBs through an inspection program, in which it routinely performs on-site procedural reviews of IRBs to determine whether they are in compliance with their own procedures and with applicable FDA regulations. The GAO reported that FDA employed about 13 full-time equivalent staff members devoted to IRB inspections in fiscal year 1995.28 FDA also has monitoring activities for individual drug studies and for clinical trials. Each involves reviewing compliance with consent requirements and other human subject protection protocols.
The GAO reviews concluded that while the FDA program is rigorous and that it detects (and corrects) problems in human subject research, “FDA’s inspection program is geared more toward protecting the eventual consumer of the drug than the subjects on whom the drug was tested.”29 If NBAC wishes to assure protection for human research subjects, this observation should trigger serious examination and consideration.
4. Nonsubscribing Federal Agencies
Subjects of research conducted or funded by federal agencies that do not subscribe to the Common Rule do not receive its core protections. There are indications that research is funded or conducted by the Nuclear Regulatory Commission, the National Endowment for the Humanities, and the Department of Labor.30 In 1995, the ACHRE found that the magnitude of research conducted by federal agencies not in compliance with the Common Rule is a significant concern and recommended that there be an assessment of the level of that research. It further recommended action to “ensure that all subjects are afforded the protections it offers.”31 Anticipating the ACHRE findings, President Clinton issued an Executive Memorandum in 1994 intended to address gaps in government coverage; specifically, he ordered that all federal agencies and departments should come into compliance with the Common Rule and to suspend noncompliant experiments immediately.32 There is no evidence that any department or agency suspended a single activity following the President’s instruction. The staff of NBAC is researching the issue of federal agency compliance with the Common Rule and this Executive Memorandum.
D-9
5. A Caveat—Not All Unregulated Research Goes Without Review
It is important to note that federal regulation is neither the only mechanism through which research is independently reviewed nor is it the only way participants in research are offered the protection of informed consent. It may not be appropriate to assume that expanding the scope of federal regulation is the only way to achieve the twin goals of assuring informed consent by subjects and objective review of protocols. Many universities extend to nonfederally funded research the same oversight required by federal regulation, mandating that all research conducted at the institution is subject to review by an IRB. Of course, in virtually all cases this voluntary extension lacks independent compliance oversight, so NBAC must confront the degree to which it considers compliance oversight to be essential to a federal protection system.
B. Documented Shortcomings of the Present System
Two recent reviews, one by the GAO in 1996 and one by the OIG of DHHS in 1998, document serious shortcomings in the functioning of IRBs across the country.33 Because our decentralized system depends upon local IRBs for review of research protocols, IRBs are the lynchpin of our human subject protection system. The two most recent reports build upon the earlier findings of the ACHRE.
These reports follow a string of earlier reports examining shortcomings in our systems of protections, and containing recommendations that have not been fully implemented. Recall Nishimi’s 1994 congressional testimony noting how many recommendations delivered over the decades have not been implemented. In that same testimony, she characterized national responses to problems as fitting a “crisis management” model, in which publicity leads to a commission, but few actual changes. A footnote to her testimony records that the President’s Commission made a follow-up report to Congress two years after its first report and called the progress in the interim “disappointing.” Nishimi, in 1994, stated that: “The Commission identified numerous deficiencies in agencies’ mechanisms to protect human subjects. It made a series of recommendations to improve Federal oversight, but to date virtually none has been implemented.”34 The ACHRE found in its 1995 report that “in comparison with the practices and policies of the 1940s and 1950s, there have been significant advances in the protection of the rights and interests of human subjects of biomedical research. However, we also find that there is evidence of serious deficiencies in some parts of the current system….” Their review found evidence of “substantial variation in the performance of institutional review boards” as well as in review of research proposal documents and in informed consent documents. Most importantly for NBAC, the committee found “evidence of confusion over the distinction between research and therapy.”35 It is worth remembering that the original National Commission spent a great deal of time in the early 1970s—and commissioned several analyses to assist its deliberations—examining the distinction between research and therapy as it set about devising a recommended definition of “research” to be regulated. ACHRE also articulated concerns about “adult subjects with questionable capacity” and research involving institutionalized children. NBAC is already addressing the concerns ACHRE identified about adult subjects with questionable capacity; the issue of the distinction (if any) between research and therapy will continue to be central to all discussions of appropriate regulatory scope.
Consistent with the comments of other observers, ACHRE recommended that IRBs give more attention to activities that pose more than minimal risk to subjects and that they seek to reduce paperwork and procedural requirements for activities posing less than minimal risk. In other words, focus resources on areas of greatest risk and concern to subjects.
GAO, in its 1996 review of human subject protections, found that “[t]he detection of recent instances of potential or actual harm to subjects both demonstrates that abuses can occur and also suggests that current oversight activities are working…[but] various time, resource and other pressures have reduced or threaten to
D-10
reduce the effectiveness of such oversight.”36 GAO found that the heavy workload of IRBs can weaken their oversight; that OPRR’s restricted site visit schedule and its location within the National Institutes of Health (NIH) hamper the effectiveness of its oversight of IRBs; and that changes in the nature of research and pressures for availability of unproven medical treatments make it difficult to protect human subjects.37 GAO also commented upon the organizational weakness in the location of OPRR within NIH that is examined in the NBAC-commissioned papers by Drs. Fletcher and McCarthy.38 This is a topic NBAC must address in its final recommendations.
The OIG reports find that “the effectiveness of IRBs is in jeopardy”39 with six major findings:
| 1. |
IRBs face major changes in the research environment, including those stemming from the expansion of managed care, increased commercialization of research, proliferation of multisite trials, new types of research, increased number of research proposals, and the rise of patient consumerism; |
| 2. |
IRBs review too much, too quickly, with too little expertise; |
| 3. |
IRBs conduct minimal continuing review of approved research; |
| 4. |
IRBs face conflicts that threaten their independence; |
| 5. |
IRBs provide too little training for investigators and board members; and |
| 6. |
Neither IRBs nor HHS devote much attention to evaluating the effectiveness of IRBs.40 While the OIG report found that OPRR’s on-site visits provide a better basis for assessment of an IRB’s |
performance than either its assurance process or the FDA inspection process, it also noted that OPRR’s resource constraints prevented it from making more than one for-cause site visit in the calendar year between April 1997 and May 1998. The OIG report stressed that it is a cardinal failing of our present system that neither OPRR nor FDA have a primary focus on assuring the effectiveness of IRBs. While the OIG report does not document any widespread abuses, the fact that we have no effective mechanism for assuring the accountability of IRBs is cause for grave concern.
The OIG report recommends “reengineering” the federal oversight process, with specific suggestions for revamping both the OPRR assurance process and the FDA inspection process for IRBs. Several recommendations focus on modifying procedural requirements in order to focus more effectively upon fundamental protections for human subjects of research. This is a theme that NBAC should embrace in all of its recommendations for change.
These findings only reinforce the sense that our existing system requires reform. While these reforms should be included in any recommendations made by NBAC, they should accompany, not supersede, additional changes to address identified risks to human subjects in presently unregulated research.
Recommendation 1: Correct Identified Deficiencies in Existing Federal Human Subjects Protection System
Before recommending that the federal government assume expanded responsibility for protection of human subjects involved in research, we should assure that it can fulfill its present obligations appropriately. We know our present review system has defects. Of those issues, the following seem most relevant to the expansion and unification questions posed by NBAC.
D-11
Recommendation 1A: Streamline the Assurance System
A number of informed observers—including some within OPRR itself—have come to believe that the existing assurance negotiation process has lost much of its original utility and has instead become unduly bureaucratic and cumbersome. While the process had important educational components in the early years of federal regulation, now that research institutions have become more sophisticated in this area, its time may have passed. Dr. Gary Ellis, testifying before the Subcommittee on Human Resources of the Committee on Government Reform and Oversight of the United States House of Representatives, acknowledged as much.41
The most consistently proposed change that is relatively easily implemented (i.e., without any regulatory modification) involves transforming the assurance system into a simplified registration system.
Streamlining the present assurance system would allow precious resources to be redirected to higher priority activities, including education and a more rigorous IRB performance-monitoring system. (Redirection of existing resources alone is unlikely to be sufficient to meet the full need but would be a good first step.) For example, if a registration model is adopted, instead of negotiating each assurance, OPRR would require each regulated entity to register with OPRR, providing the minimal amount of information required by the regulations.42 This approach would preserve the essential tether of the government to the system of institutional protections for the purposes of education and, when necessary, compliance oversight.
Recommendation 1B: Achieve Consistency Across the Government—Require Full Adherence to the Common Rule
Across the federal government the uneven application of existing regulations requires improvement: Even after President Clinton’s 1994 directive, not all federal agencies subscribe formally to the Common Rule, and among those that do the level of adherence is mixed. NBAC staff are studying current levels of compliance among federal agencies. Any recommendations formulated by NBAC should explicitly require—at a minimum—government-wide compliance with human subject protection regulations.
Recommendation 1C: Achieve Consistency Across the Government—Unify Government Oversight
In addition to requiring all government agencies to adhere to the Common Rule, NBAC should recommend unification of government oversight of human subjects in one federal agency or office. Given the uniform positive reviews from a variety of observers for OPRR’s expertise and effectiveness, this function should be assigned to OPRR, although the structure will require modification both to address the independence of the monitoring function. (See Recommendation 1D below.) Separate FDA jurisdiction over drugs, medical devices, and biologics should be retained, but FDA and the OPRR successor should enter into a Memorandum of Understanding to coordinate their functions and reduce the burden on multiply regulated entities. See further detail on this topic below.
Recommendation 1D: Assure Independence of the Government’s Monitoring Function
As noted by multiple observers from GAO to DHHS OIG to Drs. Fletcher and McCarthy, OPRR’s placement within DHHS presents serious structural problems that must not be perpetuated. A supplemental statement issued by GAO in response to congressional questions following the presentation of the GAO report noted: “…a potential weakness exists because NIH is both the regulator of human subject protection issues as well as an institution conducting its own human subject research. The Director of NIH, therefore, has responsibility for both the success of NIH’s intramural research program and for the enforcement of human subject protections by OPRR.”43 An approach for resolving these structural conflicts of interest must be incorporated into any proposed federal oversight mechanisms. The most obvious mechanism is to move OPRR (or any successor office/agency) out of NIH and place it elsewhere within the executive branch. Any successor office/agency should have the weight of authority necessary to carry out its mission, as well as the necessary resources. See Section IV below.
D-12
Recommendation 1E: Provide Adequate Resources
The current OPRR does not have enough staff or a large enough budget to meet its current mandate adequately, let alone to execute expanded responsibilities. It should be of serious concern that the financial commitment of DHHS to human subject protection, measured in financial terms, has been declining over time, even while research funding is increasing.44 While it is likely that additional resources are required to meet existing compliance oversight responsibilities, it seems without question that current resources for educational programs are inadequate. The consistency and quality of any decentralized system is necessarily dependent upon careful and continuing education of participants across sites. Documented deficiencies in the operation of IRBs call for more educational efforts and performance assessments; these tasks cannot be undertaken for research under OPRR’s current purview without additional resources. These costs should be assessed and addressed in addition to the projected costs for any new responsibilities. Mechanisms for addressing these shortcomings must be incorporated into any NBAC recommendations.
Reviews of the performance of OPRR in protecting subjects repeatedly show that it has the ability to address these shortcomings, but does not have sufficient resources for doing so. OPRR comes up short in any measure of educational activities, site visits, and timely resolution of allegations of noncompliance—to the detriment of current human research subjects.
Assuming identified deficiencies in the existing oversight system are corrected, then NBAC can move to considering expansion of federal jurisdiction in its effort to improve the federal regulatory system for the protection of human subjects. Rather than expanding regulation globally, however, and then finding mechanisms for removing low- or no-risk research from its purview, this paper recommends a different approach.
III. Issues Involved in the Expansion of Federal Oversight
Beyond the responsibility of the federal government to address known deficiencies in our system, we also know that there are human research subjects who are not receiving basic federal protections and who should be. How to provide those protections effectively—identifying the core protections to be provided around which societal consensus exists, focusing upon serious risks and with a reasonable cost/benefit ratio—is the challenge. NBAC must fully understand the gaps in current protection and practical problems that must be solved before recommending an expansion of federal oversight to encompass privately conducted research.
A. Gaps in Federal Protection
The OIG report on IRBs and the ACHRE report illustrate places where even research that is covered by federal regulation may not be receiving meaningful or accountable oversight. Beyond that, current federal regulations for protection of human subjects do not reach: research conducted or funded by federal agencies not subscribing to the Common Rule; research that is not federally funded conducted at institutions with SPAs and not covered by that institution’s assurance; and privately conducted research that is not subject to FDA jurisdiction. In none of these areas can it be assured that NBAC “twin protections” of informed consent and independent review are provided.
Dr. Gary Ellis, Director of OPRR, and others have offered examples where potentially harmful research has been reported, but where the subjects are not protected by federal regulation.45 Recent news reports about Viagra, the “male potency pill” contain references to clinics beginning their own research on its effects on women.46 (See Attachment A.) Are the participants in those efforts likely to receive the twin protections of informed consent and independent review of the risks? Do we, as a society, believe they should?
And what about the students and families about whom information would be stored in the database described in a January 1997 report in the Washington Post? (See Attachment B.) That report described a school district implementing a student database that would let schools compile medical and dental histories and
D-13
records of behavioral problems, learning disabilities, and family income. The newspaper report indicated that the new database would allow “administrators to monitor whether students of a particular ethnic background or sex were doing better or worse than others in English, algebra or any other course….a broader database would help administrators examine demographic, academic and extracurricular information in an effort to pinpoint causes and solutions.”47 Such databases could also provide a rich resource for researchers, but research uses are not currently regulated.
Other examples abound. They include research conducted at or by:
and: a December 1996 publication in a professional journal for reconstructive surgeons describing a prospective study comparing lateral and standard face lifts; there is no indication that patients were aware of or consented to their inclusion in the study.51 (See Attachment C4.)
and: “team management” research in which unsuspecting individuals were subjected to a sham robbery, resulting in significant stress, fear, and anxiety54 (see Attachment C7); another complainant to OPRR described “fright response” research in which participants were subjected to unexpected and disturbing visual stimuli.
D-14
Other research-related activities that could, and in some cases information exists to suggest they already have, present risks to human subjects include health services research and internal evaluation research. Health services research is increasingly common as managed care becomes more pervasive and typically involves efforts to measure efficacy and cost-effectiveness of various treatments in managed care organizations. Internal evaluation research involves comparisons of management techniques, labor practices, and other corporate research into how employees like or perceive their work environment. It will be a challenge to find the lines between benign surveys of employee satisfaction and more intrusive and/or coercive research that could compromise employee privacy. But while some of these examples are more egregious violations than others, none of them are currently regulated unless the research is funded by one of the Common Rule agencies.
B. Practical Problems in Expanding Federal Oversight
What might be the consequences of expanding the current definition of research and applying it globally to all research involving human subjects? More particularly, what is the wisdom, practicality, and cost-effectiveness of bringing a potentially broad range of activities under the scope of federal regulation?
1. What Should Be the Definition of “Research”?
Global applicability of the current definition of research could encompass many activities that impose very little or even no risk to subjects of that research. While the scope of federal protection is narrow, the current definition of research used for regulated activities is very broad:
D-15
‘Research’ means a systematic investigation designed to develop or contribute to generalizable knowledge.58
Many forms of polling, much market research, and arguably some forms of journalism could be considered “systematic investigation designed to develop or to contribute to generalizable knowledge” that is obtained “through intervention or interaction” with individuals or that involves “identifiable private information” about those individuals. Differentiating between activities that should be covered and those for which expanded federal regulation might be burdensome could consume significant resources and time on the part of many individuals and could prove divisive and distracting from the goal of protecting Americans from risk of serious harm through participation in research.
Should the current definition be used as is, or could it be modified to avoid such a result? The current definition was purposefully designed to be assure that subjects of research would be protected—whatever the research might be. Appendix Two of the Belmont Report (the report of the National Commission) contains a number of commissioned papers, at least four of which address the boundaries between research and therapy.59 These papers were commissioned as part of the National Commission’s formulation of its recommendations, including the definition of research in its final report.
When that definition was published in the Federal Register, only 21 comments addressed the proposed definition of research in the rulemaking process.60 The commentary accompanying the final regulation in January 1981 characterized those comments as follows: “While a few commentators favored the proposed definition because it offered flexibility to the IRB, a majority of the twenty-one opposed or raised questions about the definition. Several commentators felt that the definition is too broad and should be restricted to biomedical research.…” The DHHS Response to the comments observed that:
HHS believes that public concerns that the definitions are too broad will in most cases be met by the exemptions from the regulation. The National Commission, although not identifying specific fields of research, clearly intended to include behavioral studies in the recommended definition of ‘research.’ HHS agrees with this conclusion and does not believe that the definition of ‘research’ violates the rights of investigators given that the regulations exempt research which offers little or no risk to the rights and welfare of human research subjects.61
While one approach to the problem of sweeping low-risk research into an expanded federal regulatory scheme is to narrow the definition of research, the continuing progress of scientific advances applicable to human treatment suggests this is not a sound approach. No better definition of research than that currently used has attracted consensus support in the almost 30 years this definition has been in place. In the absence of a tested alternative, altering the definition itself seems unwise.
If the present definition is perpetuated rather than modified, it is likely that development of new exemptions should be considered to obviate unintended consequences of expanded regulatory scope and to focus government protections upon areas posing the greatest medical and ethical risks. It should be possible to craft appropriate exemptions for very low-risk “research.” In approaching such a task, the risk of harm must be balanced with the burden of regulation. On the other hand, given the extended and somewhat tortuous process required to develop and refine the current definitions and exemptions, some caution seems warranted. Before NBAC makes recommendations that might require the development of new exemption categories, alternatives should be carefully considered.
For example, not only would it be necessary to develop consensus across a broad spectrum of constituencies about new exemptions, but regulatory language would need to be carefully crafted and tested. Based on experience, this might well take a period of years. Would the entire process of expanding the twin protections of
D-16
informed consent and IRB review be delayed in the meantime, or would we go through a period in which potentially harmless or very low-risk activities would undergo unnecessary review? If the latter, what long-term effects might that have for a system that by many accounts is already overburdened and near the breaking point?
2. Who Decides an Activity Is Exempt? Conflict of Interest Questions
After the development of appropriate exemptions and embodying regulatory language, still another practical problem arises: Who will determine the applicability of the exemptions? It is fundamental that a person performing research has a conflict of interest in deciding that his or her research is exempt from review. This implies independent review, which raises a raft of troubling questions: Who will perform these reviews? How much paperwork will it require? For researchers not affiliated with universities, where will they find an appropriate IRB? Will this intensify existing incentives for a proliferation of for-profit IRBs? Might core ethical examinations be diluted by expanding the workload of IRBs along with the requirements for paperwork and review of low-risk research? At what cost might this occur?
The prospect that expansion might divert valuable resources and energy from projects needing thoughtful ethical review is troubling. It is not difficult to envision the creation of an extensive and burdensome, possibly profit-driven, rubber-stamping review system that dilutes attention to the serious ethical issues that research involving human subjects can imply. This is an outcome no one seeks. Further, the costs are potentially very large.
3. Costs
The costs involved in globally extending the current system could be significant. One indicator of the possible costs is that each (single-site) protocol review by Independent Review Consulting, Inc., (a reputable for-profit IRB that provides IRB services for unaffiliated investigators) costs $1,200.62 This does not, of course, include the costs involved in preparing materials to be reviewed by the IRB. Assuming that the direct costs of non-institutional review boards are comparable to those of academic IRBs, very large sums of money (representing the costs of creation, review, and maintenance of required information) could be at stake in a dramatically expanded system of human subject protections, especially those involving low-risk activities. The cost/benefit ratio for such an approach does not seem advantageous, especially in today’s political environment.
Recommendation 2: Expand Regulation Incrementally, Not Globally (at Least at First)
This recommendation proposes an alternative to expanding the scope of federal regulation very broadly and then crafting appropriate exemptions. It suggests adding targeted areas to the scope of federal oversight areas of research. Two possible mechanisms are proposed for NBAC’s consideration.
Recommendation 2A: Expand Jurisdiction Incrementally as “Known Risks” are Identified
As a starting place, NBAC might focus upon the goals articulated by the President of protecting subjects from unwitting participation and undue risk by focusing upon targeted areas. Given the estimate of the ACHRE that “40 to 50 percent of human subjects research poses no more than minimal risk of harm to subjects,”63 it is all the more critical to focus any new regulatory energy on activities that put human subjects at risk. While we cannot know if ACHRE’s estimate will extrapolate to presently unregulated activities, it is a reasonable starting point for thinking about these issues.
The goal should be to define areas of national concern by focusing on documented instances where human subjects have been exposed to:
D-17
| n |
where the protocols have not been subject to independent review for compliance with generally accepted standards of research involving human subjects. |
The targeted areas would focus on categories of “known risks”—research that we know puts human beings at risk, whether conducted privately or with federal support. An incremental approach seems more consistent with current trends in public policy, while still providing appropriate protections to residents of this country who participate in risky research activities. This approach would be more amenable to a documented cost/benefit analysis, and thus might be more persuasive to the public and to lawmakers.
Adopting this recommendation implies the development of categories requiring protection and procedures for invoking that protection. At first glance, likely candidate categories include:
Another, more controversial, category requiring serious examination is research that involves dignitary damage or breaches of confidentiality leaving the subject at risk.
An effort to identify and document known risks implies significant work, but this effort will likely be more productively expended—and generate greater support—than that required to extend the present regulatory system to cover “all” research.
Recommendation 2B: Explore Expanding OPRR’s Jurisdiction Without Statutory Change
Historically, OPRR has taken the position that the language of the Public Health Service Act64 requires mandatory compliance with its provisions only for research that is actually funded in whole or in part by DHHS. Thus, institutions filing an MPA voluntarily agree to apply federal regulations for human subject protections to non-DHHS research. Institutions that file SPAs have no obligation to ensure IRB review or informed consent for any other research involving human subjects. This may well be a more conservative interpretation of the Act than it requires.
NBAC should seek assistance and advice from the DHHS Office of the General Counsel to determine whether a broader reading of this statute is permissible. Specifically, “research” is not qualified in Sections 491(a) and (b)1 and refers to any biomedical or behavioral research involving human subjects. Can the Act be read to refer to all research at any institution supported by DHHS funds, not just research that is directly supported by DHHS?
Further advice and legal review will be necessary to explore this possibility. Such an expansion of OPRR’s jurisdiction will require a considerable addition of resources to OPRR. While seeking such advice may seem burdensome, the possible gains for regulated entities and for governmental efficiency warrant the effort.
IV. Possible Structures for Unified Federal Oversight
Whether NBAC decides to expand federal jurisdiction to encompass areas of known risks or to pursue more global federal jurisdiction, a different federal structure will be needed than is now in place. Deficiencies of the existing system that should be addressed in any proposed reforms should include more consistency and coordination across the government, as well as in the government’s interactions with regulated entities. Given the size of the federal government and the vast array of research sites across the country, NBAC should seek a structure that will provide a single office that works in a distributed style. Some existing agencies or offices that
D-18
currently function in this way provide models that have much to offer as exemplars. These include the Office of Governmental Ethics (OGE), the Office of Special Counsel (OSC), and the Nuclear Regulatory Commission (NRC). Although different in size and mission, each has educational and compliance-monitoring responsibilities, and each operates in a decentralized, distributed fashion.
Before considering the placement of the human subject protection monitoring system, one most important issue must be addressed—namely, in a unified federal oversight system, what should happen to the current functions represented in OPRR and FDA?
A. Unify OPRR and FDA?
Although it is always simpler from the perspective of a regulated entity to have only one federal oversight office, the missions of OPRR and FDA are sufficiently distinct that a strong case can be made that their independent functions should be maintained. Further, this is clearly the most pragmatic solution, since they currently operate under two distinct statutory authorizations, and the political ramifications of attempting a unification seem more complex and difficult than the gain would warrant. FDA and OPRR currently work in a coordinated fashion and have significant overlap in their approaches to regulated entities.
Thus, NBAC should recommend that these separate functions—drug and medical device approval and research oversight—should remain the primary province of FDA and the OPRR successor, respectively. The OPRR successor should be responsible for all regulated research involving human subjects, both government wide, and whatever private research is added to the regulatory structure.
To enhance coordination and cooperation, the two agencies should enter into a Memorandum of
Understanding that addresses interagency cooperation and jurisdiction and establishes a formal coordinating function. This should include new agreements covering IRB oversight to assure that the protection of human subjects is addressed in a reasonable, cost-effective way, especially in light of the GAO’s cautions about the substance of FDA IRB reviews and of concerns voiced by regulated entities about the sometimes burdensome nature of joint (and uncoordinated) jurisdiction by two federal agencies over the same IRBs.
NBAC or the successor agency may need to commission an examination of other special-purpose agency IRB regulations (for example, those at the Centers for Disease Control and perhaps the Department of Energy and/or those in classified settings) to determine whether other accommodations or Memoranda of Understanding might facilitate appropriate regulatory oversight.
B. Possible Models
The following existing governmental offices offer insights into possible models for an OPRR successor office that would oversee all human subject research.
1. OGE
The mandate of the OGE is to prevent ethical misconduct within the executive branch; it has responsibility for the prevention of conflicts of interest and for resolving those conflicts of interest that do arise. There are five applicable federal statutes for which it has enforcement responsibility. The Office of Public Integrity in the Department of Justice reviews OGE ethics opinions because it has enforcement authority for the underlying criminal statutes.
Created in the aftermath of the Watergate scandal, OGE was originally located within the Office of Personnel Management. During the Reagan administration, OGE became an independent agency. The Director is appointed by the President, with the advice and consent of the Senate, but that is the only politically appointed position in the agency. The remainder of the staff, about 80 people, are civil service employees. In contrast, OPRR has around 17 full-time staff members devoted to human subject protection (out of 28 total staff members). OPRR’s
D-19
FY 1995 budget was $2.25 million. Its FY 1996 budget was $2.13 million, and its FY 1997 budget was $2.10 million, a little more than half of which was spent on human subject protection activities.
OGE promulgates standards of conduct based on 14 fundamental principles. Its advisory opinions and ethical guidance are widely disseminated in the federal ethics community to assist in keeping officials informed and up to date. OGE oversees a broadly decentralized program in which each federal agency names a Designated Agency Ethics Official (DAEO); these 144 officials report jointly to the head of the agency and to OGE. This model seems particularly relevant when considering a government-wide human subject protection function.
OGE supports the DAEOs by developing educational materials and conducting training workshops for them and the other staff in each agency with responsibility for ethics compliance, who together comprise what is known as the federal “ethics community.” There are close to 12,000 part-time members of the federal ethics community, with about 400 of them serving on a full-time basis. While OGE audits their performance on a regular basis, the DAEOs hold significant responsibility within their agencies for educational programs and for compliance with congressional and presidential directives. This model of distributed responsibility dovetails nicely with the local control philosophy of federal oversight for research involving human subjects.
Although OGE focuses its efforts on education and providing positive guidance in response to questions, it also maintains a significant audit program, with 27 full-time auditors. These auditors review advice provided by DAEOs, the content of agency ethics training programs, and required financial disclosure forms. When violations of the standards of conduct are substantiated, they can lead to administrative sanctions (including reprimands, time off without pay, and/or demotion). Violations of the five applicable statutes carry higher penalties. OGE has 77 full-time employees and an annual budget of $7.6 million. See Attachment D for further information on OGE.
OGE’s independence from other government agencies presents an example that would cure the structural deficiencies found in OPRR’s placement within an agency that it must also monitor for compliance, as cited by GAO and Drs. Fletcher and McCarthy. At the same time, the joint reporting status of the DAEOs presents an interesting model that balances working within each agency’s individual culture while achieving consistent policy interpretation. Further, its independent standing emphasizes the importance of the issue it monitors and insulates it from political pressures. Finally, the distributed model could prove equally strong in the setting of regulated institutions.
On the other hand, OGE’s independent status and relatively small size may also reduce its leverage in budgetary processes, as it may not always have a seat at the table when budgetary compromises are reached. Embedded within a larger federal agency, budgetary negotiations have a different complexion. It is difficult to predict the quality and consistency of top-level attention to issues of human subject protection if those responsibilities are placed in an independent agency or department, especially in periods lacking in public focus on these issues.
2. OSC
The OSC was originally part of the U.S. Merit Systems Protection Board, but became “an independent federal investigative and prosecutorial agency” in July 1989. The principal responsibilities of the OSC are three-fold: 1) investigating allegations of prohibited personnel practice; 2) interpreting and enforcing the Hatch Act (political activities of federal employees); and 3) operating a whistleblower disclosure hotline to receive information “about wrongdoing in government.” The OSC’s role was expanded in 1994 to include investigation and prosecution of cases involving the denial of federal employment rights to veterans.
The President appoints the head of the agency, the Special Counsel. The remainder of the staff, about 95 civil service employees, report to the Special Counsel to carry out OSC’s responsibilities. OSC’s 1998 budget was $8.4 million.
D-20
Although OSC’s responsibilities are primarily executed within the executive branch, it serves as a useful model for NBAC because of its ability to work in a distributed, decentralized way across the full range of federal agencies. For example, OSC has jurisdiction to investigate allegations of prohibited personnel practices within any executive branch agency. These investigations are frequently conducted in conjunction with other government agencies. This model is particularly useful when thinking about oversight of intragovernment activities. See Attachment E for further information on OSC.
3. NRC
Holding wide regulatory and compliance responsibilities, the NRC operates on a completely different—and much larger—scale than the previously discussed offices. Established as an independent agency in 1974 by the Energy Reorganization Act, the purpose of the NRC is to “ensure adequate protection of the public health and safety, the common defense and security, and the environment in the use of nuclear materials in the United States.” The NRC’s responsibilities include regulation of commercial nuclear power reactors; medical, academic, and industrial uses of nuclear materials; and the transport, storage, and disposal of nuclear materials and waste. The NRC adheres to five Principles of Good Regulation that encourage ethical performance, openness to the public, efficient management and administration, clear regulations, and reliability.
Five commissioners are appointed by the President and confirmed by the Senate for five-year terms. One of the appointed commissioners is designated by the President to function as the chairman. A civil service staff reports to an executive director, who executes the directives of the commission. The overall structure and organization of the NRC provide NBAC with another established model of an independent agency that works in a distributed way within federal agencies and at diverse academic and private sites throughout the country. Further, it provides a model to examine when considering suggestions, such as Dr. Fletcher’s, that OPRR (or its successor) needs a citizen advisory panel.
Divided into divisions with specific responsibilities, the NRC has educational and compliance responsibilities similar to those of the OPRR, albeit on a much larger scale. Among its multiple divisions are one with responsibility for regulatory programs and another with responsibility for oversight and investigations. An Office of State Programs coordinates NRC activities with state and local governments as well as with other federal agencies and the sovereign Indian nations. NRC has 3,000 employees and an annual budget of $468 million. See Attachment F for further information on NRC.
Given the magnitude of NRC, it is somewhat difficult to make relevant comparisons to how this model might operate if translated into the human subject protection area. One possibility is that NBAC, or some similarly constituted commission, could serve as the policymaking body, with OPRR and FDA staffs carrying out their present roles. In such a configuration, perhaps the OPRR (research-oversight) function would fall under the NBAC successor function while the FDA staff would remain in that agency but have dual policy guidance.
If NBAC or a successor commission were to serve as the policymaking or advisory body for an OPRR successor, two issues must be addressed: 1) NBAC’s present expiration date (authority for human subject protection cannot be allowed to expire) and 2) the need for a revised charter to provide formal regulatory authority.
Recommendation 3: Explore Existing Models of Federal Offices/Agencies with Both Educational and Compliance Responsibilities—Design NBAC’s Recommendations Based Upon Those Models
Devising an improved governmental structure for a unified human subjects protection system will take expertise beyond the scope of this paper. Aside from explorations of the policy and political implications of its recommendations, NBAC will need to commission legal analyses of what enabling legislation or regulation will be necessary to effect any structure it suggests. NBAC must also address—perhaps through additional commissioned papers or through advice from established governmental mechanisms—reasonable resource allocations for the expanded functions it envisions.
D-21
Because this problem has been so intractable for so long, I encourage NBAC to provide specific instruction and draft legislation as part of its final report to the executive and legislative branches. Otherwise, its recommendations could well become just one more report sitting on a shelf.
V. Conclusion
In its June 1998 report, the OIG of DHHS found significant cause for concern in the current operation of our human subject protection system. While the OIG found no “widespread abuses of human research subjects,” its report identified aspects of our current system in pressing need of reform. This report does not stand alone: The observations of the OIG echo and reinforce those of multiple other observers of the current system, including many inside the government who hold responsibilities for protecting human subjects of research.
The challenge for NBAC is to devise recommendations for assuring substantive ethical consideration of the serious issues present in human subject research that can be enacted in the current political environment. This means addressing identified deficiencies in our current regulatory scheme, filling in some of the known gaps representing areas of real risk to residents of this country who participate in research, and assuring true accountability for this regulatory system in a cost-effective manner.
Responding to these challenges requires retooling the existing federal structure to provide cleaner lines of authority, uniform implementation of existing rules across the government, and streamlined links between the government and local IRBs.
Research subjects—particularly those who are not told they are participating in experimental activities or those participating in research that has not received prior independent ethical review—are among the most vulnerable of our population. In permitting their rights, welfare, and dignity to be compromised, we compromise our own.
It is time to finish the job of protecting human subjects that began more than three decades ago.
Acknowledgments
I am very grateful for the kind and generous help accorded to me by many people during the course of this process. Ada Sue Selwitz (University of Kentucky), Joan Rachlin (PRIM&R), Paula Knudson (University of Texas Health Science Center at Houston), Eric Meslin (NBAC), Gary Ellis (OPRR), and Michael Walker (University of Illinois) reviewed drafts of this paper in progress and provided many helpful comments; any errors that remain are my own.
A number of people gave graciously of their time and expertise in response to requests, including Stuart Gilman of the Office of Government Ethics, Victor Baird of the Senate Ethics Committee, Michelle Russell-Einhorn and Tom Puglisi of the Office for Protection from Research Risks, Allan Shipp of the American Association of Medical Colleges, Sal Giorgiani of Pfizer Pharmaceuticals, Mark Frankel and Al Teich of the American Association for the Advancement of Science, Erica Heath of Independent Review Consulting, Inc., and Robert Levine of Yale University.
Melanie Marino provided research support and editorial assistance throughout the development of this paper. Debra Kincaid was her usual cheerful self in helping to make room for this project amongst many others.
D-22
Attachments
| A: |
Kolata, G. “Doctors Debate Use of Drug to Help Women’s Sex Lives.” New York Times National. April 25, 1998. |
| B: |
Robberson, T. “Plan for Student Database Stirs Opposition in Fairfax.” Washington Post. January 9, 1997. |
| C1: |
OPRR Compliance Oversight Log. Letter to Dr. Melody Lin, Compliance Oversight, NIH, received June 18, 1993. Obtained from the OPRR under the Freedom of Information Act. |
| C2: |
OPRR Compliance Oversight Log. Regarding the Newark Beth Israel Medical Center, received September 28, 1994. Obtained from the OPRR under the Freedom of Information Act. |
| C3: |
OPRR Compliance Oversight Log. Regarding the St. Vincent Hospital Medical Center, Portland, Oregon, received April 13, 1995. Obtained from the OPRR under the Freedom of Information Act. |
| C4: |
Ivy, E.J., Lorenc, Z., and Aston, S.J. “Is there a Difference? A Prospective Study Comparing Lateral and Standard SMAS Face Lifts with Extended SMAS and Composite Rhytidectomies.” Plastic and Reconstructive Surgery. December 1996. |
| C5: |
“Adolf Coors Foundation Grants $240,000 to UCCS to Study Effects of Mercury Fillings.” University of Colorado at Colorado Springs, Office of Public Relations. November 9, 1995. Obtained from the OPRR under the Freedom of Information Act request by C.K. Gunsalus, 1998. |
| C6: |
OPRR Compliance Oversight Log. Received January 29, 1993. Obtained from the OPRR under the Freedom of Information Act request by C.K. Gunsalus, 1998. |
| C7: |
Romano, L. “A Night at the Office Became a Nightmare.” Washington Post. January 29, 1997. |
| C8: |
Letter from Dr. Gary Ellis to Dr. Curt Tompkins with attachments, April 28, 1997. Letter from Dr. Gary Ellis to Sung M. Lee, July 21, 1997. Obtained from the OPRR under the Freedom of Information Act request by C.K. Gunsalus, 1998. |
| C9: |
Kolata, G. “Selling Growth Drug for Children: The Legal and Ethical Questions.” New York Times National. August 15, 1997. |
| D: |
U.S. Office of Government Ethics. www.usoge.gov. Accessed June 11, 1998. |
| E: |
U.S. Office of Special Counsel. www.access.gpo.gov/osc. Accessed June 11, 1998. |
| F: |
U.S. Nuclear Regulatory Commission. www.nrc.gov. Accessed June 11, 1998. |
Notes
1 NBAC, Full Commission Meeting, Arlington, Virginia, May 17, 1997.
2 William Jefferson Clinton, Morgan State University Commencement Address, May 18, 1997.
3 To be fair, research involving human subjects encompasses a much broader range of activities than does research involving animals. Few of the difficult issues raised by behavioral research, violations of confidentiality, or invasion of privacy arise when working with animals, for example, which makes the prospect of more broadly regulating research on humans more complex in some ways than devising regulations for the appropriate treatment of animal subjects of research.
4 OIG, DHHS, Institutional Review Boards: A Time for Reform (OEI-01-97-00193), U.S. Government Printing Office, Washington, D.C., June 1998. U.S. Government, Human Radiation Interagency Working Group, Building Public Trust: Actions to Respond to the Report of the Advisory Committee on Human Radiation Experiments, March 1997. Final Report: Advisory Committee on Human Radiation Experiments, U.S. Government Printing Office, Washington, D.C., October 1995.
D-23
5 Statement of Robyn Y. Nishimi, Ph.D., Senior Associate, OTA, Hearing Before the Legislation and National Security Subcommittee of the Committee on Government Operations, House of Representatives, September 28, 1994, U.S. Government Printing Office, Washington, D.C., p. 164, referencing statements of Dr. Charles R. McCarthy, retired Director of OPRR. The DHHS OIG and GAO studies reinforce this conclusion.
6 Estimates of the number of IRBs operating in the United States range from around 3,000 to more than 5,000. OPRR oversees 700 IRBs associated with MPAs; about 1,250 associated with Cooperative Project Assurances; and around another 3,000 associated with SPAs. Personal communication from Tom Puglisi, OPRR, to C. K. Gunsalus, September 1998.
7 Report of GAO to the Ranking Minority Member, Committee on Governmental Affairs, U.S. Senate, Scientific Research: Continued Vigilance Critical to Protecting Human Subjects, U. S. Government Printing Office, Washington, D.C., March 1996, p. 17.
8 Rothman, D. J., Strangers at the Bedside: A History of How Law and Bioethics Transformed Medical Decision Making, Basic Books, 1991, pp. 168–189.
9 Beecher, H.K., 1966, “Ethics and Clinical Research,” New England Journal of Medicine 274:1354–1360.
10 Report and Recommendations of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research, U.S. Government Printing Office, Washington, D.C., 1978.
11 The Model Federal Policy for the Protection of Human Subjects, 56 Federal Register 28002, June 19, 1991.
12 Nishimi testimony, pp. 149–150.
13 45 CFR 46, Revised June 18, 1991 (Effective August 19, 1991) Subpart A—Federal Policy for the Protection of Human Subjects (Basic DHHS Policy for Protection of Human Research Subjects):
“Unless otherwise required by department or agency heads, research activities, in which the only involvement of human subjects will be in one or more of the following categories, are exempt from this policy:
| 1. |
Research conducted in established or commonly accepted educational settings, involving normal educational practices, such as (i) research on regular and special education instructional strategies, or (ii) research on the effectiveness of or the comparison among instructional techniques, curricula, or classroom management methods. |
| 2. |
Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures or observation of public behavior, unless: (i) information obtained is recorded in such a manner that human subjects can be identified, directly or through identifiers linked to the subjects; and (ii) any disclosure of the human subjects’ responses outside the research could reasonably place the subjects at risk of criminal or civil liability, or be damaging to the subjects’ financial standing, employability, or reputation. |
| 3. |
Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures, or observation of public behavior that is not exempt under paragraph (b)(2) of this section, if: (i) the human subjects are elected or appointed public officials or candidates for public office; or (ii) federal statute(s) require(s) without exception that the confidentiality of the personally identifiable information will be maintained throughout the research and thereafter. |
| 4. |
Research, involving the collection or study of existing data, documents, records, pathological specimens, or diagnostic specimens, if these sources are publicly available, or if the information is recorded by the investigator in such a manner that subjects cannot be identified, directly or through identifiers linked to the subjects. |
| 5. |
Research and demonstration projects, which are conducted by or subject to the approval of department or agency heads, and which are designed to study, evaluate, or otherwise examine: (i) public benefit or service programs; (ii) procedures for obtaining benefits or services under those programs; (iii) possible changes in or alternatives to those programs or procedures; or (iv) possible changes in methods or levels of payment for benefits or services under those programs. |
| 6. |
Taste and food quality evaluation and consumer acceptance studies, (i) if wholesome foods without additives are consumed, or (ii) if a food is consumed that contains a food ingredient at or below the level and for a use found to be safe, or agricultural chemical or environmental contaminant at or below the level found to be safe, by the Food and Drug Administration, or approved by the Environmental Protection Agency, or the Food Safety and Inspection Service of the U.S. Department of Agriculture.” |
D-24
14 45 CFR 46.110. Expedited review procedures for certain kinds of research involving no more than minimal risk, and for minor changes in approved research.
15 Report of the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research, Protecting Human Subjects, U. S. Government Printing Office, Washington, D.C., 1981.
16 Nishimi testimony, pp. 162–163.
17 21 CFR. 56 155 (b) states: “The records required by this regulation shall be retained for at least 3 years after completion of the research, and the records shall be accessible for inspection and copying by authorized representatives of the Food and Drug Administration at reasonable times and in a reasonable manner.” OPRR’s authority to investigate derives from the Public Health Service Act, as amended by the Health Research Extension Act of 1985, Public Law 99-158, November 20, 1985, Section 491(2) which states: “The Secretary shall establish a process for the prompt and appropriate response to information provided to the director of NIH respecting incidences of violations of the rights of human subjects of research for which funds have been made available under this Act. The process shall include procedures for the receiving of reports of such information from recipients of funds under this Act and taking appropriate action with respect to such violations.”
18 Nishimi testimony, p. 162.
19 Final Report: Advisory Committee on Human Radiation Experiments, Chapter 18, Recommendation 13, Commentary.
20 45 CFR Part 46.103(a).
21 Scientific Research: Continued Vigilance Critical to Protecting Human Subjects, p. 6.
22 There is one MPA institution in Canada (McGill). Statistics on OPRR assurances and oversight in personal communication from OPRR to C.K. Gunsalus, August 10, 1998.
23 Statistics on OPRR caseload from personal communication, Gary R. Ellis to C.K. Gunsalus, April 1998.
24 Scientific Research: Continued Vigilance Critical to Protecting Human Subjects, pp. 19–20. Institutional Review Boards: Their Role in Reviewing Approved Research, p. 13.
25 Scientific Research: Continued Vigilance Critical to Protecting Human Subjects, pp. 8, 19. McCarthy, C.R., Report for NBAC, Reflections on the Organizational Focus of the Office for Protection from Research Risks, 1996, p. 10. Institutional Review Boards: Their Role in Reviewing Approved Research, p. 12.
26 Final Report: Advisory Committee on Human Radiation Experiments, Chapter 18, Recommendation 9.
27 Testimony of Dr. Gary B. Ellis, Director, OPRR, Office of Extramural Research, NIH, DHHS, before the Subcommittee on Human Resources, of the Committee on Government Reform and Oversight of the U.S. House of Representatives, June 11, 1998.
28 Scientific Research: Continued Vigilance Critical to Protecting Human Subjects, p. 12.
29 Scientific Research: Continued Vigilance Critical to Protecting Human Subjects, p. 21.
30 Ellis testimony.
31 Final Report: Advisory Committee on Human Radiation Experiments.
32 President Clinton’s Order directs that all departments and agencies of the government “cease immediately sponsoring or conducting any experiments involving humans that do not fully comply with the Federal Policy.” Memorandum for the Vice President, the Heads of Executive Departments and Agencies, Subject: Review of Federal Policy for the Protection of Human Subjects, February 17, 1994.
33 OIG, DHHS, Institutional Review Boards: Their Role in Reviewing Approved Research (OEI-01-97-00190); Institutional Review Boards: Promising Approaches (OEI-01-98-0091); Institutional Review Boards: The Emergence of Independent Boards (OEI-01-97-00192);
Institutional Review Boards: A Time for Reform (OEI-01-97-00193), U.S. Government Printing Office, Washington, D.C., June 1998.
Scientific Research: Continued Vigilance Critical to Protecting Human Subjects.
34 Nishimi testimony, p. 157, footnote 3.
35 Final Report: Advisory Committee on Human Radiation Experiments, Chapter 17, Finding 22.
36 Testimony of Sarah F. Jagger, Director, Health Financing and Public Health Issues, Health Education and Human Services Division, U.S. GAO, before the Committee on Governmental Affairs, U.S. Senate, March 12, 1996.
D-25
37 Ibid.
38 Scientific Research: Continued Vigilance Critical to Protecting Human Subjects, p. 20.
39 Institutional Review Boards: A Time for Reform, p. ii.
40 Ibid.
41 Ellis testimony.
42 45 CFR Part 46.103(b) requires that each institution provide certain specific information to OPRR.
43 Supplemental Submission by Sarah F. Jagger, Director, Health Financing and Public Health Issues, U.S. GAO, contained in letter to the Honorable Ted Stevens, Chairman, Committee on Governmental Affairs, U.S. Senate, March 20, 1996, in the Proceedings of the Hearing Before the Committee on Governmental Affairs, U.S. Senate, U.S. Government Printing Office, Washington, D.C., March 12, 1996, p. 399.
44 Nishimi testimony, 1994.
45 Letter from Dr. Gary B. Ellis, Director, OPRR to James F. Childress, Ph.D., Chairman, Human Subjects Subcommittee, NBAC, April 10, 1997.
46 Kolata, G., “Doctors Debate Use of Drugs to Help Women’s Sex Lives,” New York Times, Sec. A, April 25, 1998.
47 Robberson, T., “Plan for Student Database Stirs Opposition in Fairfax,” Washington Post, Sec. A, January 9, 1997.
48 OPRR Compliance Oversight Log, Letter to Dr. Melody Lin, Compliance Oversight, NIH, received June 18, 1993. Obtained from the OPRR under the Freedom of Information Act request by C.K. Gunsalus, 1998.
49 OPRR Compliance Oversight Log, Regarding the Newark Beth Israel Medical Center, received September 28, 1994. Obtained from the OPRR under the Freedom of Information Act request by C.K. Gunsalus, 1998.
50 OPRR Compliance Oversight Log, Regarding the St. Vincent Hospital Medical Center, Portland, Oregon, received April 13, 1995. Obtained from the OPRR under the Freedom of Information Act request by C.K. Gunsalus, 1998.
51 Ivy, E.J., Lorenc, Z., and Aston, S.J., “Is There a Difference? A Prospective Study Comparing Lateral and Standard SMAS Face Lifts with Extended SMAS and Composite Rhytidectomies,” Plastic and Reconstructive Surgery, December 1996.
52 “Adolf Coors Foundation Grants $240,000 to UCCS to Study Effects of Mercury Fillings,” University of Colorado at Colorado Springs, Office of Public Relations, November 9, 1995. Obtained from the OPRR under the Freedom of Information Act request by C.K. Gunsalus, 1998.
53 OPRR Compliance Oversight Log, received January 29, 1993. Obtained from the OPRR under the Freedom of Information Act request by C.K. Gunsalus, 1998.
54 Romano, L., “A Night at the Office Became a Nightmare,” Washington Post, January 29, 1997.
55 The National Human Genome Research Institute, Final Report of the Task Force on Genetic Testing: Promoting Safe and Effective Genetic Testing in the United States, September 1997.
56 Letter from Dr. Gary Ellis to Dr. Curt Tompkins with attachments, April 28, 1997. Letter from Dr. Gary Ellis to Sung M. Lee, July 21, 1997. Obtained from the OPRR under the Freedom of Information Act request by C.K. Gunsalus, 1998.
57 Kolata, G., “Selling Growth Drug for Children: The Legal and Ethical Questions,” New York Times National, August 15, 1997.
58 45 CFR 46.102: “Human subject” means a living individual about whom an investigator, (whether professional or student) conducting research obtains (a) data through intervention or interaction with the individual, or (b) identifiable private information.
“Intervention” includes both physical procedures by which data are gathered (for example, venipuncture) and manipulations of the subject or the subject’s environment that are performed for research purposes.
“Interaction” includes communication or interpersonal contact between investigator and subject.
“Private information” includes information about behavior that occurs in a context in which an individual can reasonably expect that no observation or recording is taking place, and information which has been provided for specific purposes by an individual and which the individual can reasonably expect will not be made public (for example, a medical record). Private information must be individually identifiable (i.e., the identity of the subject is or may readily be ascertained by the investigator or associated with the information), in order for obtaining the information to constitute research involving human subjects.”
D-26
59 Levine, R.J., ”The Boundaries Between Biomedical or Behavioral Research and the Accepted and Routine Practice of Medicine,” July 14, 1975; London, P. and Klerman, G., “Boundaries Between Research and Therapy, Especially in Mental Health;” papers by David Sabiston, M.D. and John Robertson, J.D., 1975. Commissioned Papers for the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research, U.S. Government Printing Office, Washington, D.C., 1978.
60 Federal Register, Vol. 46, No. 18, January 26, 1981, pp. 8372–8373.
61 Ibid. At the same time, other changes were made in the Belmont Report’s proposed definition. In response to other concerns about the breadth of the proposed definition of research, DHHS inserted the term “living” into the definition of human subject to clarify that historical and biographical research were not covered. The final regulation also used “private” to modify “information” to make it clear that the “regulations are applicable only to research which involves intervention or interaction with an individual or identifiable private information.” Private information was clearly defined, with the following concluding comment: “It is expected that this definition exempts from the regulations nearly all library-based political, literary and historical research, as well as purely observational research in most public contexts, such as behavior on the street and in crowds.”
62 Expedited reviews cost $200 each. Independent Review Consulting, Inc., Institutional Review Board, Fee Schedule, 1997.
63 Final Report: Advisory Committee on Human Radiation Experiments, Commentary following Finding 22, Chapter 17, 1995.
64 Public Health Service Act, as amended by the Health Research Extension Act of 1985, Public Law 99-158, November 20, 1995.
D-27

THE HISTORY, FUNCTION, AND FUTURE OF INDEPENDENT INSTITUTIONAL REVIEW BOARDS
Erica Heath
Independent Review Consulting, Inc.
E-1
I. Executive Summary
The National Bioethics Advisory Commission (NBAC) has requested information about the philosophical and practical issues related to the role of independent institutional review boards (IRBs) in the current medical research community. This paper provides a working definition of independent IRBs. It describes their role within a broader framework of protections for human subjects. It addresses their history and development and describes the strengths and weaknesses of independent IRBs.
As the term suggests, an independent IRB is a subset of a wider universe of IRBs; as such, it exists for the same purpose as all IRBs—to review clinical research plans to ensure that adequate human subject protections have been incorporated. An independent IRB is subject to the same federal and state regulatory requirements applicable to all IRBs. Although it is difficult to produce a single definition of an independent IRB, due to the diversity of these entities, the following description is offered:
An independent IRB is one that reviews research for the purpose of assuring adequate protection of human subjects for entities that generally are not part of the same organizational structure as the IRB.
Beginning in 1966, the federal government established requirements for protection of human subjects in institutions receiving federal funding. Centers conducting research entered agreements called Multiple Project Assurances (MPAs) with the Department of Health, Education, and Welfare (DHEW) through the Office for Protection from Research Risks (OPRR) (now the Office for Human Research Protections—OHRP).1 That the system was decentralized and was institutionally based is a reflection of the organization of research in that era. Academic medical centers were the locus of most research, research was predominantly single site, and most sites acted independently and interacted rarely.
Over time, the research landscape has dramatically changed. In order to meet the demands of the new research environment, independent IRBs were born. The Food and Drug Administration’s (FDA’s) recognition that IRBs need not be located in an institutional setting created the first gateway for the use of independent IRBs throughout the 1980s. In 1995, OPRR began granting Single Project Assurances (SPAs) for projects reviewed by independent IRBs.
Although the greatest need for independent IRBs remains outside the academic and hospital settings, independent IRBs have been used in many institutional settings including institutions that contract for outside review, institutional IRBs that accept the review of an independent IRB for multicenter studies, and institutions that use an independent IRB as a bridge to an improved internal review system.
The benefits of independent IRBs continue to emerge: 1) independent IRBs fill a void by providing review to centers that might not otherwise have adequate IRB review, 2) independent IRBs have provided significant advantages in reviewing multisite research, 3) independent IRBs provide structured and efficient reviews, and 4) independent IRBs’ independence from the institutions for which they provide reviews frees them from the conflicts of interest associated with the institutions.
Several perceived weaknesses have been identified as inherent in the structure of independent IRBs: 1) conflict of interest, 2) the possibility of “shopping” for IRB approval, and 3) lack of physical presence at the performance site. All of these concerns can be addressed through proper organizational structure and/or implementation of standard operating procedures.
Because independent IRBs evolved out of a changing research environment, they are well suited to ensure that the needs of investigators, sponsors, and government regulators are met, while maintaining human subject protection.
E-3
II. Independent IRBs Defined
A. IRBs in General
Before discussing the definition of an independent IRB, a general review of the essentials of an IRB is offered along with a review of some of the elements that all IRBs share and some elements that distinguish them:
and practice. This diversity is reflected in the number of names used to describe them (see Exhibit 1). Many of these adjectives can apply to one IRB or to several. A few examples follow:
Exhibit 1
Various adjectives, illustrative of the wide variety among IRB form and function, have been used to characterize IRBs:
E-4
B. Similarities Among IRBs
The class of independent IRB is a subset within this general description of IRBs. Similarities exist between independent and institutional IRBs:
| n |
They exist for the same purposes: protection of human subjects of research. |
| n |
They are guided by the same federal and state legal and ethical requirements, including both the Department of Health and Human Services (DHHS) and FDA regulations, as applicable. |
| n |
They must have an organizational structure and written operating policies and procedures. |
| n |
Their membership composition must meet the same regulatory standards. |
| n |
They are subject to external audit by both FDA and OPRR (for SPA approved work). C. Differences Among IRBs |
Although there is a splendid variety within both independent and institutional structures for IRBs, there are several key features that distinguish the independent IRB:
Just as an institutional IRB is part of an institution, an independent IRB is always a part of an organization that can be defined as an institution within the Common Rule.3 As defined, institutions of either type may be large or small, for-profit or nonprofit, professional or volunteer, professional medical practices, hospitals, nonprofit foundations, or contract research organizations. The independent IRB may also be part of a corporation unaffiliated with any other organization.
D. A Suggested Definition
The definition suggested here is intended to highlight both the similarities and differences. An independent IRB is
…an IRB...which reviews research...for the purpose of assuring adequate protection of human subjects...for entities that generally are not part of the same organizational structure as the IRB.
This definition suggests that an independent IRB performs the same function as any IRB. It reviews research for the same purpose as other IRBs. The defining difference is that the institution conducting the research and the institution supporting the IRB are different organizational entities.4
E-5
III. An Environment Engendering Independent IRBs
The traditional institutional IRB was created in response to the research environment. When that environment changed it was necessary to create a legal and ethical alternative. The independent IRB arose to fill the need created by this change.
A. The Early Regulation of Medical Research and the Public Health Service Response
As long as man has been interested in scientific learning, people have conducted experiments to determine how the human mind and body respond to certain stimuli, from machinery and electricity to sounds and chemicals. Gradually such experiments evolved from single anecdotal studies to more formal experiments, to research in which groups of subjects were studied in an organized manner to systematically answer a broader question.
Many research studies have lead to groundbreaking discoveries that have benefited humankind. The public has known, however, that these research projects also have the capacity to damage human participants and may present unacceptable risks to society as a whole. This negative side is evidenced by the horrific experiments conducted during World War II or by the later revelations concerning American studies such as those performed at the Jewish Chronic Disease Hospital5 or at Willowbrook State Hospital.6 In 1966, an article by Henry Beecher7 brought prominent attention to human research abuses in medical schools and hospitals, citing 22 cases involving highly questionable ethics.
In recognition of the potential risks to human subjects inherent in scientific research, and knowing that the U.S. government was actively funding such research, U.S. Surgeon General William Stewart issued an important policy statement on February 8, 1966,8 related to the administration of federal grants and contracts supporting research, development, and related activities involving human subjects. Key elements of this policy were:
The first assurances, issued in 1966, were very short and dealt only with fundamental issues. Later assurances have become complex and reflect many subsequent interpretations of the initial basic premises.
The concepts outlined in Surgeon General Stewart’s policy statement were refined over the next few years, and by 1971 they had made their way into the Grants Administration Manual of DHEW. The concepts were made available to the newly formed reviewing committees through distribution of a pamphlet readers called “The Little Yellow Book.”9 This pamphlet instructed that studies involving human subjects needed committee review. The review was to address three concerns: 1) whether benefits of the study exceeded risks, 2) whether the rights, safety, and welfare of subjects were protected, and 3) whether adequate provisions were made to obtain informed consent. Human subjects were persons placed “at risk” by their participation. (Interestingly, if an investigator decided that his subjects were not at risk, review was not required.) In May 1974, the first regulations requiring IRB review for protection of human subjects were issued as Title 45 of the Code of Federal Regulations (CFR), Part 46. For the first time the committees conducting research reviews became known as “institutional review boards.”10 The new regulations provided revised definitions of research, human subject, and assurance, provided criteria for IRB review, and expanded the elements of informed consent.
E-6
In June of 1974, the National Research Act (Public Law 93-348) was signed into law creating the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission). The National Commission was charged with making recommendations particularly about inclusion of various vulnerable populations in research. Its best-remembered report dealt with the “ethical problems” precepts underlying Western research. The National Commission’s work resulted in the Belmont Report and in an affirmation of the basic requirements of the IRB system.
Over time, the regulatory system evolved to include more types of research and to increase the importance of IRBs and the amount of work they were asked to perform. Social and behavioral research funded by the PHS was brought within the jurisdiction of the regulation and IRB review.
Critically, an increasing number of assurances contained a statement that all research conducted within the institution must be reviewed using the single standard set forth in 45 CFR 46. This meant that, in an institution with an MPA, all studies were reviewed under 45 CFR 46 regardless of the source of funding or other regulatory controls.
Other federal agencies were actively developing human subject protection programs, most of which adopted the same basic requirements involving IRB review and informed consent. However, each agency had slightly different requirements. For instance, the Department of the Navy required signatures of all IRB members on approval letters, while the Department of Energy had other elements of consent. FDA requirements were more voluntary and did not require consent if the doctor determined it was not in the subject’s best interest. This conflicting hodgepodge of regulations caused substantial confusion.
In 1978, the National Commission concluded that IRBs should be governed by uniform federal regulations.11 This very well received recommendation eventually resulted first in the 1981 regulation, which harmonized FDA with DHHS, and then in the 1991 issuance of the Common Rule.12 The National Commission also recognized that flexibility must be maintained in creating IRBs. For example, the National Commission explained that an IRB may be located in the institution where the research is being conducted or outside of it and may review research for one institution only or for several institutions.13
B. The FDA Response
Although the FDA was an agency within DHEW and later DHHS, its history with regard to human subjects protection developed independently of its sister agency, the National Institutes of Health. FDA regulations developed in response to other incidents, congressional actions, and its own regulatory responsibility.
The FDA’s history of regulation of human subjects research started in 1962 with the Kefauver Amendment to the Food, Drug, and Cosmetic Act. This act included the requirement that informed consent should be required unless it “was deemed not to be feasible,” or it was “contrary to the best interests of such human beings.”14 In 1971, the FDA required IRB review if the study was to be conducted with institutionalized subjects or in an institution with an IRB; for sites with no IRB, IRB review was not required.15
E-7
FDA regulations requiring IRB review for FDA regulated products were first published on January 27, 1981 (see Exhibit 2). They closely resembled the DHEW regulations in the description of an IRB and in the review criteria used.
Exhibit 2
“Physicians who practice in their offices and who wish to conduct clinical investigations for a sponsor or as sponsor-investigators are required to comply with these regulations to obtain a research permit. The agency recognizes, however, that in some instances such physicians (and other health professionals who would otherwise qualify for a research permit) may not be affiliated with an institution or have direct access to an IRB. In those instances, FDA advises that several options are available to the physician. A sponsor-investigator who is unaffiliated with an institution with an IRB can comply with this requirement by obtaining review at an institution whose IRB conforms with the regulations or by submitting the research proposal to an IRB created under the auspices of a local or State government health agency, a community hospital, a private or public medical school, a county or State medical society, the State medical licensing board or an IRB created by the sponsor.”
46 Fed. Reg. 8962 (Jan. 27, 1981) • Preamble comment #17
Recognizing that many products were tested at sites without an IRB, FDA nevertheless required IRB review for all studies. In the preamble to the 1981 regulations, FDA recognized the gap in coverage by IRBs and suggested that local governments, medical societies, or the sponsor itself might form IRBs for these studies.
FDA accepted the Common Rule on June 18, 1991, although the agency published deviations from the Common Rule for purposes of meeting its statutory mandate to regulate health-related products.16 While the regulations presumed that clinical investigators were affiliated with medical institutions that had an IRB, the FDA recognized that there may be circumstances for which there was no IRB available and that contracting with an IRB might be possible.
C. The Changing World of Medical Research
When Dr. Stewart issued his policy statement in 1966, research was conducted typically by a single investigator working in an academic medical center on a federally funded project, with a small number of human subjects. Most exceptions to single-site studies were federally funded cancer studies carried out by groups such as the Eastern Oncology Group or the Pediatric Oncology Group, which conducted multicenter studies centered in academic centers. The world of research was poised for change. Several events transformed the face of research in the United States.
First, Medicare’s introduction of Disease Related Groups as a basis for reimbursement led to a decreasing number of hospital admissions, shortened hospital stays, and a resulting lower hospital census. This led to a corresponding increased need for delivery of ambulatory care and thus for research in that setting.
Second, federal legislation imposed the requirement that sponsors provide evidence that their pharmaceutical products were effective—evidence that would be provided primarily through human research studies.17 The number of human subjects needed to show efficacy grew quickly. Large multicenter trials became more standard. This led to discontent with the inconsistency associated with review of one protocol by many IRBs under the decentralized IRB system.
A third change was environmental. Specialty equipment and medical and scientific expertise could increasingly be found in community settings. Laboratory tests could be performed quickly and efficiently in-house. Magnetic resonance imaging and other diagnostic tests became available at for-profit diagnostic centers. Other business tools, such as courier services, fax and modem transmission, and affordable computers, allowed the placement of research studies in smaller, less costly, and more responsive community medical centers.
E-8
Fourth, academic institutions oriented to research covered by government grants were often not attuned to the needs of the pharmaceutical industry for timely and validated study data. Some institutions were more intrigued with basic research than in conducting the directed work necessary to support a drug sponsor’s protocol. Pharmaceutical sponsors, wishing to achieve speedy reviews of proposed studies and uniformity among many study sites, often perceived the academic community as impractical in producing data.
Gradually, more pharmaceutical studies were placed in secondary care hospitals and, eventually, in private medical practices. It was no longer considered mandatory, or even wise, to test a pain reliever, a metered-dose inhaler, or a vaccine in expensive large academic medical centers—especially if the eventual users would be treated in ambulatory settings.
In 1991, with most federal agencies adopting the Common Rule, the FDA adopted similar regulations under Title 21 CFR, Parts 50 and 56 to provide protections for human subjects participating in commercially funded research. Although the FDA adopted most of the Common Rule’s research review requirements, it also crafted carefully designed provisions that deviated from the Common Rule. These deviations from the Common Rule created regulations that would better fit FDA’s mission to protect the public health in reviewing and approving new pharmaceuticals, medical devices, and related technologies. The deviations accommodated diverse medical settings regulated through control of the product rather than study funding and were particularly suited to research intended to support product marketing applications.
With all of these changes, new means of addressing research review requirements were necessary. Investigators who were asked to conduct studies in community settings found little infrastructure available. Services available in an academic environment were nonexistent outside of that environment. There was no investigator training, little information on accounting or budgeting issues, little available liability insurance, and few trained coordinators. There were few resources sufficient to create or manage an internal IRB. Moreover, institutionally based IRBs generally were unwilling or unable to review clinical studies outside of their particular institution. The chronic problem of under-resourced IRBs combined with liability concerns led to courtesy reviews being offered only rarely by institutional boards to community-centered studies.
As previously stated, the FDA acknowledged this problem in 1981 when it promulgated regulations on the protection of human subjects in research. It anticipated that medical societies and medical boards would step forward to create regional boards but acknowledged that other solutions were possible.18 In reaction to the changes in the medical research environment, the community of independent IRBs was born.
IV. The Development of Independent IRBs
Between the late 1960s and today, many independent IRBs were established19 (see Exhibit 3) to meet the needs of the changing research environment. They developed in response to different environments. They served a broad range of needs, from the review of food ingredient proposals, to psychological studies, to physiological and surgical protocols. Each was unique.
| Exhibit 3. Founding Dates | |||||
| 1968 | Western IRB | 1985 | Essex IRB | 1993 | Chesapeake |
| 1981 | St. David’s | 1985 | RCRC | 1996 | Copernicus |
| 1983 | Biomed | 1986 | Ethical Review Board | 1999 | IntegReview |
| 1983 | Schulman Associates IRB | 1989 | New England | 2000 | Goodwyn.com |
| 1984 | IRC Independent Review | 1991 | Quorum | ||
| Consulting | |||||
| E-9 | |||||
Although no accurate count is currently possible due to differing definitions and the lack of any central counting method for IRBs, a list of some independent IRBs that review FDA regulated studies is included as Appendix A.
The FDA’s recognition that IRBs need not be located in an institutional setting created the first gateway for the use of independent IRBs. Thus, between 1981 and 1995 independent IRBs primarily were used to review FDA-regulated clinical studies in small clinics, community hospitals, and private practices. Today, independent IRBs are responsible for review of a wide variety of studies conducted in a wide variety of settings.
For many reasons, including the increase in regulatory requirements for premarket clinical testing and the market “exclusivity” granted to drug sponsors for such tests, the number of research studies funded both by public and private sources has increased dramatically.20 As a result, multisite studies involving thousands of human subjects have become much more common. Because independent IRBs are not limited in their review to a single site, they have proven their value in the area of multicenter or national trials.
The greatest need for independent IRB review remains outside the academic and hospital setting. However, some hospitals that conduct little research and are too small to support their own IRB engage the services of independent IRBs. Additionally, independent IRBs are now serving as IRBs for some institutions where the IRBs connected with the institutions have chosen not to review some or all the research conducted at their institution. Further, independent IRBs also provide their review services to investigators performing research not subject to federal regulation. While not federally regulated, such research may be funded or conducted by foundations or private institutions that require IRB review.
OPRR was the federal entity responsible for regulating the conduct of research funded by the DHHS and for signing assurance agreements. For a long time the OPRR did not sign any assurances for institutions that wished to contract with independent IRBs as review bodies for DHHS-funded research. However, in 1995 the OPRR began accepting SPAs for projects that involved review by “a separate institution with an IRB.”21 The OPRR’s acceptance of independent IRB reviews was based on the IRB’s commitment to stay well informed about local sites and community opinions and to comply with all applicable OPRR requirements.22 Many independent IRBs now review projects subject to SPAs, often for small companies or companies with little research experience that are seeking Small Business Innovation Research grants.
An organization of independent IRBs was formed in 1993 to provide a central discussion area concerning public policies and issues. The Consortium of Independent IRBs (CIRB) recently was incorporated as a nonprofit corporation and has its headquarters in Washington D.C. One of its first actions was the adoption of a code of ethics. For its members, the code makes clear that the major priority of the independent IRB is the protection of the research subject (see Appendix B).
V. The Mechanics of Operating an Independent IRB
Many questions have been raised about how independent IRBs work, how members are recruited, how clients are found, and how money is handled. Whatever is said must be applied to the majority, but never to all, independent IRBs. There is no single model
A. Separation of Business and Review
In any business there are departments or units to accomplish different tasks. Human resources, marketing, legal affairs, insurance, and finance and accounting are essential to a business but are not central to the product being generated, be they legal opinions, medical care, IRB reviews, computer chips, widgets, or aircraft.
Most institutions, independent or not, make an effort to shield the IRB from the business of the institution. Few academic IRB members know the amount of the grant budget requested.23 Few independent IRB members know the business relationship between the business and the client.
E-10
The majority of institutions with independent IRBs maintain a distinct separation between the operation of running the business and the administration of an independent committee capable of rendering professional decisions. Members convene and render decisions and then return to their external lives and to prepare for the next meeting. Meanwhile, administrative employees translate those decisions for applicants, prepare the IRB correspondence, write the minutes, and make sure that the files are filed and the bills are paid.
B. Recruitment of IRB Members
Many academic institutions are able to assign faculty to the IRB and to define it as a part of their duties as professional staff. They also reach out to their community in order to obtain members unaffiliated with the institution or whose interests are not in the sciences. Appointments are often made at the CEO or vice president level.
Recruitment of members for an independent IRB is usually from a broader pool. Some of the best members of independent IRBs are retired professionals who have the expertise, time, and dedication to serve. Members may be from the same town as the IRB or may live elsewhere in the country. This allows independent IRBs to choose the best-qualified members. Appointments are made according to the policies and procedures of the organization.
One hallmark of a typical independent IRB is that most members will have no other affiliation with the institution. Members are generally independent contractors.
C. Retention of IRB Members
Institutional IRBs work diligently to keep members interested, involved, and attending. Some provide parking or meals, while others provide educational opportunities. Release time is occasionally provided, usually to the chair.
Independent IRBs generally pay their members. The amount and schedule of payment to members differ with each entity. Some payment schedules are on a flat fee basis with a different amount paid for initial, continuing review, and modifications or specialty reviews, such as of adverse events or investigator’s brochures. Others pay a flat fee per meeting. Some pay members for the amount of work reviewed. Some pay all members on an equal basis, while others pay physicians more. The payment is never contingent on the decision of the member to approve or disapprove. Most members of independent IRBs find the fascinating variety of studies and the problems presented intellectually stimulating and enjoy being involved in questions that are presented in the daily news.
D. Setting and Collection of Fees
The fees collected for study review must be sufficient to cover the costs of running the business. These costs include, but are not limited to, salaries and benefits to principals and staff, fees to members, overhead (copiers, lights, janitorial, phones, computers, computer service experts), insurance (professional liability, workers compensation), marketing (trade shows, advertising), travel (lectures, site audits), education (for staff, members, and investigators), and, of course, taxes. Fees can be set to encourage submission of multisite or single-site studies. They can be flat fees (better for longer studies) or fees per action.
E. Liability Concerns
Actuaries have found it difficult to determine the potential liability faced by the company supporting an independent IRB. Initially there was no liability insurance available. Currently there are several brokers who have found companies willing to write liability insurance. Institutions with independent IRBs must also protect the members through indemnification agreements and insurance.
E-11
F. Professional Reputation24 Concerns
The reputations of IRBs are known by and shared among sponsors. Some IRBs are known to question everything or nothing, to meet frequently or rarely, to be distant and unapproachable or open and communicative. Independent IRBs stress quality and professionalism as well as timeliness and pricing in their marketing; their reputations for meeting these claims are known by and shared among sponsors.
G. Effect of Warning Letters/Closures
One protection against inadequate IRB review for all IRBs is the reality of federal oversight by the FDA and OHRP compliance programs. It has been amply demonstrated that an IRB can be closed by OPRR or by the FDA and that it can be days or months before reinstatement.
The effect of IRB closure on the supporting institution is considerable. Since the user community is relatively small, and since FDA warning letters are published on their website, adverse decisions or actions such as a warning letter about an IRB can become quickly known.
Although the institution receiving a warning letter may suffer damage, it can recoup and reenter the research world often relatively unscathed. Re-entry is more difficult when the applicant has the ability to select an IRB that has not been cited for future reviews.
H. Diversity of Services
As with academic IRBs, most independent IRBs can review studies from a variety of disciplines. In order to distinguish its IRB from other independent IRBs, most companies supporting independent IRBs offer specialty areas. One IRB offers quality assurance monitoring, one is known for education, one specializes in review of studies with vulnerable populations, and another specializes in review of medical devices.
VI. Strengths of the Independent IRB
Because the independent IRB emerged as a result of the changing research environment described earlier in this paper, its development closely matches the needs created by that change. While the benefits of independent IRBs continue to emerge with a still-changing environment, several benefits are apparent.
A. Independent IRBs Provide Review for Studies at Sites Without an Internal IRB
Small organizations (e.g., private practice corporations, small clinics, and research centers) conducting research often have several choices for IRB review: they may form an institutional IRB, use the services of a neighboring (perhaps competing) IRB, or contract for IRB services.
Forming an internal IRB in this environment is frequently inappropriate. Few members of small organizations are versed in the regulations, issues, and ethical requirements. There may be too few employees to provide appropriate IRB member diversity. There may be too little research to gain experience with IRB review. The time and cost associated with establishing an in-house IRB, if done moderately well, can be prohibitive in smaller research settings. In small organizations, there is also a substantial conflict of interest as all salaries are dependent upon approval, and frequently many employees are also equity holders. Although some institutionally based IRBs provide review for studies conducted outside their institutions, most do not.
Thus, the evolution of research with IRB review into the ambulatory setting probably could not have occurred without the emergence of independent IRBs to fill the void. To this day, the primary focus of independent IRBs remains sites without other sources for IRB review.
E-12
B. Independent IRBs Are Structured to Provide Efficient Reviews
Development of new drug and device products is costly and time consuming. Yet, patent laws restrict the period of time during which the proprietary company can prevent the entrance of generic copies of new drugs into the market. This time can be whittled away during the research and development phase. Thus, commercial study sponsors always seek means to reduce the research and development time. While these commercial sponsors expect IRBs to perform research reviews properly, they also expect that such reviews will be performed quickly and efficiently.
Independent IRBs are geared to meet these multiple needs because they have IRB members who understand the need to meet to discuss and decide on research proposals. Most independent IRBs meet weekly; some meet even more often. As a result, independent IRBs can often provide research sponsors with a decision quickly—sometimes in a matter of days. In contrast, because most academic medical center IRBs are volunteer based and meet on a less regular schedule, their review may take much longer.
C. Institutional Independence Supports Objective Reviews
IRB board members connected with the institution for which they provide review are subject to the influences associated with such connections. Specifically, they often have a collegial relationship with the investigators for whom they provide review, or they may share office space with the institutional arm that obtains grants and contracts. They may also be concerned about the financial well-being and prestige of the institution that employs them—factors that are often driven directly by research-related revenues. Further, they may develop specific viewpoints because they are limited to working within the institution. These factors could result in biases that affect an IRB member’s decision whether to approve or disapprove a study. They can also affect the vigilance with which the IRB conducts continuing review.
Because independent IRBs are not connected with the organizations for which they provide review, they can avoid such influences. The avoidance of such influences, in turn, may lead to greater objectivity in review.25
D. Independent IRBs Provide Consistency of Review in Multisite Studies
Because independent IRBs are not limited in their review to a single site, they are uniquely suited to review and oversee multicenter or national trials. A unified review eliminates the problems (e.g., conflict of modification requirements, uniformity of advertising methods, central knowledge of adverse events) associated with multiple IRB review of a single sponsor’s research plan. A further advantage of an independent IRB reviewing a multi-center or national trial is that it can develop a better understanding of the overall safety profile of the drug, device, or biologic involved, since it receives a broad spectrum of serious adverse event reports and other medical data from multiple sites. Such a diverse information base may not be available to single-site IRBs.
E. Independent IRBs Provide Review for Unregulated Research
As an indication of the acceptance of IRB review as an ethical imperative, researchers who have graduated in the last two decades and have moved into positions of responsibility assume that their research should be IRB reviewed. This is supported by peer reviewed journal requirements. Independent IRBs report an increasing number of requests for voluntary review of social and behavioral research that is not otherwise regulated by the federal government.
F. Independent IRBs Allow Institutional IRBs Breathing Room
Research review demands are increasing both within and without the hospital setting. Recently, independent IRBs have demonstrated their ability to provide support to overburdened institution-based IRBs. Independent IRBs are now assisting a number of institution-based IRBs in meeting their increasing demands by conducting
E-13
initial and continuing review of a percentage of the institution’s research plans. It is reported that in at least one instance, this was an OPRR-recommended resource.
G. Independent IRBs Provide a Bridge Between the Worlds of the IRB and Industry
Although communication and mutual recognition of basic principles of research are beneficial, there is little communication between those proposing studies and those reviewing them. It is unusual for members of either profession to communicate with the other.
The independent IRB often provides a bridge to understanding. Most IRB speakers at industry events are from independent IRBs. Invitations to IRB events made to industry are often extended by independent IRBs. Better understanding among all the parties to research can help avoid errors from miscommunication.
VII. Perceived Disadvantages
Independent IRBs are not traditional and have been criticized on several fronts. It has been suggested that independent IRBs have several disadvantages that are inherent in their structure:
While independent IRBs must be diligent in assuring that these perceived weaknesses do not become realities, they all can be addressed through proper organizational structure and/or implementation of standard operating procedures.
A. Internal Procedures Can Ensure That IRBs Identify and Consider Local Issues and Attitudes
When the IRB structure was developed, it was recognized that local IRB review was important for the proper protection of human subjects. Clearly, independent IRBs must meet their regulatory responsibilities to be sensitive to local issues and attitudes. However, in our current global village, the term “local” has evolved. It no longer means that an IRB’s physical presence in the community is necessary to meet this requirement. Independent IRBs have developed novel and effective approaches for assuring accurate and up-to-date knowledge of local issues and attitudes. Site-specific questionnaires are employed by many independent IRBs. Regular site telephone contact and written reports are also useful. The Internet and other technological advances now allow for almost instantaneous flow of information between communities. Site visits, if necessary, can be arranged. At least one independent IRB employs local consultants, while another has a contract with a professional monitoring group employing local monitors. Nonlocal IRBs realize that local issues are often, in fact, national issues. Information and issues often transcend small communities.
The FDA has recognized local review alternatives in its Non-Local IRB Review Information Sheet.26 The OPRR and the FDA have facilitated participation of an individual IRB member or consultant from the local community by sanctioning IRB meetings by teleconference or other technologies that allow real-time interaction. With OPRR’s recent issuance of a policy statement that allows IRBs to conduct meetings by phone, IRBs conducting federally funded research now have the capability of appointing an IRB member who lives in the local community.27
E-14
Although it is important to maintain a system that addresses local attitudes and concerns, when multicenter trials are involved, the local community is provided enhanced protections. With a central perspective, the central IRB has the ability to work with a number of sites involved in a particular study. Knowledge gained at one or more sites (e.g., serious adverse event reports) can be applied to all sites.
| B. |
Conflict of Interest Associated With the Fee for Service Can Be Addressed Through Organizational Structure |
Another frequently cited concern is that independent, for-profit IRBs might compromise the review process in order to advance the financial well-being of the firm. It has been alleged that such independent IRBs are paid to approve studies. On the other hand, those in the community of independent IRBs consider their reviews provided to be equally—or more—stringent that the institutional boards.28 To view this concern in the proper light, it should first be understood that all IRBs are subject to the same regulations. Thus, independent IRBs have a responsibility to ensure that each and every research plan meets the ethical, legal, federal, or state requirements for protecting human subjects.
Putting aside the independent IRB’s legal responsibilities to safeguard the rights and welfare of human subjects, the concern over profit motives is addressed through organizational structure and internal policies. First, fees are based on the review function itself and not on the review outcome. Most IRB fee schedules set fees for different aspects of the review process (initial review, continuing review, modifications). The fee is the same regardless of the review outcome.
Second, most independent IRBs are structured so that administrative and review functions remain separate. IRB members are not involved in the business end of the IRB. For example, management policies are implemented to ensure that IRB members are not privy to financial information regarding the company.
Finally, many independent IRBs ensure that the IRB membership contains very few, if any, members who are part of the IRB’s management structure or have an equity interest. Although the regulations require at least one external member, most independent IRBs have only one internal member.
Of course, conflict of interest is not unique to the independent IRB. Many institution-based IRBs are subject to similar economic pressures to approve research contracts. In recent years, many university IRBs have instituted fees for their service. Some of these fees are equivalent to the fees of independent IRBs, although their overhead costs are often much less. In addition, in an institution there are a number of interests, including departmental conflicts, the need to publish, power struggles, and the importance of very large grants, that could affect votes of individual members or the pressures placed on the IRB. Conflicts of interest come dressed in many costumes only one of which is green.
| C. |
IRB “Shopping” Can Be Addressed Through Regulations and Due Diligence to Assure That IRBs Have Knowledge of Previous IRB Reviews |
The concern regarding “IRB shopping” has also been raised as a problem associated with independent IRBs. The specific concern is that if a research plan is questioned or rejected by one IRB, the investigator may contract with another IRB without informing it about the prior board’s determination. Many independent IRBs support the implementation of an effort (ranging from concerted but voluntary IRB requirements or federal regulations) that would require study sponsors and investigators to inform IRBs about any prior review of the study plan, along with the findings, if known, of the prior review. Such a provision would largely eliminate the concerns associated with IRB shopping.
However, in the absence of such regulation many independent IRBs already have procedures or policies in place to determine if a particular research plan has been previously reviewed by another IRB. These policies may involve direct questions to the research site or, where necessary, discussions with an IRB assumed to have
E-15
primary jurisdiction over review of the particular study. For example, if an investigator is associated with a particular institution that has an in-house IRB, the independent IRB will question why the in-house IRB was not utilized. This question would be presented to both the investigator and the in-house IRB. These due diligence inquiries go a long way toward addressing the concerns related to IRB shopping.
VIII. The Future
What does the future hold for independent IRBs? The independent IRB will always have as its primary goal the protection of human subjects involved in research. The future will require independent IRBs to continually review the means of meeting that goal.
The IRB system will certainly change. Some changes are already occurring and others—many suggested by NBAC—will be the subject of future discussion. While federal action to improve IRBs and human research protection is effective in warning the research community of what is expected by the regulatory authorities, it may be that the rules and issues are changing more rapidly than they can be learned.
Methods of IRB Operations
As fast as the worlds of medicine, patient reimbursement, clinical research, and industry are changing, IRB operations will also change.
Independent IRBs will continue to fill areas of need created by new technologies, new populations, and new demands.
E-16
To handle the ever-changing research environment, independent IRBs are well suited to adjust, diversify, and meet the needs of investigators, sponsors, research institutions, and government regulators, while maintaining the protection of human subjects as their focus.
Independent IRBs will continue to respond to the legitimate needs of human subjects and the ever-changing research community. The future will find independent IRBs playing a critical role—as members of the larger community of IRBs—in the protection of human subjects of a wide range of research.
IX. A Personal Evaluation
In the request for this paper, I was asked to include a personal reflection. As a participant in this world from almost its inception, I have been associated with a large academic IRB, an independent IRB, and several community hospital IRBs. Making the adjustment from the academic IRB world to the independent IRB world necessitated learning new methods for ethically reaching the same goal of protecting human subjects. In each instance, the IRB has been a source of personal pride and of growth. As in most facets of life, there is a continuum with a normal distribution. Most studies of IRBs have demonstrated that there is an inevitable distribution of practices and quality. There are excellent IRBs and poor IRBs among all families of IRBs: academic, hospital, college, industry, government, independent, and others.
I have occasionally pondered—and there is no evidence to prove or disprove—whether the median quality independent IRB is somewhat better than the median quality institutional IRB. The program closures in academic centers in the past several years demonstrate that institutions will certainly lose money, time, contracts, and reputation. But in each case the institution has been able to eventually rebound and improve. Most of us involved with independent IRBs innately understand that if the IRB were found to be equally deficient, liability insurance premiums would soar, prestige would plummet, the client base would disappear, and the business would be dead. This is a very large incentive to maintain quality.
Change has been a hallmark of the protection of human subjects. Every few decades the thinking about ethics has evolved: Nuremberg, Helsinki, Belmont, ICH, and Helsinki again. Every decade or so the regulations have changed. There have always been new issues: extending of clinical rules to broader social sciences, using computers to enhance IRB operations, working with regulations from different organizations, debating waiver of IRB or consent or privacy needs and genetics issues, and fighting inappropriate changes to the IRB job and bureaucracy. Personally, about the time that boredom hit or I became fed up with the fighting the same fights, new issues have emerged to engage me or to allow me to develop new skills.
E-17
Although the world of research continues to present new challenges, the pace of change seems to have quickened. Communication now spreads ideas, news, dangers, and gossip more quickly than our capacity to verify the information, challenge it, or change ideas or procedures to meet it.
There are currently new issues, new risks, new sites, and new organizations to direct us as well as new requirements and new kinds of research. Each of these presents challenges to those of us whose focus is the human subject. Offering innovative channels for building protection of human subjects must happen if the needs of this century are to be met.
X. Acknowledgments
This report reflects the experiences of one person. I became involved in IRBs in late 1970 at the University of California, San Francisco (UCSF), assisted in the writing of UCSF’s second MPA in 1974, and co-wrote the first faculty guidance document. In 1984, I resigned from UCSF, and, as no regional medical societies had answered the call for regional IRBs, I founded IRC Independent Review Consulting.
IRC, incorporated in 1994, currently specializes in review of medical device studies, use of biological specimens, biotechnology, and social and behavioral studies. IRC has worked with several institutions as the IRB of record for their SPA.
The thinking and opinions in this paper are mine and do not necessarily reflect those of other independent IRBs. I would like to offer appreciation for the many people who provided their editorial assistance, their time, and their kindness.
Notes
1 OPRR was recently relocated and renamed the Office for Human Research Protections (OHRP).
2 See 21 CFR Part 56.107 or 45 CFR 46.107.
3 Title 45 CFR 46.102( c) and 21 CFR 56.102(f). As used in this Part…institution means any public or private entity or agency (including federal, state, or other agencies).
4 Heath, Erica, “The Noninstitutional Review Board: What Distinguishes Us from Them,” IRB: A Review of Human Subjects Research 20(5):8–11 (1998).
5 Katz, Jay, “Experimentation With Human Beings; The Authority of the Investigator, Subject, Professions, and State in the Human Experimentation Process.” New York: Russell Sage Press, pp. 9–65 (1972).
6 Ibid. pp. 1007–1010
7 Beecher, Henry, “Ethics and Clinical Research,” New England Journal of Medicine 274:1354 (1966).
8 Katz, p. 855.
9 DHEW, Institutional Guide to DHEW Policy on Protection of Human Subjects, (NH 72-102) Dec 1, 1971.
10 Although many new boards took the name IRB, many other boards became known by other names such as the Committee on Human Research (CHR), The Human Experimentation Committee (HEX), the Committee on Protection of Human Subjects (CPHS), etc. This has led to some confusion when investigators are asked if there is an IRB at their institution.
11 43 Federal Register 56174.
12 Although 45 CFR 46 is referred to as the Common Rule, each signatory agency that adopted the same rule uses the Common Rule with its published distinctions. Thus, FDA’s regulatory variant without reference to an assurance and split into two sections as 21 CFR 50 and 21 CFR 56 remains one version of the Common Rule.
13 43 Federal Register 56177.
14 FD&C Section 505(I)(4) 1962 PL 87-781, 10/10/62.
E-18
15 21 CFR 312(a)(2) 10C; FDA Compliance Program Guidance Manual March 15, 1977.
16 56 Federal Register 28025.
17 FDCA Efficacy 505(b)(1)(A).
18 46 Federal Register 8962. Preamble comment #17.
19 www.himanet.com, businesses supporting research: independent IRBs.
20 OIG, DHHS, “Recruiting Human Subjects: Pressures in Industry-Sponsored Clinical Research,” OEI-01-97-00185, June 2000.
21 Report of the OIG, DHHS, “Institutional Review Boards: The Emergence of Independent IRBs,” June 1998.
22 Ibid.
23 Whether this separation of money and review will continue if IRB members are asked to evaluate the effect of budgets on recruitment is an interesting question.
24 During the writing of this sentence, my teen walked in with a food treat saying, “You want a bite of this? It’s from (brand name) so you know it’s healthy and good.” What more can be said for the value of one’s reputation?
25 Ibid.
26 FDA, Non-Local IRB Review, Information Sheets: Guidance for Institutional Review Boards and Clinical Investigators (1998 update).
27 See Memorandum from J. Thomas Puglisi, Director of the Division of Human Subject Protections, OPRR (March 28, 2000).
28 A complaint frequently heard is that “77 other IRBs have approved this, why can’t you?” In most cases, the 77 other IRBs are academic or hospital based.
29 An informal survey of independent IRBs showed that most had experienced at least one external audit from a nongovernmental compliance auditor.
30 Kowalczuk, Liz, “Medical Schools Join Forces: Harvard, Others Aim to Give Drug Firms Faster OK’s on Clinical Trials,”
Boston Globe, July 2, 2000.
E-19
| APPENDIX A | ||||
| List of Independent IRBs Gathered from Various Sources | ||||
| ARENA | HIMANET | CIRB | ||
| member* | member | |||
| Allendale Investigational Review Board | NJ | X | ||
| Argus IRB | AZ | X | ||
| Biomed IRB | CA | X | X | X |
| Chesapeake Research Review, Inc. | MD | X | X | X |
| CHSD | CA | X | X | |
| Clinical R&D Services IRB | NJ | X | ||
| Copernicus Group IRB | NC | X | X | X |
| Essex Institutional Review Board, Inc | NJ | X | X | X |
| Ethical Review Committee | MO | X | X | X |
| Goodwyn IRB | OH | X | X | X |
| Independent Investigational Review Board | FL | X | ||
| IntegReview | TX | X | X | X |
| Internet IRB | FL | X | ||
| IRB Services (Canada) | ONT | X | ||
| IRC Independent Review Consulting, Inc. | CA | X | X | X |
| New England IRB | MA | X | X | X |
| Quorum IRB | WA | X | X | X |
| Reliable IRB | CA | X | X | |
| Research Consultants Review Committee | TX | X | X | X |
| Schulman Associates IRB, Inc | OH | X | X | X |
| Southwest Independent IRB | TX | X | ||
| St. David’s Human Research Review Board | PA | X | X | X |
| Sterling IRB | X | |||
| Triad IRB | IL | X | ||
| Western Institutional Review Board | WA | X | X | X |
| Wyle Laboratories IRB | TX | X | ||
*ARENA: Applied Research Ethics National Association is a membership organization. This column is marked if the CIRB representative or a known principle of the company is an ARENA member.
E-21
APPENDIX B
Code of Ethics
The Consortium of Independent Review Boards
Each member IRB of the Consortium of Independent Review Boards (CIRB) pledges to follow the articles of the CIRB Code of Ethics, as contained in this document.
| 1. |
The primary mission of CIRB members is to protect the interests, rights and welfare of human subjects in IRB reviewed studies. |
| 2. |
CIRB members will be guided by the fundamental principles of research ethics put forth in the Belmont Report (The National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, 1979)—Respect for Persons, Justice and Beneficence. |
| 3. |
CIRB members will adhere to the letter and the spirit of laws and regulations requiring the protection of human subjects. |
| 4. |
CIRB members will protect against conflicts of interest. |
| 5. |
CIRB members will develop and follow a plan for its training IRB members. |
| 6. |
CIRB members will protect the confidentiality of subject information and sponsor proprietary information to the extent allowed by law. |
| 7. |
CIRB members will promote ethical recruitment practices for clinical research subjects. |
Concerns regarding ethics violations shall be communicated in writing to CIRB’s Executive Committee, and if appropriate, to CIRB’s legal counsel for review and recommendations.
E-23

THE DANISH RESEARCH ETHICS COMMITTEE SYSTEM1—OVERVIEW AND CRITICAL
ASSESSMENT
Commissioned Paper Søren Holm University of Manchester
F-1
Summary2
The Danish Research Ethics Committee (REC) system was legally established in 1992, but its history goes back to the late 1970s. In 1977 an agreement was made between the Danish Medical Association, the Association of Danish Counties (representing the hospital owners), the Danish Medical Research Council, and others to establish a REC system in accordance with the Helsinki II declaration of the World Medical Association (1975). The system began working in 1980 and was fully established in 1982. The overwhelming majority of Danish hospitals are publicly owned, and most doctors are salaried employees of the public health service. From its inception the system therefore included the major players on the Danish health care scene. The legislation passed in 1992 merely formalised the features of the earlier system with a few changes, and made it absolutely clear that what is important in deciding whether a research project should be assessed is not the profession of the researcher, but the nature of the project. According to the law, all biomedical research projects must be submitted for approval, and the official commentary on the law makes it clear that “biomedical” should be read very broadly to include all research projects involving present and former patients. The law was revised in 1996, and some uncomplicated questionnaire and register based studies are now exempt from the requirement of REC approval.3 Already from the beginning the Danish REC system was characterised by four main features which set it apart from most systems elsewhere:
| 1. |
RECs are regional and not institutional. |
| 2. |
RECs are relatively small, and there is a large proportion of lay members. |
| 3. |
There is a relatively easy mechanism for the approval of multi-centre trials. |
| 4. |
There is a central national REC above the regional RECs. Denmark has eight regional RECs each covering one or more of the Danish counties. All biomedical |
research protocols within this geographic area must be submitted to the REC for approval. It is illegal and punishable by up to four months in prison to begin a biomedical research project without REC approval. A REC can make four different decisions about a project: a) approval, b) approval on the condition that certain changes are made (resubmission not necessary), c) approval denied, but changes proposed for a possible resubmission, and d) approval denied. Total denial of approval is rare, but it is very common that researchers are asked to change parts of their projects, most often the patient information sheet. All decisions about approval of a research project must be unanimous in the regional committees, otherwise the project is referred to the Central Research Ethics Committee (CREC). A researcher who is not satisfied with the decision of the regional REC can appeal to the CREC. Decisions made by the CREC are final and cannot be appealed to any other administrative body.
A committee has between 7 and 15 members, with a majority of lay members of one (i.e., if there are 7 members, 4 are lay and 3 are professional). The professional members are appointed by the Danish Health Sciences Research Council, and the lay members are appointed by the County Councils. Although the lay members are politically appointed, they do not represent their respective political parties in the REC. The lay members are not usually lawyers, clergy or philosophers, but “true” lay people. The members are appointed for four year periods, and can be reappointed once. Each REC has a secretariat, usually staffed by a lawyer, but there are no bioethics advisors attached to the RECs. One problem in the system is that few RECs have access to expertise within research methodology, and therefore they may have problems in assessing certain kinds of projects (e.g., projects involving very advanced statistical methods or qualitative research methods).
F-3
The number of research projects assessed by each REC varies between 120 and 500 per year, with the RECs covering university hospitals having the largest work load. Some RECs debate all submitted projects in a meeting of the full REC, whereas others only debate those projects that at least one member finds problematic.
The reason for having regionally and not institutionally based RECs is that this removes some of the pressures that an institutionally based REC may encounter. In an institution there may be pressure applied on the REC to approve or disapprove certain kinds of research, disregarding the ethical status of the research. A regional REC is far less likely to succumb to such pressures because the members are not all associated with one single institution.
The RECs have a legal right to control whether research projects are conducted in accordance with the permission given, and to have access to the final results of a trial. This right is very seldom used at present, because the RECs lack the manpower to perform active controls. Prior to the last revision of the legislation in 1996 the RECs and a number of commentators in the public debate advocated more funding for the RECs in order to enable them to take on an active controlling role, but this proposal did not find favour with the politicians.
Multi-Centre Trials and the CREC
A major problem in the function of RECs in many countries is the approval of multi-centre trials, i.e., trials taking place in many centres and therefore under the jurisdiction of many different RECs. The Danish REC systems have developed a simple mechanism for handling the assessment and approval process of such trials. According to the Danish regulations the protocol for a multi-centre trial should be submitted to the committee in whose area the leading investigator works, along with information about which other hospitals/clinics are involved in Denmark. This REC will then distribute the protocol to the other RECs in whose area there are hospitals/clinics involved in the trial, and ask for their comments on the project within a timeframe of 30 days. The REC to which the multi-centre project is submitted is responsible for final approval of the project, on behalf of all the RECs involved, and will take care of coordinating the various comments that are submitted from the other RECs. In this way a multi-centre project can be approved within 60 days, almost as fast as a single-centre project. If one of the RECs involved does not think that the project should be approved, but the others think it is acceptable, the coordinating REC will try to negotiate a compromise, but if no compromise can be found the project will be referred to the CREC for final decision (this happens less than ten times per year).
The CREC consists of two members appointed by each REC (one professional and one lay member), two members appointed by the Minister of Health, and two members appointed by the Minister of Research. The CREC appoints its own chairman among the members. The CREC has five functions: 1) it acts as an appeal body for researchers who are dissatisfied with the decision of their local REC, 2) it makes decisions about multi-centre trials in cases where there are irresolvable disagreements among RECs, 3) it develops guidelines for specific areas of research ethics assessment (e.g., use of radioactive material, remuneration of research subject, trials performed by Danish researchers in third world countries), 4) it ensures uniformity of decisions in the local RECs, and 5) it publishes a yearly report highlighting some of the current problem areas in research ethics.
Background Information About the Danish Health Care System
Danish RECs are embedded in Danish society in general and in the Danish health care system in particular. This influences their structure and mode of operation, and some knowledge of this context is therefore necessary for a full understanding of the RECs.
F-4
Denmark is a small country in the northwest of Europe. It has a population of 5.2 million inhabitants, of whom approximately 300,000 are first and second generation non-Scandinavian immigrants (mainly from Turkey, Pakistan, the former Yugoslavia, Vietnam, Sri Lanka, and Somalia). The state religion is Lutheran Christianity, but Denmark is one of the most secularised countries in Europe. There has traditionally been a very strong labour movement and a strong social democratic political party, and concepts like “equality” and “solidarity” still play a large role in Danish political debate.4 Since the 1930s an extensive welfare state has developed with the provision of social security, unemployment benefits, pensions, education and health care being seen as the responsibility of the state. The development of the welfare state has been supported by almost all political parties after the Second World War.
A Short History of the Danish Health Care System
The health care system is a major component of the Danish welfare state. The first mention of health insurance can be found in 1403 when the guild of bakers in Copenhagen decided to pay illness benefits to its members. This was followed by similar systems in other guilds and most guilds had some form of health insurance at the end of the 18th century.5 When the guilds were abolished during the end of the 19th century many small cooperative health insurance funds emerged either as continuation of the funds set up by various guilds or as one branch of a cooperative movement which also included cooperative dairies, banks, shops, breweries, etc. These funds reimbursed general practitioners and practising specialists, although there was often a small co-payment on the part of the patient. These small local funds soon formed local negotiation consortiums negotiating fixed prices with the local physicians and excluding physicians not willing to restrict themselves to the negotiated fees. By the beginning of the 20th century each region in the country had what was in effect a one-payer systems for a large part of the population, with only a small upper-class still paying directly out of its own pocket. Later in the century after the Second World War many of the smaller funds merged and only a few large funds continued to operate. Payment to these funds was graded according to broad income bands, but there were also substantial state subsidies, and the state paid for those who could not afford membership themselves.
In the hospital sector the system of payment was different, because almost all hospitals in Denmark were established by municipal authorities or the state, the exception being a few hospitals established by religious orders. In this sector payment has therefore always been predominantly tax-based although extras like single rooms were traditionally available for those who could pay.
In the 1960s it was decided politically to move to a purely tax-based system with the counties (administrative units with 50,000 to 600,000 inhabitants) as the administrative units responsible for planning, running, and funding both primary and hospital care, and this decision was finally implemented in 1973. This decision created an integration of many previously separate parts of the health care system, but at the same time it upheld a division in 14 small geographical units.
The Present Structure of the Health Care System
The 1973 structure continues to operate although some changes have been made in recent years. In this structure the counties are responsible for planning, managing and running hospitals, general practice, practising specialists, community physiotherapy, and responsible for paying for the subsidies on subscription medicine.
The state has no direct responsibility for health care, but the Ministry of Health establishes general guidelines for the quality of the services to be offered. The Ministry of Health was established in 1987. Previously the health area had been managed by a department in the Ministry of the Interior, and the late establishment of a separate Ministry of Health can be seen as a reflection of the limited formal influence of the state in this area. All employees in hospitals are salaried. All groups, including junior doctors, work 37 hours a week.
F-5
Until 1989 Denmark did not have any real private hospitals,6 and the range of fee-for-service care available was therefore confined to the procedures which could be carried out in the surgery of individual doctors. In 1989 the first private hospital was established under the guise of catering primarily for foreigners or Danish nationals living abroad. This publicly announced target-group turned out to be small, and the hospital soon diverted its efforts towards elective orthopaedic surgery for Danes who wanted to jump the waiting lists. A number of similar small clinics and three larger hospitals have since been established, but the total number of available beds in the private sector is still below 150 compared to approximately 22,500 somatic beds in the public sector. Along the way all three of the large private hospitals have gone bankrupt (one of them with accumulated losses of 182 million Danish kroner). One of these hospitals was economically reconstructed, but this one remaining large private hospital has not returned any profit in six years of operation.7 Health insurance covering treatment in private hospitals is now available, but payments are not tax-deductible, and it is mainly bought by companies for their top executives. About 23,000 Danes have comprehensive hospital insurance.7 The number of insured is predicted to rise, especially in the form of so-called “catastrophic illness insurance” which pays out a lump sum if the insured person gets a serious illness. This sum can be used for treatment at a private hospital, but can also be used for other purposes. A number of private firms have included this type of insurance in their benefit package.
Community Services/Primary Care
General practitioners, practising specialists, dentists, and community physiotherapists are all (at least in theory) private businesspeople. Reality is however somewhat different. Reimbursement to general practitioners and practising specialists is dependent on the practice being authorized by the county. Each county can unilaterally decide on the number of practising specialists it wants to authorize, whereas the number of general practitioners is regulated by a formula relating the size of the population to a minimum number of general practitioners.
Each patient has to register with a specific general practitioner. The doctor then receives a small yearly fee for each patient, but the main part of the income of general practitioners is based on fees received for specific services. There is no patient co-payment, and it is illegal to charge the patients extra fees. A general practitioner can only have a certain number of registered patients.
Patients can only see a practising specialist or a community physiotherapist within the public system if they are referred by their general practitioner. In that case treatment is free, but if the patients themselves seek a practising specialist directly they have to pay the full fee out-of-pocket.
Danish Medical Research Before the Helsinki II Declaration
From the beginning of modern medicine in the 1800s Danish medicine developed a strong research culture. Like in most other countries research came to be seen as a natural extension of the physician’s obligation to treat and cure patients, and no sharp distinction was made between therapeutic interventions and research interventions. The mechanism for the control of research was the conscientious doctor’s careful consideration of the best interest of his or her patient, and this was supposed to be covered by the provision in the legislation on licensing of medical practitioners which contained an explicit duty to show care and conscientiousness in the performance of one’s medical practice (Law no. 72, 1934). This view was prevalent until well into the 1960s. Immediately after the Second World War there was some discussion in Ugeskrift for Læger (the Danish Medical Journal) about the Nazi experiments, but on a general level this discussion and interest soon petered out. The only interest which remained was in the fate of the Danish doctor Carl Værnet who had performed experiments on homosexual men in the Buchenwald concentration camp. He was arrested in Denmark immediately after the war, but was then allowed to go to Sweden for specialist treatment of a heart condition. He
F-6
escaped from Sweden and eventually made his way to Argentina where he died in the late 1950s. The Nuremberg Code never had any major impact on Danish research practice or the legal regulation of research in Denmark.8 During the 1950s the randomised, controlled trial (RCT) began to be used by Danish medical researchers, and in the Danish Medical Journal from the mid and late fifties there are many reports of RCTs performed without the consent and knowledge of the patients in the trials. Some of these studies involved the new neu-roleptic drugs that were being developed at that time, and in some cases the researchers felt justified in not informing the nursing staff at the relevant units that some patients were receiving a new drug, whereas others were receiving placebo. It was feared that the results would be biased if the nurses had this knowledge.
The first Helsinki Declaration of the World Medical Association (WMA) in 1964 had no impact in Denmark,9 and the unveiling of research scandals in other countries was scarcely mentioned in the Danish medical press.
The Helsinki II Declaration and the Establishment of Research Ethics Committees
The draft for the Helsinki II Declaration of the WMA was written by three Scandinavians, professor Erik Enger from Norway, docent Clarence Blomquist from Sweden, and professor Povl Riis from Denmark.10 It was passed by the WMA general assembly in October 1975 and adopted by the board of the Danish Medical Association (DMA) in December 1975. It thereby became binding on all members of the DMA (about 98 percent of all active Danish doctors).9 Article I.2 of the original Helsinki II declaration specifies that the research plan for a medical trial should be put to an independent committee for review and guidance. Immediately after the adoption of the declaration the DMA therefore initiated efforts towards establishing such independent committees. These efforts were primarily led by professor Povl Riis, who at that time was also editor of the Danish Medical Journal. A series of meetings were held between representatives of the DMA and the other main interested parties, the Danish counties as the hospital owners and employers of most doctors, the Danish universities, the Danish Medical Research Council, and a number of others. This led to a proposal in 1977 recommending the provisional establishment of a system of RECs for a trial period of unspecified duration.11 This proposal was adopted by the DMA, the Danish counties, the Danish universities, the medical scientific associations, the pharmaceutical industry, the Danish medical and health care journals, the Danish Dental Association, the Danish Association of Pharmacists, and the Ministry of the Interior.12 Although the proposal thus had the backing of the counties and the Ministry the system of RECs established was still of an extra-legal nature and researchers had no legally binding obligation to submit research protocols to the RECs.
The first RECs were established in 1980, and the whole country was covered by 1982.13 The system was based on regional RECs, each covering one or more counties and being responsible for all biomedical research in that area. The committees had between six and ten members. Half of these were professional members appointed by the Danish Medical Research Council after consultation with local representatives of the medical and other health care professions, and the other half were lay members appointed by the County Council(s). All members were appointed for four year terms, with the possibility of reappointment without limits the number of terms. The terms followed the election term of the County Councils. Although the lay members were politically appointed they were appointed in a personal capacity and not as representatives of their party. Each REC elected a chairman and a vice-chairman (one professional and one lay). All decisions to approve a research project in a REC had to be unanimous.
The system further contained a Central Research Ethics Committee covering the whole of Denmark. The CREC was constituted of the chairmen and vice-chairmen of the regional RECs14 and a number of members appointed by the Ministry. The CREC was first formed in 1981, and its first chairman was professor Povl Riis, who continued in this post until 1998.
F-7
The CREC had two functions. The first of these was to act as an appeal body in cases of disagreement in a REC or between RECs, or in cases where a researcher appealed a decision made by a REC. The second was to issue general recommendations to RECs concerning the evaluation of research projects. These recommendations indirectly became normative for the conduct of researchers and the planning of research projects, since it became clear that projects not respecting the recommendations would not be approved.
Given the extra-legal status of the committees no formal sanctions existed that could be applied to researchers who either did not submit their research projects or did not conduct their research projects in the form in which they had been approved. There were, however, a range of informal sanctions which seems to have been sufficient to ensure compliance. These included the threat that nonapproved research would not be published, and the belief that employers would view a breach of research ethics rules as a serious breach of the employment contract.
In the early period of the function of this system researchers were asked to submit a research protocol and a self-declaration concerning compliance with the Helsinki II Declaration. If researchers declared that the project was in full compliance with the declaration they could initiate the project before the REC had evaluated it (projects involving children and other incompetent patients could not be initiated without explicit approval). In these cases the REC did not often perform a substantial evaluation of the projects, but only of the patient information material. This gradually changed, and in the late 1980s all project were actively evaluated before approval.
Putting Research Ethics Committees on a Legal Footing
During the 1980s there were a small number of public “research scandals” in Denmark, and there was an at times heated public discussion about the effectiveness of the RECs which were claimed to be too medically dominated.15 This lead to a political debate about the status of the REC system, and to the appointment of a commission to consider a legal establishment of RECs and a revision of the system. The commission published a report in 1989 recommending that a law should be passed establishing a national REC system.16 After some further political discussion a law was passed in 1992 (Law no. 503, 1992, On a scientific ethical committee system and the consideration of biomedical research projects). Minor changes were made to this law in 1996 (Law no. 499, 1996). The REC system established by this law is to a very large extent identical to the previously existing extra-legal system. The regional RECs and the CREC, and the division of labour between the RECs and the CREC, are retained.
The composition of RECs is slightly changed to give more lay representation. According to the law a REC has between 7 and 15 members, with a majority of lay members of one (i.e., if there are 7 members, 4 are lay and 3 are professional). The professional members are appointed by the Danish Health Sciences Research Council, and the lay members are appointed by the County Councils. A practice has developed so that at least one of the professional members is a general practitioner. Although the lay members are still politically appointed, they do not represent their respective political parties in the REC. There are no specific requirements as to who the lay members should be. The lay members are not usually lawyers, clergy or philosophers, but “true” lay people, although teachers and clergy are probably over-represented among the lay members.17 The members are appointed for four-year periods, and can be reappointed once. Each REC has a secretariat, usually staffed by a lawyer, but there are no bioethics or research methodology advisors attached to the RECs. All decisions about approval of projects have to be unanimous, otherwise the project must be referred to the CREC for decision.
The law opens a possibility to establish more than one REC in a given region if the number of research projects submitted becomes too great for one committee. This situation has arisen for the committee covering
F-8
the Copenhagen and Frederiksberg municipalities, and thereby the University of Copenhagen and the National Hospital. In this region two RECs have been formed, and research projects are distributed between them on a consecutive basis, REC-1 getting the unevenly numbered and REC-2 the evenly numbered projects.
Members of the RECs are not paid for their work, except the chairman and vice-chairman (25,000 and 20,000 Danish kroner per year, respectively), but can get reimbursement for lost earnings while attending meetings (330 Danish kroner per meeting). RECs are directly funded by the counties, and research projects based in county institutions are handled free of charge. There is a charge of Danish kroner 2,500 per project for projects based in noncounty institutions (including projects initiated by the pharmaceutical industry). The RECs have tried to have the charge abolished because it creates a large administrative burden for very little financial gain, but have as yet not been successful.
The Constitution and Function of the Central Research Ethics Committee
According to the legislation the CREC consists of two members appointed by each REC (one professional and one lay member), two members appointed by the Minister of Health, and two members appointed by the Minister of Research. Three of the members appointed by the government should represent handicap groups or social interest groups, the remaining government appointee represents the research interests of the state. The CREC appoints its own chairman among the members. Members of the CREC are appointed for four-year terms and can be reappointed once. The CREC has five main functions: 1) it acts as an appeal body for researchers who are dissatisfied with the decision of their local REC,18 2) it makes decisions about multi-centre trials in cases where there is irresolvable disagreements among RECs, 3) it develops recommendations for specific areas of research ethics assessment (e.g., use of radioactive material, remuneration of research subject, trials performed by Danish researchers in third world countries), 4) it ensures uniformity of decisions in the local RECs, and 5) it publishes a yearly report highlighting some of the current problem areas in research ethics.
The CREC further has an obligation to cooperate with the Danish Council of Ethics through common meetings and the publication of common reports.19 This cooperation has for some years been characterised by a state of “armed neutrality,” with the CREC feeling that the Council of Ethics wanted to interfere with the CREC’s handling of specific research projects, and the Council of Ethics feeling that the CREC was very reluctant to discuss major general problems in research ethics. Part of the problem seems to be that the two bodies have not fully understood the roles they each have. Despite this turf war one major report on health science information banks was published in 1996.20 In the CREC decisions can be made about a project by majority vote, but there has to be a majority of both the professional and the lay members. Decisions made by the CREC cannot be referred to any higher administrative authority, so the only recourse for a researcher who is dissatisfied with a CREC decision is to go to court claiming that the decision made was illegal and should therefore be made void. No such case has been before the Danish courts yet.
Besides the CREC an informal coordination mechanism between RECs has also emerged in form of the Secretariat Council (“Sekretariatsrådet”) where the administrators from the REC secretariats meet regularly to discuss common problems. It is also mainly through this informal body that information is exchanged with the RECs in the other Nordic Countries.21 The recommendations issued by the CREC are available in English translation, but this publication is unfortunately now very out-of-date.22 Work is under way to consolidate most of the recommendations into two guidance documents, one directed at researchers and one directed at RECs, but this work has not been finished at the present time, but should finish sometime in the summer or autumn of 2000.23 These guidance documents will be issued as common guidance from the CREC and the Minister for Research.
F-9
No Danish RECs Outside of Biomedicine
The Danish REC system only covers biomedical research (although the definition of biomedical is rather wide, see the section “What projects should be submitted”). There is no REC system for research outside the health sector. In the mid-1990s it was discussed whether RECs were needed for the social sciences, the humanities, or other areas of research. A working group was established by the Danish Social Sciences and the Danish Humanities Research Council to consider this question, and it reached the conclusion that RECs were not necessary in these areas.24 Seen from a research ethics perspective the arguments presented seem more than a little dubious. In the working group there was clearly a worry that RECs would mean a requirement for informed consent, which would be difficult to obtain in some social science and psychology projects. It was further argued by some members of the group that (some kinds of) social science are very important for policy formation and administration in a modern society, and that they should therefore not be too constrained by ethical demands. Some also claimed that the potential for harming research participants in the social sciences is much lower than in the health sciences.
After the report from the working group the discussion about RECs outside the health area has died down in Denmark.
Punishment for Breaking the Law
A researcher who is in breach of the law, for instance by not submitting a project or by carrying out a project in an illegal manner, can be reported to the police, charged and if found guilty by the courts receive a punishment consisting of a fine or imprisonment up to 120 days. A number of researchers have recently been reported to the police, but none of the cases have yet been before the courts.
For a registered health professional (a physician, nurse, physiotherapist, etc.) the Danish Board of Health (“Sundhedsstyrelsen”) can also initiate professional disciplinary procedures, since breach of the REC legislation is viewed as a breach of professional duty.
The RECs do not have any formal relationship with the institutions from which the researchers come, or with the public funding bodies for research. They cannot ask institutions to initiate disciplinary procedures, or funding bodies to withdraw funding, if they find evidence of breach of the law.
This does, however, not mean that institutions or funding bodies are uninterested in the decisions of RECs. The Danish Health Sciences Research Council requires REC approval as one of the necessary conditions for funding, and the same policy is followed by the major private funders in the health research area.
There are also a number of cases where researchers have resigned after REC critique of their research. In some of these cases it has been evident that the institution has leaned on the researcher.
What projects should be submitted?
Paragraph 6 in the law makes it clear that all biomedical research projects should be submitted for approval to the regional REC and that a project can only be initiated once approval has been obtained. This includes pilot-experiments. According to the official comments on the legislation “biomedical” should be interpreted very broadly to include not only interventional or diagnostic studies involving some kind of bodily intrusion or the use of drugs, but also basic biological research on humans, health related epidemiology, health related sociology, and all projects were people are studied because they are patients or have been patients.
No distinctions are made with regard to the legal status of the institution where the research is taking place (i.e., public/private), with regard to the funding source (i.e., public/private), or with regard to the profession of the researcher. All projects must be submitted. What decides whether a project should be submitted is solely its content.25 A project carried out in a private institution, without any kind of public funding, and by a researcher who is not a health professional will thus require REC approval, if it falls within the legal definition of “biomedical.” On the formal/legal level the REC system functions in exactly the same way as the system for
F-10
issuing building permits, which also covers all kinds of buildings, no matter how they are funded or by whom they are built.
There are, however, still a number of grey areas with regard to the demarcation between research and quality assurance/development activities, between research and educational projects performed as part of the studies of medical and other students, and most significantly between research and “experimental” use of new treatments. The last grey area concerns the established right of medical practitioners to try new and untested treatments in patients for whom this seems to be the best (or in some cases the last or only) option. There have been a number of cases where doctors have used this therapeutic privilege on substantial series of patients, and where the situation has looked more like research than last resort therapy when viewed from the outside. The Danish Board of Health has tried to clarify the situation by issuing official guidance to Danish doctors, but this has not totally resolved the unclarities.26 A REC can make four different decisions about a project: a) approval, b) approval on the condition that certain changes are made (resubmission not necessary), c) approval denied, but changes proposed for a possible resubmission, and d) approval denied. Total denial of approval is rare, but it is very common that researchers are asked to change parts of their projects, most often the patient information sheets. Amendments to approved projects also have to be submitted for approval and cannot take effect before they have been approved.
A REC can also decide that a project falls outside the legal definition of a biomedical research project and that it therefore does not need REC approval. When such a decision is made researchers sometimes ask whether they cannot get approval anyway, because they feel that they need it, either for future publication of results, or in order to get access to patients (this last factor is most often important for nonmedical researchers), or in order to document approval to non-Danish research sponsors. RECs are, however, not able to approve projects outside their remit.
Certain kinds of projects need REC approval, but also need approval from other bodies before they can be initiated. Projects involving the use of pharmaceuticals for nonregistered indications need approval from the Danish Pharmaceuticals Authority (“Lægemiddelstyrelsen”) which is also responsible for official inspection of GCP-compliance in Denmark. Projects involving the establishment of a register or a bio-bank containing person identifiable information needs approval of the register and its data-protection provisions from the Registry Authority (“Registertilsynet”).
Assessment of Multi-Centre Projects
A major problem in the function of RECs in many countries is the approval of multi-centre trials, i.e., trials taking place in many centres and therefore under the jurisdiction of many different RECs.27 Often researchers have to communicate with a large number of RECs, and they may be met with mutually contradictory demands for changes in the project design or patient information by different RECs. The researcher may in the end have to activate different versions of the protocol in different centres, in order to meet the irreconcilable demands of the different RECs.
The Danish REC system has developed a simple mechanism for handling the assessment and approval process of such trials. According to the Danish regulations the protocol for a multi-centre trial should be submitted to the committee in whose area the leading investigator works, along with information about which other hospitals/clinics are involved in Denmark. This REC will then distribute the protocol to the other RECs in whose area there are hospitals/clinics involved in the trial, and ask for their comments on the project within a timeframe of 30 days. The REC to which the multi-centre project is submitted is responsible for final approval of the project, on behalf of all the RECs involved, and will take care of coordinating the various comments that are submitted from the other RECs. In this way a multi-centre project can be approved within 60 days and almost as fast as a single-centre project. If one of the RECs involved does not think that the project should be
F-11
approved, but the others think it is acceptable, the coordinating REC will try to negotiate a compromise, but if no compromise can be found the project will be referred to the CREC for final decision (this happens less than ten times a year).
The results of the Danish system for multi-centre trials are 1) that the researcher is only in correspondence with one REC, 2) that demands for changes in design or patient information will be co-ordinated so that the researcher is never required to reconcile contradictory demands him- or herself, and 3) that all Danish centres in a multi-centre project conduct the trial according to one approved protocol and not according to a number of regionally specific versions.
Consequences of Legalisation
The legalisation of the REC system has had a number of consequences. It has meant that biomedical research in Denmark is no longer controlled by the Helsinki II Declaration, or any other subsequent declarations that the WMA may pass. A REC can take the Helsinki II Declaration into account in areas where present Danish legislation is unclear or gives no specific guidance, but the declaration can never supersede Danish legislation. This has created problems for a number of researchers holding foreign grants (including NIH grants) since the Danish RECs are no longer able (or willing for that matter) to certify that the project is performed in accordance with the Helsinki II Declaration (see the standard letter from one of the RECs in Appendix A28).
Another consequence has been that Danish RECs are now subject to the same rules about public access to the administrative process as other parts of the public administration. This does in principle give public access to all research protocols, except those that contain commercially sensitive information (and even in such cases the public should have access to those sections of the protocol that are not commercially sensitive). The CREC has actively tried to obstruct public access, and has therefore been in protracted conflict with the parliamentary Ombudsman. The CREC has argued that it is necessary to protect the researchers intellectual property rights in new scientific ideas or methods and that public access must therefore be restricted (as the only way to restrict access by competitors), but this argument has been firmly rejected by the Ombudsman.29 The present situation is unclear. Technically the CREC and RECs have accepted the Ombudsman’s ruling, but in practice some requests for access are apparently still being obstructed. Some RECs now ask researchers to specify in advance what parts of their protocols they consider commercially sensitive, and what parts can be open to immediate public access.
The 1996 Revision of the Law
The 1996 revision of the 1992 law was initially aimed at clarifying which projects should be submitted to the RECs for approval (Law no. 499, 1996). Under the 1992 law every project with a biomedical component, or dealing with patients or previous patients should be submitted. This included a large number of research projects based on questionnaires, or on anonymised analysis of already existing health databases.30 Most of these projects contained no research ethical problems of any kind. The purpose of the revision of the law was thus to exclude such unproblematic projects in order to reduce the work load of RECs. During the parliamentary debate about this revision an initially unrelated media debate flared up concerning the amount of money researchers were paid by pharmaceutical firms, and especially about the problems caused by researchers receiving large per capita payments for each person included in a trial. This debate was initiated by a single television program concerning the payments received by oncological researchers in connection with trials of Taxol (Paclitaxel), but was quickly broadened by the printed press. This debate was picked up by some politicians and the government rapidly added a new clause to the bill before parliament stating that the patient information has to contain explicit information about the support received from firms and private and public foundations. This exemplifies
F-12
a fairly common feature of the Danish political debate about research ethics and the regulation of research. “Research scandals” are always met with a demand for tighter regulation, even in those cases where the researcher in question is breaching existing regulation, and where the response should therefore be punishment and perhaps increasing control of compliance with existing regulations.
The Function of the System
How does the Danish REC system then work in practice? This is a difficult answer to answer in abstraction, and only a limited amount of research has up to now been done on the function of the system. In this section I will try to give an overview of what is known, as well as of the views held in the Danish biomedical research community.
Knowledge About the System in the Population and Among Researchers
The Danish population is in general positive towards medical research, and accepts that it is necessary, although negative views about medical researchers are also prevalent.31 The general knowledge about the existence of the REC system in the Danish population is poor. In a telephone survey of a random sample of the adult Danish population (N=1,137) it was found that only 30 percent were aware of the existence of an official body which controls medical research, and that only a very small minority was aware of the composition of RECs and that the lay members outnumber the professional members. Most believed the lay representation to be small.32 Other studies have, however, shown that the fact that a project has been approved by an independent REC is an important factor in determining the willingness to participate in the project, and that this holds for the public, for out-patients, and for actual trial participants.33 With regard to medical and other researchers we only have indirect evidence. The number of submitted research projects rose rapidly during the first ten years of the existence of the REC system, but now seems to have stabilised around 2,400 projects per year with only very little annual growth. This is probably an indication of a situation where those projects that should be submitted are submitted. Courses about research ethics is an obligatory part of medical undergraduate education, and of some doctoral programs.
A recent survey of Danish doctors’ knowledge about the content of various national and international declarations and oaths showed that the Helsinki II Declaration was the declaration that was best known.34 The knowledge about the content of the Helsinki II Declaration was even better than the knowledge about the Danish Physician’s Oath (“Lægeløftet”) which every Danish doctor solemnly swears at the graduation ceremony.
The Evaluation of Projects in RECs
Because of the large differences in number of projects submitted each year, each REC has slightly different procedures for evaluating projects. All RECs operate a system of designated pre-evaluators where a project is allocated to two members (one professional and one lay) for specific scrutiny after it has been checked for completeness and legality by the secretariat. Most RECs have 6 to 12 meetings per year, and in some RECs with the smallest number of projects every project submitted is discussed in a meeting where the pre-evaluators briefly outline the project and give an opinion. In other RECs with larger numbers of projects only certain kinds of projects are discussed in a meeting, this will include those projects where the pre-evaluators have identified problems, but also certain generic types of projects. In the two RECs for Copenhagen and Frederiksberg municipalities which handle 30 percent of all research projects in Denmark the types of projects that will always be discussed in a meeting includes projects involving: 1) inmates in prisons, 2) fetuses, embryos, or gametes, 3) radiation above a certain level, and 4) a desire expressed by the researcher for discussion in a meeting.35 Projects involving children and other incompetent persons are circulated to all members of the REC
F-13
and only approved without discussion in a meeting if no member has any queries about the project. All in all five to ten percent of all projects are discussed in a meeting in these two RECs, whereas the rest are managed simply by consensus between the two pre-evaluators. This consensus may involve requirements for changes in the research design or patient information.
RECs do perform a rudimentary scientific review of the projects that are submitted, and projects that are clearly methodologically substandard will be rejected. The argument here is the obvious one that people should only participate in research which is methodologically sound and able to answer the research question asked. Because of the composition of the RECs and their secretariats it is, however, impossible to perform an in-depth scientific review (see the section below, “Problems in the Constitution and Membership of RECs”). The Danish system does not contain any specific mechanism to ensure that such a scientific review does take place. Most research projects will be vetted in the institutions where they originate, but there is no guarantee that this happens, and no way of documenting it formally.
Chairman’s action does not take place in the primary evaluation of a project, but can take place in cases where a research project is resubmitted with the requested changes, or in cases where a researcher submits minor amendments to an already approved project. The meetings of RECs are not public and the minutes are viewed as internal working papers and are therefore not open for public access.
The Effects of REC Evaluation
It has been shown that the research protocols submitted for REC approval contain very few ethical considerations, even in those cases where the project contains substantial ethical problems.36 Another study has shown that the patient information sheets that are submitted to Danish RECs are difficult to read, and that they often lack important information. The REC process rectifies some of these problems, but even after REC approval not all patient information is satisfactory.37 It is, however, the general impression that the “ethical standard” of research protocols has gone up over the years.
One positive unintentional side-effect of having a REC system which requires submission of all research protocols is that the scientific quality of the protocols has improved considerably over the years. The mere fact that somebody else outside the research team is going to read the protocol diligently forces the researcher to state his or her considerations about design, number of patients, etc., very explicitly, and thereby forces the researcher to think in a more structured and explicit way.
Problems in the Constitution and Membership of RECs
The majority of lay members in Danish RECs, which seems to be a unique feature of the Danish system, causes no problems. Just like the professional members the lay members have problems in the beginning finding out “what it is all about,” but they soon settle in and are able to make a constructive contribution. Lay members do not only contribute to the vetting of the readability and content of patient information but can and do make comments on all parts of the protocols. The fact that there is more than one lay member has two positive effects. It refutes the charge of tokenism, and it reduces the chance that the individual lay member can be silenced by the professional members. The way lay members are appointed also removes any possibility for research institutions to influence the process and recruit “tame” lay members to the RECs. In general those who are appointed have an interest in the area prior to their appointment and are used to committee work from previous experiences on political committees of various sorts.
A greater problem is the potential lack of certain kinds of expertise in the RECs. Part of their remit is to ensure that the research projects submitted are scientifically/methodologically sound and worthwhile. This in many cases requires an expertise in research methodology and/or statistics that is not necessarily present in the RECs. The professional members may possess this expertise, but then again they may not. This has been
F-14
exemplified by a number of research protocols using qualitative research methodologies that have been rejected initially, mainly because no one on the RECs in question possessed the necessary knowledge about this particular form of research. This problem could be solved either by enlarging the secretariats of the RECs by the establishment of a post for a research methodologist who could screen projects, by stating more specific requirements for members of RECs in this area, or by establishing some form of peer review.
Another potentially problematic lack of expertise among the members is in the area of research ethics/law and bioethics/biolaw in general. In the present system this expertise is mainly held by the secretariats, at least as far as the legal knowledge is concerned, but this potentially reduces the possibility for really in-depth ethical discussions of problematic projects.
The Control Function of RECs
According to Danish legislation the RECs have a legal duty and right to monitor that the approved projects are carried out in compliance with the approved research protocol (§ 9, sect. 1 & 2). In discussions before the initial legislation in 1992 and before the revisions in 1996 it was pointed out by the RECs, the DMA, the Danish Council of Ethics, and many others that such a control and monitoring function could only work if the RECs were given additional resources. The members of RECs are, as mentioned above, not paid, and most of them feel that they are already devoting considerable time and energy to REC work, and the secretariats are not excessively well staffed. There is thus simply no available resource in the form of person hours to perform any active control. The politicians did, however, show themselves to be completely resistant to these arguments.
Most RECs now require researchers to submit a final short report when a project is finished or abandoned, but this only gives a very superficial picture of the actual conduct of the research in question. At present the only real control occurs in cases where the RECs are alerted to potential irregularities by research participants, relatives, or health professionals. In such cases RECs do perform site-visits or summon the researcher to explain him or herself.
Discussions about the control function are presently underway between the CREC and the Ministry of Research, but the outcome is uncertain. The plan involves site visits to 5 to 10 percent of all projects performed by two members of an especially established team of monitors. The present idea is that these monitors should be recruited among former REC members.
Advantages and Problems in a Regional REC System
The main reason for having regionally and not institutionally based RECs is that this removes some of the pressures that an institutionally based REC may encounter. In an institution there may be pressure applied on the REC to approve or disapprove certain kinds of research, disregarding the ethical status of the research or its compliance with national rules or international declarations. A regional REC is far less likely to succumb to such pressures because the members are not all associated with one single institution. The experiences in the Danish REC system is that the professional members do not feel themselves to be representatives of their institution, just as the lay members do not feel themselves to be representatives of their party. In a given region there will usually be many more research active institutions than there are professional members of the REC, and since the professional members are appointed not based on advice from the institutions but from the professions, any idea of representing the institution and its interests is effectively suppressed. The downside is that the REC may sometimes lack knowledge of very specific, but important institutional factors influencing a given research project.
There is, however, a problem in applying the exact same structure across Denmark. Even though several counties may elect to have one joint REC the differences in research activity between different regions is so large that the work load and experience of RECs vary widely. If multi-centre projects are discounted some
F-15
RECs see less than 50 new projects a year, whereas others see more than 400. With multi-centre projects included the discrepancy becomes less (100 versus 500) but it is still substantial. In general those RECs that cover universities and/or university hospitals get the largest load of projects.
Four Simple Improvements to the Danish REC System
Following from the description above there are a number of simple improvements which could be implemented while maintaining the strengths of the system, which I take to be:
| 1. |
The regional and not institutional RECs. |
| 2. |
The large number of lay members in RECs. |
| 3. |
The existence of the CREC with the role described above. |
| 4. |
That all projects have to be submitted, both public and private. The first of these improvements would be to upgrade the secretariats of the RECs with expertise in research |
methodology and statistics, so that the methodological soundness of the submitted projects could be screened, prior to the RECs’ consideration of the projects. A formal mechanism for peer review could be another option, but peer review is notoriously open to a range of biases. Above I have also identified the lack of bioethics/biolaw expertise as a problem, but I do not think that it is a problem of the same magnitude as the possible lack of methodological expertise.
The second improvement would be to require researchers to submit clear justification for the importance of their projects, preferably in the form of a structured review of the already available knowledge in the area. Due to the work of the Cochrane collaboration the methodology for performing structured reviews (and meta-analyses) is rapidly developing, and it is now clear that the traditional unstructured review which often forms the “Background” section of a research protocol is inadequate.
The third improvement, which is discussed in more detail below, would be to develop the monitoring role of RECs so that they could really fulfil their mandate. This would, like the first improvement mentioned require increased funding.
The fourth and final improvement would be a change in REC culture, so that RECs are more actively engaged in public discussion about difficult research projects. There have been a few instances where the CREC, the researchers, and the affected patient groups have actively sought to create public debate and awareness, but this could be developed more. This would hopefully have the beneficial side-effect that the public become more aware of the existence and role of RECs.
Areas of Possible Future Development
In the following section I will discuss two possible future developments of the Danish REC system. One of these is an extension or accentuation of its function as a democratic institution, and the other is an extension of its monitoring role with regard to already approved research.
RECs as Democratic Institutions
Biomedical research involving human subjects is a social practice which relies on social acceptance for its continuation and flourishing. This social acceptance has to encompass both the goals of the activity and the way the activity is conducted. In a very early paper on medical research ethics Hans Jonas pointed out that research and development is always an optional goal.38 It is not incoherent or irrational to think that no more
F-16
medical research should be performed, as long as one is willing also to accept that no more medical progress will be made. But then it is not irrational not to wish for progress! The RECs probably have only a minor role to play in explaining the general goals of biomedical research to the public, but they do have potentially very important roles to play with regard to the social acceptance of the goals of specific projects and the conduct of research. We know that recruitment rates to biomedical research have been falling steadily over the last 10 to 20 years,39 and unless this trend is reversed it will lead to serious problems concerning both the pace of biomedical progress and the generalisability of those results that are generated.
Although the approval procedure could be analysed purely in terms of protection from problematic research, the presence of lay members on most RECs point to another possible function. What are the lay members there for? The most minimal interpretation of their role is that they are there simply to ensure that the information given to prospective research participants is understandable by “ordinary people” and not too filled with medical jargon. On this minimal interpretation the role of the lay person would be purely as a “linguistic sounding board.” However, some countries have a majority of lay members on their RECs, and in most countries lay members are not chosen on the basis of their ear for language, so it is not unreasonable to suggest that they also perform other roles. But what roles?
If we reconceptualise RECs not only as formal approval bodies, but as institutions within a democratic framework which at the same time regulates and legitimises biomedical research we may become clearer about the role of both the RECs themselves and their lay members. When a REC approves a project it is not a neutral administrative act, it is also an implicit endorsement of the project and its qualities; or that is at least the way it will seem to the outside observer. RECs carry the honorific “ethics” in their name, and something that is approved by an ethics committee must ipse facto be ethical! RECs may not want their approval to have this implication of endorsement, but it is difficult to avoid, and it is worth considering whether it cannot be used constructively.
Can we imagine a situation where REC approval actually functions as a partial legitimisation of the specific research project?
The most common public worries about biomedical research are that research is only carried out to promote the career of the researchers or to promote the interests of the pharmaceutical industry. The researchers are not really interested in helping patients, or solving those health problems that are important seen from the point of view of society. Many research projects are therefore performed that are really unimportant, and where the participation of research subjects is therefore wasted (This is a simplified and thereby slightly caricaturised version of the public worries). How would a REC have to look like, and what would it have to do in order to be able to allay these public worries?
First, it would probably have to be (and be seen to be!) totally independent of research interests. This points towards that the members of RECs should not be appointed by the research institutions themselves, but through some independent mechanism. It further points towards a very substantial representation of non-researchers on the RECs. It is “common knowledge” that doctors (and other researchers) are as thick as thieves, and this common knowledge will affect the perception of RECs, whether or not it is actually true! In this context it is not enough to argue that researchers are honourable persons who would never let their own interests or the interests of their colleagues influence their decisions on RECs, if the public is not fully convinced by the argument.
Second, the nonresearchers would have to be “elected” to the REC by a mechanism that is transparent and accepted in the society where the REC is operating. The nonresearchers will have to be independent, to be beyond reproach, and to be people who are seen as truly representing the public interest. Different methods may suit different societies but just co-opting the “great and the good,” or the local vicar does not add much democratic legitimation.
F-17
Third, RECs would have to be very open about their methods of working and the reasons for specific decisions. Only by aiming at complete transparency can the necessary confidence be developed in the public.
Fourth, many RECs would have to become tougher in their rejection of research protocols that are deemed to be methodologically poor, or to give only very limited benefit to society. People who are willing to become research subjects are a scarce resource, and just like other scarce resources it should be protected and used wisely and not squandered on projects without clear benefit. A potential research subject should not have to worry about whether or not the project he or she is being asked to participate in is of good scientific quality and likely to produce beneficial scientific results. The fact that is has been approved by a REC should be conclusive evidence of scientific quality and expected benefit.
Fifth, RECs would have to engage in public discussion and consultation concerning contentious research projects and contentious justifications for research projects. Whether a research project is socially acceptable in a certain society, and whether it will add or detract from the general acceptance of biomedical research is not always a question which can be answered by pure conceptual analysis, or by applying a set of rules and guidelines. Some societies may accept certain kinds of research which would be deemed unacceptable in other societies, and certain justifications for research may be acceptable in some societies but not in others (e.g., research with the primary aim of benefiting the national pharmaceutical industry). As democratic institutions RECs would have to consult those people on whose behalf the decisions are made, in order to be able successfully to claim that they represent these people.
These five requirements that RECs would have to fulfil before they could gain a stronger role in the democratic legitimation of research would in many instances necessitate radical changes in the structure and function of existing RECs, and it is therefore doubtful whether RECs will take on this role in the future.
Monitoring the Conduct of Research40
In many countries RECs have a right and an obligation to monitor how the approved research projects are actually conducted, but this monitoring role is in many cases much less developed. If monitoring is performed it is often based only on annual or final reports from the researchers themselves, or is only activated when there are complaints about specific projects. We are thus in most countries in a situation where it is ensured that the research protocols are ethically acceptable, but where it is never in reality controlled that the research is conducted according to the protocols and that there are no ethically problematic breaches of the protocols. The situation can in certain respects be compared to a situation where sensible speed limits are imposed, the quality of cars inspected, but the speed of motorists never measured and speeding tickets only issued in cases where an accident has occurred.
Other agencies than RECs may in some cases perform monitoring of biomedical research. This is for instance the case with all GCP-compliant research, where the sponsor (often the pharmaceutical industry) is required to ensure both adequate monitoring and auditing of the research. The aims of this monitoring are, however, not primarily to ensure an ethically acceptable conduct of the research, but to ensure the scientific validity. The GCP rules do contain provisions about ethics and ethics review, but their main raison d’etre is not the maintenance of ethical standards. There are also many biomedical research projects that are not subject to the GCP rules, since their purpose is unrelated to the development and registration of new pharmaceuticals.
How can the monitoring role of RECs be developed in the future?
There seem to be two possible ways to go. The first of these possible developments involves more and more detailed regulation of specific aspects of the research design, patient information, etc. When research “scandals” are unveiled a standard response from politicians is “We must have stricter regulation,” but it is doubtful whether this is actually a correct and useful response. Many of the “scandals” concern research projects that have either never been approved by a REC or are conducted in breach of the approved protocol. It is, to
F-18
say it mildly, unclear why and how stricter regulation can help in such cases. The more reasonable response seems to be to punish the transgressors (partly for reasons of future deterrence) and to ensure better control in the future, so that no unapproved research can be conducted, and breaches of the approved protocols can be detected and rectified. Stricter regulation without increased control may even in some circumstances be counterproductive because it can increase the incentive to try to circumvent the REC system, either by redescribing research as “quality control” or “routine data collection for statistical purposes,” by cutting corners in the actual conduct of research, or by carrying out the study in another country/jurisdiction with less constraining regulation.
What would be involved if RECs took the second route and began to monitor research projects?
Many models can be envisaged, but a comprehensive monitoring of research projects must involve at least three components:
| 1. |
The researchers’ self-assessment of compliance with the protocol. |
| 2. |
Site visits to control documentation and data-protection issues. |
| 3. |
Surveys of patients. The first of these components would be the easiest to implement, but would give the least reliable data. |
Researchers could simply be sent a standardised questionnaire at the end of their project, asking simple questions about consent and information procedures, etc. Although such a process will not generate absolutely reliable data because of problems of self-incrimination, it is not worthless. It becomes important because if researchers are asked about their consent procedures, their recruitment problems, their data protection measures, etc., they are given a chance to reflect upon their own practice and the practice of their co-workers, and this can, at least in some instances, lead to beneficial changes in practice. In the long run the mere fact that researchers know that they will be asked such questions may also lead them to proactively ensure that they comply better with the regulations than in situation where they know that no control is going to happen.
The second and third component are more difficult to implement and require a much greater investment of resources, but they are never the less important because they give a more accurate picture of the ethical conduct of research. By implementing direct control of a proportion of all research projects the REC will be able to detect if there are clear breaches of the rules and regulations governing research. The REC will furthermore be able to get a better feel for how the research is conducted within the different research active institutions in a given area, and this information may be valuable in the assessment of future research protocols.
It could be argued that the monitoring function should be separate from the RECs, and that it is a natural function of, for instance the bodies that authorise health care professionals. The conduct of unethical or unapproved research is a breach of professional duty, and should be controlled and sanctioned as any other kind of professional misconduct or malpractice (e.g., by official censure or removal of authorisation). This argument is not unreasonable but if a separation between the approval and monitoring functions was implemented in this way it would probably lead to an underutilisation of the information produced by the monitoring exercise. The authorising bodies are usually only interested in clear cases of professional misconduct, since it is only such cases that can form the basis for action against individual health care professionals. The RECs are (or should be) interested in a much broader range of information including the clear cases of misconduct, but also cases of exemplary or innovative research practice, and cases where the rules are not clearly broken but just bent in problematic ways. It is this broad range of information which will allow a REC to identify areas of research practice where intervention or guidance is necessary.
In order for RECs to fulfil such a monitoring role, and to utilise the information gained constructively, they must be given certain powers. The legislation or regulations governing RECs must clearly state that 1) RECs have a duty to monitor approved projects, 2) RECs have a right to access and collect the information that is
F-19
necessary to fulfil the duty, including a right to perform the necessary inspections at premises where research is taking place, and 3) RECs are given authority to apply a range of sanctions to researchers who perform research that contravenes the regulations or the approval that has been given. RECs would also need more staff and more money, since good monitoring of research performance is very labour intensive.
If institutionally based RECs took on a more active monitoring role they might very easily come into conflict with some of the (perhaps more superficial?) interests of the institution. Regionally based RECs could more easily handle such conflicts of interest.
Can the Danish REC System Be Transferred?
Although a number of problems in the current Danish REC system have been identified in this paper, the overall assessment is that the system functions satisfactorily, and that with a few modifications it could be brought to function really well. But can its structure be transferred elsewhere and the same level of functionality be expected?
One consideration to take into account is simply the matter of size. The system at the regional level is size-independent in the sense that a region can be subdivided, or more RECs established in a region if the number of research projects in the region grows too large. At the national level there is however size dependency. A CREC with representation of all regional RECs can only work if the number of RECs is reasonably small, otherwise the CREC will simply be too large. This problem can be handled as long as the increase in number of RECs is not large, for instance by only having one representative from each REC, but if there are 500 to 600 RECs there is no way to give all direct representation on a CREC. In such a situation it would seem reasonable to split the function of the CREC in two and establish two kinds of bodies. The first kind of body would deal with the appeal function of the CREC, and would be regional appeal-RECs each covering a number of RECs. On these A-RECs it would still be possible to have direct representation of the involved RECs. The second kind of body would be a national body issuing legally binding recommendations for the evaluation of research proposals in RECs.
The size problem also plays a role in considering the transferability of the Danish system for handling multi-centre trials, but here a further consideration also comes into play. Denmark is a fairly culturally homogeneous country, and although there are regional differences, it is still a viable assumption that a project that is acceptable in Copenhagen is also acceptable in the west of Jutland. Regional values are not so different that the approval of one REC cannot in most cases be extended to other RECs. This situation may not obtain in other countries where either regional differences are larger, or where certain sectors of the health care system are based on specific, for instance religious, value systems.
There is no reason to believe that the majority of lay members on Danish RECs could not be implemented successfully elsewhere, although the mode of appointment would probably have to be modified according to local political customs.
Similarly there seems to be no reason why regional RECs, with the advantages described earlier, could not be transferred to other contexts than the Danish. There may be some institutions that are so special, either because of their area of work, or because of the value system on which they are based, that they would require their own institutional RECs, but the number of such institutions must be fairly small. It is also important to note that even if the regional RECs in Denmark approve a research project this does not give the researcher any positive claim right against his or her institution to be allowed to perform the project at the institution. Under the current system REC approval only entails that the project fulfils a general societal set of ethical rules. If an institution wants to implement its own more stringent set of rules that option is still open.
F-20
Acknowledgements
This paper builds on a number of my previous published papers in this area. Some of these have been published in collaboration with others, and I gratefully acknowledge the invaluable contributions of my co-authors Søren Madsen, Povl Riis, Peter Rossel, and Henrik R. Wulff.
I gratefully thank Rikard Vrogaard, Head of Secretariat, The Scientific-Ethical Committee for Copenhagen and Frederiksberg Municipalities, for many helpful comments to a draft of this paper.
Finally I thank my mother Edith Holm, current member of the Danish Central Scientific-Ethical Committee, for many informative and helpful discussions over the years.
Notes
1 The committees are called “Videnskabsetiske Komitéer” in Danish and this literally translates to “Scientific-Ethical Committees.” This is also the term the committees themselves use in English language correspondence and publications. Here I will, however, use the more broadly recognised standard term in English “Research Ethics Committees.”
2 I have previously published two short papers on the Danish REC system: Holm S. How many lay members can you have in your IRB? An overview of the Danish system. IRB: A Review of Human Subjects Research 1992; 14(6):8–11.
and
Holm S, Wulff HR. Os Comitês de Ética na Dinamarca. Bioética 1998; 6(2):171–175.
3 A REC may also decide that a certain project falls outside the law’s definition of biomedical research, and therefore does not need approval.
4 Holm S. Private hospitals in public health systems: Social and ethical considerations. The Hastings Center Report 1989; 19:16–20.
Holm S. Solidarity, justice and health care priorities. In Szawarski Z, Evans D (eds.). Solidarity, Justice and Health Care Priorities (Health Service Studies 8). Linköping: Linköping University Press, 1993 (p. 53–64).
5 Hansen HC. Historien om Sygekasserne. København: De samvirkende centralforeninger af sygekasser i Danmark, 1974. Ito H. Health Insurance and Medical Services in Sweden and Denmark 1850–1950. In: Heidenheimer AJ, Elvander N (eds.). The Shaping of the Swedish Health System. London: Croom Helm, 1980 (p. 44–67). Mahler E. Københavnerordningens historie. København: C.A. Reitzels forlag, 1991.
6 There are a number of hospitals owned by religious orders, and a few sanatoria owned by patient organisations. These nonprofit institutions are in a formal sense private, but all their beds are contracted for by the public health care system on long-term contracts, and they are thus an integral part of the public health care system.
7 Brunsted B, Kolbye T. Sygdom er en sløj forretning. Dagens Medicin 2000; 17 (torsdag den. 25. maj):14–16. Health Care in Denmark. Copenhagen: Ministry of Health, 1997.
8 The same lack of impact of the Nuremberg Code can be found in many other countries, see for instance: Herranz G. The Inclusion of the Ten Principles of Nuremberg in Professional Codes of Ethics: An International Comparison. In: Tröhler U, Reiter-Theil S (eds.). Ethics Codes in Medicine: Foundations and Achievements of Codifications since 1947. Aldershot: Ashgate Publishers, 1998 (p. 127–139).
Winslade WJ, Krause TL. The Nuremberg Code Turns Fifty. In: Tröhler U, Reiter-Theil S (eds.). Ethics Codes in Medicine: Foundations and Achievements of Codifications since 1947. Aldershot: Ashgate Publishers, 1998 (p.140–162).
9 Rossel P. Medicinsk Etik. København: Gads Forlag, 1979.
10 Blomquist C, Enger E, Riis P. Nordic proposal concerning new ethical rules for biomedical research. Nordisk Medicin 1975; 90(3):79–80.
11 Riis P. Letter from Denmark. Planning of scientific-ethical committees. Br Med J 1977; 2(6080):173–174.
Riis P, Gjørup S, Vagn-Hansen P, Winkler K. Helsinkideklarationen og videnskabsetiske komiteer i Danmark. Ugeskrift for Læger 1977; 139(40):2404–2409.
Karlsson Y. Network of research ethical committees created in Denmark, Finland and Norway. Nordisk Medicin 1977; 92(8–9):222–223.
F-21
12 Betænkning om information og samtykke i forbindelse med forsøg (betænkning nr. 1335). København: Forskningsministeriet og Sundhedsministeriet, 1997.
It is worth noting that the Danish Nursing Council and the organisations representing other professions allied to medicine were not invited to participate in the negotiations leading up to the establishment of RECs and were not parties to the final agreement.
13 Greenland and the Faeroe Islands are parts of the Kingdom of Denmark, but have extensive home rule. The initial extra-legal REC system was not implemented in Greenland or the Faeroe Islands and the legal regulation in 1992 explicitly excluded these two parts of the kingdom from the provisions in the law. A REC has been established on the Faeroe Islands in 1999 in accordance with a Royal Decree in the form of a Regulation issued by the Ministry of Research activating certain parts of the Danish legislation on the Faeroe Islands. The regulation establishes a REC for the Faeroe Islands with similar composition as Danish RECs, and with observers (not members) in the Danish CREC. It is likely that the same will happen with respect to Greenland from the 1st of July, 2000 (Rikard Vrogaard, personal communication).
14 Formally the only requirement is that each REC should appoint a professional member and a lay member to the CREC, but in practice this has always been the chairman and the vice-chairman of the REC.
15 Scocozza L. Forskning for livet: den medicinske forskningsetiks forudsætninger og praktikker. København: Akademisk Forlag, 1994.
Scocozza L. Forskning for Livet. In: Schou I (ed.). Patienten i lægemiddelforskningen. København: MEDIF & MEFA, 1995 (p. 33–35).
16 Forskning på mennesket: etik/jura (betænkning 1185). København: Sundhedsministeriet, 1989.
17 With regard to membership of the RECs the Danish system differs substantially from the Norwegian system, which is otherwise similar in many ways. In Norway the composition of the regional RECs is precisely specified as: One medical member from the medical faculty in the region.
One medical member from the public health authority in the region.
One member with psychological expertise from the psychological institute or faculty in the region. One member who is a registered nurse.
One member appointed by the hospital owners (i.e., the counties in the region). One member with ethical expertise.
One lawyer.
One lay representative.
Three other differences between the Norwegian and the Danish system are: 1) that Norwegian RECs formally only give advice on projects, 2) that Norwegian RECs are not classed as public administrative bodies, and 3) multi-centre projects are approved by only one REC without consulting other RECs.
Mandat for de regionale komiteer for medisinsk forskningsetikk. Kirke-, utdannings- og forskningsdepartementet 19. Januar 1989 (med endringer senest 5. Mars 1999).
18 The law also gives “anyone with a special interest in a project” leave to appeal to the CREC, but this very rarely happens.
19 Denmark also has a national advisory bioethics committee. This committee, called The Danish Council of Ethics (“Det Etiske Råd”), was established by law in 1987 with the double task of 1) advising the Danish parliament and government on ethical issues related to health care, and 2) promoting public debate on bioethical issues (see: Cushman R, Holm S. Death, democracy and public ethical choice. Bioethics 1990; 4:237–52 and Holm S. New Danish law: Human life begins at conception. Journal of Medical Ethics 1988; 14:77–78). The Council consists of 17 members with an equal gender distribution. The chairman and eight of the members are appointed by a subcommittee of the Danish parliament, and the remaining eight members are appointed by the Minister of Health. Members either have to be experts in the subject areas of the Council, or they have to have participated in the public debate on ethical issues. Members are appointed for a three-year period and can be reappointed once.
The Council advises government and parliament, both through answering questions put to it by the government, and through developing reasoned statements on ethical issues which the Council itself sees as important. The reports of the Council are mainly published in Danish, but English translations of the most important can be found in the annual reports, and on the Council website (http://www.etiskraad.dk).
The Council has a very broad range of activities aimed at creating public debate. It organises its own public meetings, and also sponsors meetings on bioethical issues organised by local groups all over Denmark. It produces videos and more traditional teaching materials for use in the public schools as well as in higher education.
During its 10 years of existence the Council has managed to generate a sustained and broad public debate about bioethical issues in Denmark. Few Danes are unaware of the existence of the Council, although many seem to impute much greater power to the Council than it really has. This half of the Council’s activities have thus been very successful.
F-22
The Council’s success in its advisory role has been less conspicuous, at least if it is measured as the direct impact on legislation. In most cases the Danish parliament has not directly implemented the regulations proposed by the Council, but regulations that are more liberal. This has, for instance, been the case in the area of assisted reproductive technologies. The Council has, however, had some influence even in these cases by pointing to areas for which some kind of regulation should be developed.
All in all, it is probably fair to say that the Danish Council of Ethics has been a success in the sense that its existence and activities have put much more focus on the ethical issues inherent in many developments in biomedicine than would otherwise have been the case.
20 Health Science information banks – Biobanks. Copenhagen: The Danish Medical Research Council, the Danish Central Scientific-Ethical Committee and the Danish Council of Ethics, 1996.
21 There are also twice yearly meetings between the REC in Lund, Sweden and the RECs of Copenhagen and Frederiksberg municipalities, and the Copenhagen county REC. With the opening of the Malmö-Copenhagen bridge in July 2000 the research collaboration between the universities and hospitals in the Øresund region is expected to increase very rapidly, and this will create a need for coordination between the involved RECs in Sweden and Denmark.
22 Collection of Annexes. København: Den Centrale Videnskabsetisk Komité, 1994.
23 Rikard Vrogaard, personal communication.
24 Hartlev M (ed.). Den gode samfundsforsker: om etik i samfundsforskningen. København: Akademisk Forlag, 1996.
25 Prior to the legislation a number of nonmedical researchers complained about having to submit their projects to a system on which they had no influence. The RECs were by some seen as an attempt to enforce a medical hegemony on other groups.
26 Sundhedsstyrelsen. Vejledning om indførelse af nye behandlinger i sundhedsvæsenet (99.07.02). København: Sundhedsstyrelsen, 1999.
27 Evans D, Evans M. A Decent Proposal: Ethical Review of Clinical Research. Chichester: John Wiley and Sons, 1996.
28 Kindly provided by Rikard Vrogaard.
29 The CREC’s view of the case can be found in the Annual Report 1996 in a section with the slightly misleading title “Cooperation with the Parliament’s Ombudsman.” Annual Report 1996. København: Den Centrale Videnskabsetiske Komité, 1997. See also Skou E-M. Det Videnskabsetiske Komitésystem. In: Schou I (ed.). Patienten i lægemiddelforskningen. København: MEDIF & MEFA, 1995 (p. 66–74).
30 Every Danish citizen and every permanent resident of Denmark is allocated a unique Central Personal Register number. All health information is stored with linkage to this number, and this creates an extremely good environment for register-based epidemiological research since different registers can easily be linked and index persons easily traced. See Forslag til en national strategi for sundhedsvidenskab (betænkning 1284). København: Forskningsministeriet, 1995.
31 Rossel P, Holm S. How does the public perceive the motives of medical researchers for doing research? Bulletin of Medical Ethics 1999; 146(March):16–7.
Saurbrey N, Jensen J, Elmegaard-Rasmussen P, Gjørup T, Guldager H, Riis P. Danish patients’ attitudes to scientific-ethical questions. An interview study focusing on therapeutic trials. Acta Med Scand 1984; 215(2):99–104.
32 Holm S, Rossel P. Hvad ved den danske befolkning om det videnskabsetiske komitésystem. Ugeskrift for Læger 1996; 158:4383–4384.
33 Madsen S, Holm S, Riis P. Ethical aspects of clinical trials: the attitudes of the public and out-patients. Journal of Internal Medicine 1999; 245:571–579.
Madsen SM, S. Holm, B. Davidsen, P. Munkholm, P. Schlichting, and P Riis. 2000. “Ethical aspects of clinical trials: The attitudes of participants in two non-cancer trials.” Journal of Internal Medicine 2000; 248(6):463–474.
34 Fabrin A, Hasman A, Kristensen K, Rabøl LI, Holm S. Do doctors know the content of the Hippocratic oath and other medical oaths and declarations. Bulletin of Medical Ethics 2000; 154 (January):13–16.
35 De Videnskabsetiske Komitéer for Københavns og Frederiksberg Kommuner - Årsberetning 1996–1997. København: De Videnskabsetiske Komitéer for Københavns og Frederiksberg Kommuner, 1998.
36 Holm S. Moral reasoning in biomedical research protocols. Scandinavian Journal of Social Medicine 1994; 22(2):81–85.
37 Holm S. Skriftlig patient information: en analyse af danske biomedicinske forsøgsplaner. Ugeskrift for Læger 1992; 154:2432–2435.
F-23
38 Jonas H. Philosophical Reflections on Experimenting with Human Subjects. In: Freund PA (ed.). Experimentation with Human Subjects. London: George Allen and Unwin, 1972 (p. 1–38).
39 Blichert-Toft M, Mouridsen H, West Andersen K, From the Danish Breast Cancer Cooperative Group (DBCG). Clinical trials. Sem Surg Oncol 1996; 12:32–38.
Hunter C, Frelick R, Feldman A, Bavier A, Dunlap W, Ford L, et al. Selection factors in clinical trials: results from the Community Clinical Oncology Program Physicians Patient Log. Cancer Treat Rep 1987; 71:559–565.
Jack W, Chetty U, Rodger A. Recruitment to a prospective breast conservation trial: why are so few patients randomized? BMJ 1990; 301:83–85.
DeVita V. Breast cancer therapy: Exercising all our options. N Engl J Med 1989; 320:527–529.
Zelen M. Strategy and alternate randomized designs in cancer clinical trials. Cancer Treat Rep 1982; 66:1095–1100.
Anonymous. DBCG (Danish Breast Cancer Cooperative Group) 1977 – 1997 Jubilee publication. Copenhagen 1998:19–20,
48 –49.
Fisher B. On clinical trial participation (editorial). J Clin Oncol 1991; 9:1927–1930.
Antman K, Amato D, Wood W, Corson J, Suit H, Proppe K. et al. Selection bias in clinical trials. J Clin Oncol 1985; 3:1142–1147.
40 In this section I assume that there are separate mechanisms for dealing with the sub-class of scientific misconduct that involves direct scientific fraud. This is not within the remit of Danish RECs, and I believe that this reflects a proper division of labour.
There is a Danish national system for the investigation of research fraud, not only in biomedical research but in all branches of research.
F-24


VULNERABILITY IN RESEARCH SUBJECTS: A BIOETHICAL
TAXONOMY
Commissioned Paper Kenneth Kipnis
University of Hawaii at Manoa
G-1
The concept of vulnerability appears to have been grandfathered into the lexicon, lore, and literature of research ethics without undergoing stringent certification. And yet the need for some such notion has long been appreciated. More than 50 years ago, reflecting on the ethical implications of the Nazi medical experiments, the authors of the Nuremberg Code emphasized the necessity of the subject’s informed consent, too hastily ruling out, as it quickly became apparent, medical research on children and those with cognitive impairments.
In the United States, widely studied episodes such as Willowbrook,1 the Brooklyn Jewish Chronic Disease Hospital Case,2 and the Tuskegee Syphilis Study3 provoked debates that eventually gave birth to our current methods for ensuring the ethical conduct of research. But despite the remarkable circumstances of the subjects involved in those studies—institutionalized children, hospitalized elderly, and impoverished and poorly educated black Alabama males—it is not much of an exaggeration to say that in the minds of many investigators the paradigmatic research subject remains more or less a mature, respectable, moderately well-educated, clear-thinking, literate, self-supporting U.S. citizen in good standing—that is, a man who could understand a 12-page consent form and act intelligently on the basis of its contents. While I shall assume in what follows both that the existing guidelines are sufficient to deal ethically with the paradigmatic research subject, and, further, that all those standard protections are reliably in place, the vulnerable research subject nonetheless requires ethical consideration going beyond that baseline.
More recently, in the wake of the Nuremberg Code’s shortcomings, systematic attention has been accorded to a motley collection of vulnerable subpopulations. In 1979, for example, the seminal Belmont Report4 briefly considered children, the institutionalized mentally ill, and prisoners, mentioning dependency and compromised capacity for consent as representative hallmarks of vulnerability. There was no effort to be comprehensive. The more recent Federal Regulations on the Protection of Human Subjects (45 CFR 46) implement the requirement that Institutional Review Boards (IRBs) take into account the “special problems of research involving vulnerable populations, such as children, prisoners, pregnant women, mentally disabled persons, or economically or educationally disadvantaged persons” (46–111). Criteria for vulnerability are not discussed although subparts are included with supplementary regulations for some of these groups. Finally, the Final Report of the Advisory Committee on Human Radiation Experiments,5 after reviewing patterns of unethical misconduct in military research, recommended special protections for enlistees.
Though this recent subpopulation focus is an improvement over earlier approaches, it is surely reasonable to register comparable concerns when contemplating research on, for example, drug abusers, the desperately ill, Ugandan women, illegal aliens, the impoverished homeless, women in the process of miscarrying, psychology undergraduates, and the elderly in the early stages of dementia. Though commentators may speak as if there were something common to these disparate groups, it is not now clear what that characteristic (or that set of characteristics) is. And even if such criteria were articulated, one would surely want to know what it was about those features that made those who possess them “vulnerable.” Finally, it is not generically apparent what researchers should do when confronted with a vulnerable subject. These are some shortcomings of the current subpopulation focus.
Regrettably, the term “vulnerable” too often gets played as a bioethical trump card, summarily tossed on the table in the course of debate, sometimes with the stern admonition that it would not be decent to exploit such subjects. Given the absence of agreed-upon standards for identifying and responding to vulnerability, such a move too often serves as a conversation-stopper, abruptly ending dialogue rather than furthering it. It may be possible to do better.
The aim of this paper is, broadly, to provide a needed overview and analysis of the concept of vulnerability and, narrowly, to develop a useful taxonomy. I am here challenging the current subpopulation focus that is evident both in the writings on such research and in the efforts to draft subparts for each designated group.
G-3
I am arguing that the current conceptualization be supplemented or supplanted by something like the analytical approach that I will set out here. My aim is to tease out and consider circumstances that directly signal the vulnerabilities researchers should take into account. In a list that is intended to be exhaustively applicable to research subjects, six discrete types of vulnerability will be distinguished—cognitive, juridic, deferential, medical, allocational, and infrastructural. If the listed subpopulations are groups deemed to be vulnerable, the six circumstances described here are intended to represent the ethically relevant features that bespeak vulnerability, not only in the designated subpopulations but in other groups as well.
Each of these vulnerabilities is conceived, not as a flashing red light ordering researchers to stop, but rather as a cautionary signal, calling for proper safeguards. Accordingly, having ascertained that a candidate-subject (C-S) is vulnerable in one or more of those discrete ways, researchers would then be required 1) to conduct further inquiries and, if necessary 2) to implement compensating measures in the design of the protocol as a condition for proceeding. While some examples of these measures are sketched or referenced, it is not possible to set out here, much less resolve, all of the pertinent ethical problems. Rather the general aim is to provide a needed map of the conceptual geography, one that offers usable guidance while organizing and sharpening issues that might be fruitfully engaged later. First, however, as a prerequisite to understanding vulnerability, one must reflect on the Nuremberg Code’s foundational concern: the concept of consent.
Consent as an Ethical Power
Consent is usefully understood as an ethical power: something we do with words. Philosophers have found it remarkable—even “magical”—that we have the ability, merely by intoning the proper words under the right circumstances, to alter the systems of obligations and permissions that envelope us.6 Ordinarily it is a wrong—even a criminal offense—for you to remove my lawnmower from its place in my garage. But if you ask, “Can I take your lawnmower?” and I reply, “You can take my lawnmower,” an action that would have been wrong thereby becomes—Lo!—one that is unexceptional. Merely in saying, “You can take my lawnmower,” I can bring it about that you can take my lawnmower. In giving permission, an act can become permitted.
Note that consent does not always effect permissibility. If I say you can take my neighbor’s lawnmower, it may not be permissible for you to take it. And if I consent to your killing me, you would not thereby be permitted to do so. That some deed is okay with me does not always mean it is okay.
Notwithstanding the occasional misfire, this amazing ability to give or withhold permission constitutes a critically important ethical power. The connections between a contextually appropriate utterance, its dramatic effect on the permissibility of action, and the various circumstances that can impair that connection, causing a misfire: these three elements constitute the focus of the present study. Accordingly, we can define the vulnerabilities that concern us as those special circumstances of the C-S that call into question the efficacy of consent in effecting the permissibility of research. Despite the presence of consent and the standard baseline protections, vulnerability, in conjunction with other circumstances, can occasion a misfire. Absent compensating measures, it may still be impermissible to conduct research.
We can conceive ourselves as surrounded by a zone of privacy the boundaries of which are, characteristically and for the most part, subject to our will. Though the zone’s dimensions vary with law and culture, our capacity to exercise sovereign authority over such domains as physical property, certain categories of personal information, our immediate physical environs, our body, our intellectual creations, and so on, is reasonably conceived to be constitutive of a developed sense of self, at least in part.7 Boundary crossings—physical touching is a ready example—characteristically require an antecedent consent. In the most dramatic case, an act of sexual intercourse is, absent consent, the crime of rape. It is, I think, fair to say that, since the ascendancy of research ethics as a loose body of theory and doctrine, both of which are broadly coupled with implementing
G-4
organizations (IRBs and national and international agencies), there has emerged a near global appreciation of the relevance of that ethical power in the context of research on human subjects. The entitlement not to be treated as a laboratory animal may be as close as humanity has come to a genuinely secured human right.
Before moving on, it will be helpful to mark a potential confusion involving two types of consent. The consent that is of importance here—I have called it grantive consent elsewhere8 —constitutes a giving of permission. In consenting, something not permitted may become permitted. But there is a different type of consent that generates obligations. In consenting to the terms of a contract, for example, both parties typically assume reciprocal obligations. Having agreed to terms, you may come to have an obligation to mow my lawn, and I may come to have an obligation to pay you. For the purposes of the present inquiry, the consent pertinent to research ethics is not assumed to encompass this second type consent—we can call it contractive consent. Notwithstanding the difference, investigators have sometimes fixated on the separate question of what their research subjects owe to them: strict adherence to a protocol’s requirements, for example. My concern here is, rather, with the C-S expressed willingness to be studied as part of a scientific investigation and with the efficacy of that consent in granting permission. I am setting aside questions regarding the duties of the subject following consent.
Vulnerability and Biomedical Research
The concept of vulnerability points in two directions. By definition, it is a distinctive precariousness in the condition of the subject: a state of being laid open or especially exposed to something injurious or otherwise undesirable. A vulnerability is, so to speak, an avenue of attack. But, in the second place and in the contexts where we use the term, we are characteristically mindful of certain others who are disposed to capitalize on such weakness, exploiting open avenues of attack—intentionally or negligently—and taking unfair advantage to the subject’s detriment. The wrongfulness of using others in this way, selfishly and unfairly—Kant would say “merely as a means”—characteristically grounds humanity’s severe condemnation of research on unconsenting subjects.
To avoid confusion, it is important to mark the difference between the everyday sense of “vulnerability” and the special use pertinent to the context of human research. Consider, for example, the distinctive vulnerability of blind people: they are characteristically less able to protect themselves, and, accordingly, it is easy for wrongdoers to victimize them in certain ways. But this vulnerability is unlikely to be of consequence in the context of most research. Investigators are not lurking out there, waiting to pounce upon and exploit the sightless. Notwithstanding the vulnerabilities of many handicapped persons, the absence of a common capacity does not in itself signal a need for special precaution on the part of researchers. The vulnerabilities that concern us here are only those that call into question the efficacy of consent in effecting permissibility. A person who is plainly vulnerable in the everyday sense may not be a vulnerable research subject. Our focus is on the sense of the term pertinent in the research context.
A second ambiguity may also be a source of confusion. While we can, for example, speak of men as vulnerable to testicular cancer, we are talking about a type of harm that only affects males: we are not referring to a way of being peculiarly laid open to that harm. Being male is not a way of being especially exposed to testicular cancer: it is a precondition for having it. On the other hand, weakened immune systems make people vulnerable to infection. Lacking normal protection, they are at heightened risk. It would perhaps be less confusing to say that males are generically susceptible to testicular cancer, meaning merely that the disease is a harm only they can suffer. Vulnerability, conversely, connotes unusual exposure to some type of injury, and, accordingly, I shall reserve the term exclusively to describe conditions that heighten the risk of harm.
Thus, while only a pregnant woman may lose her fetus, she is not, on that account alone, a vulnerable research subject. When a research protocol heightens the risk of this loss, investigators would surely have to
G-5
disclose that to her, but she would still not be a vulnerable research subject as we are using these terms. However, assuming both that she will carry the fetus to term and that the protocol can cause fetal malformations, then, depending on one’s metaphysics, one could describe as vulnerable either the fetus or the person it will become. Notwithstanding the pregnant woman’s informed consent, research might still be impermissible.
A usable analysis of vulnerability will serve at least three purposes. In the first place it will provide a checklist of circumstances that, along with other conditions, can invalidate the permissibility of research. Each of these circumstances generates its own problems. Is it possible, researchers will want to know, to conduct ethically responsible research on these subjects notwithstanding their vulnerability? A usable analysis of vulnerability would have to suggest responses to that question. In the second place, it will provide an intellectual basis for treating a subpopulation as vulnerable and—equally important—for determining, generically, what specific supplementary measures are called for in the light of their vulnerabilities. And, finally, it will provide a basis for a warranted finding that some researcher has, knowingly or negligently, taken unfair advantage of vulnerable research subjects. Though discussion of the range of corrective responses to such misdeeds would also take us beyond the scope of this paper, the setting of standards, in the nature of the case, provides researchers with usable guidelines for the responsible crafting of protocols even as it generates a basis for criticism, condemnation, and discipline following a showing that there has been a serious breach of those same standards.
Foreshadowing the analysis that follows, each of the six types of vulnerability is distinguished by a positive response to a unique question. Summarizing, these are as follows:
Cognitive: Does the C-S have the capacity to deliberate about and decide whether or not to participate in the study?
| Juridic: | Is the C-S liable to the authority of others who may have an independent interest in that |
| participation? | |
| Deferential: | Is the C-S given to patterns of deferential behavior that may mask an underlying unwillingness |
| to participate? | |
| Medical: | Has the C-S been selected, in part, because he or she has a serious health-related condition for |
| which there are no satisfactory remedies? | |
| Allocational: | Is the C-S seriously lacking in important social goods that will be provided as a consequence |
| of his or her participation in research? |
Infrastructural: Does the political, organizational, economic, and social context of the research setting possess the integrity and resources needed to manage the study?
It is important, in the discussion that follows, to be mindful that participation as a subject in medical research generates benefits as well as risks. Well-designed studies produce knowledge that can help similarly situated patients. But, more important, where there are no satisfactory treatments, participation in a clinical trial may be a patient’s best chance. For example, during the early trials of antiretrovirals for HIV infection, prisoners justly complained that the existing protective rules were barring their access to the only treatments offering a hope of benefit. As has been observed, it would be toweringly wrong to let sailors drown solely because the available life rafts had not been approved by the Coast Guard. We need to be exquisitely careful not to allow a misguided solicitude to load further and unjust disadvantages upon the shoulders of those who are already disproportionally burdened.
G-6
Cognitive Vulnerability
Lawyers make a useful distinction between arm’s length relationships and the much closer ties fiduciaries have with their clients. The former is exemplified in the purchase of a used car. While sellers may not lie (or create a misleading impression by, say, setting back the odometer), neither are they bound to disclose all the pertinent information they have. Buyers are thrown upon their own resources. Fiduciaries, on the other hand, have to take their client’s interests as primary, working to reduce, as much as possible, the knowledge differential that marks that distinctive type of cooperation. Where a critical choice must be made, an ethical attorney must ensure that the client fully understands what is at stake. The lawyer’s objective is that, regardless of what happens, the client will continue to acknowledge ownership of the decision. Here they must become educators, intelligibly conveying a usable sense of the situation, explaining all the options, and—especially—setting out the risks and possible benefits attaching to each option.
With respect to the consent of the C-S, the traditional requirement of informed consent points in the direction of the fiduciary model. The burden on the researcher is not merely to state the pertinent facts, but to ensure they have been appreciated.
Of the six types of vulnerability catalogued here, cognitive limitations are the most familiar. The researcher must ask, “Does the C-S have the capacity to deliberate about and decide whether or not to participate in the study?” Circumstances that suggest the presence of this type of vulnerability would include some degree of immaturity, dementia, certain types of mental illness, and mental retardation. But educational deficits and unfamiliarity with the language may also play a role. Also included would be C-Ss who cannot be sufficiently informed and/or who cannot complete effective deliberation within the available timeframe. For example, some years ago I interviewed patients and clinicians involved in an early trial of tocolytic treatment for preterm labor. At the time the standard treatment was ethyl alcohol. While this could arrest uterine contractions briefly, it was plainly not a satisfactory treatment. Pregnant women brought to the hospital in the process of miscarrying had to make a decision about a complex clinical trial without the time to learn all that was involved or to deliberate effectively. Even apart from the time problem, the C-Ss were in the midst of crisis and not in what educators would describe as a teachable moment. The conception of a cognitive limitation that is commended here is intended to apply to situations like these as well as to the other more familiar cases. Vulnerability is present precisely because the measures ordinarily taken to ensure that the C-Ss are adequately informed will not do in the face of such circumstances.
It would take us too far afield to set out a comprehensive review of the measures researchers might take to address cognitive limitations. We are familiar enough with most of the standard strategies: plain-language consent forms, advance directives (where incapacity is anticipated), supplementary educational measures, and the proper use of surrogates and advocates.
Juridic Vulnerability
Juridic vulnerability calls attention to the formal authority relationships that often characterize social structures. The most striking examples are prisons and the military, where wardens and officers have legal authority over prisoners and enlistees. But the category also includes children under the authority of their parents, psychology students subordinated to their college professors, institutionalized persons (including institutionalized children and their parents) subject to the authority of custodians, and certain third-world woman who may be legally subject to their husbands. Related issues can arise when the C-Ss are engaged in illicit activities. This catalogue is not exhaustive.
In these cases researchers must ask, “Is the C-S liable to the authority of others who may have an independent interest in that participation?” The worry is that the “consent” of the C-S might be merely a reflection of the wishes of those in authority. This distinctive vulnerability—the juridic fact of their subordination to the authority
G-7
of another—can call into question the validity of their consent. This is especially a concern when those in authority are also those who are conducting, commissioning, or somehow benefiting from the research.
In its extensive review of human subjects research in the military, the Final Report of the Advisory Committee on Human Radiation Experiments recommended9 that officers be specifically excluded from recruitment sessions and that an ombudsman be present to ensure that the voluntariness of participation is adequately stressed. Likewise, children can be questioned separately from their parents and confidentially. The task for the researcher is to devise a consent procedure that will adequately insulate the C-S from the hierarchical system to which he or she is subject.
Deferential Vulnerability
While juridic subordination directs our attention to objective features of the formal hierarchical context within which the C-S functions, deferential patterns are, instead, subjective responses to certain others. To be sure, the two are often present together. With respect to military officers, enlistees are generally both deferential and juridically subordinated. But when, in the presence of colleagues, friends, loved ones, and so on, one is exhorted to stand up on behalf of a popular charitable project, one may care deeply about the opinion of those significant others even though they do not, like officers, occupy formal positions of authority.
A researcher needs to understand these powerful social and cultural pressures and devise consent procedures that take them into account. There are peoples, for example, who commonly display a ready agreeableness on the surface that may mask an inner reticence. There are children who are uncomfortable taking issue with adults and third-world women who may find it hard to turn down requests from men, especially if they are respected doctors in white coats. Also included here is the Stockholm syndrome usually thought of in connection with the behavior of hostages, but also perhaps present in some heavily institutionalized subjects.
The question the researcher must ask is, “Is the C-S given to patterns of deferential behavior that may mask an underlying unwillingness to participate?” The distinctive vulnerability of these subjects consists in their readiness to accede to the perceived desires of certain others notwithstanding an inner reticence to do so. Those involved in subject accrual need to be selected with care, perhaps with the advice of local informants or consultants in psychology and anthropology. The conversational setting may require attention. The challenge is to devise a process that eliminates as much as possible the social pressures that a C-S may feel even if, in reality, they are not being imposed.
Medical Vulnerability
As defined here, a medically vulnerable C-S has a serious health-related condition for which there are no satisfactory remedies. Metastatic cancers can fall into this category, as can severe spinal cord injuries, Parkinson’s disease, multiple sclerosis, Alzheimer’s disease, end-stage AIDS, and so on. Also included are illnesses for which there are treatments that are not suitable for particular patients. For example, because it requires the use of blood products, rescue therapy for cancer, though effective, would not be a satisfactory treatment for most Jehovah’s Witnesses. The question for the researcher is, “Has the C-S been selected, in part, because he or she has a serious health-related condition for which there are no satisfactory remedies?” A medically vulnerable research subject knows he or she has been chosen, in part, because of such an illness.
What makes these patients vulnerable is their medically exigent state. Having run out of options, they will be willing—even eager—to undergo risks that would ordinarily be foolish. As Christiaan Barnard observed, it makes sense to leap into a crocodile-infested river to escape from a lion, but not if there is no lion.10 There is an unfortunate tendency to see these patients as coerced. A gunman says, “Your money or your life.” In handing over your wallet, it is important to observe that title to it does not thereby pass to the mugger. While he now has it in his possession, the wallet is still not his even though you gave it to him. Analogously, it
G-8
is assumed that the infirmities of medically exigent patients strong-arm them into submission, thereby giving rise to the broadly held view that consent extorted under such duress cannot effect permissibility.
This view is seriously misconceived. For facing a potentially fatal infection, I can properly consent to antibiotic treatment even though it is an equally forced choice. And having been cured, I cannot then avoid the obligation to pay my doctor’s bill on the grounds that the imminent threat of death made me consent to the treatment. The deal with the doctor certainly was “your money or your life,” but plainly I am obligated to pay anyway. But now observe that if my physician were to exact an exorbitant price for the antibiotic, I might properly claim that he took unfair advantage of my precarious circumstance. He exploited me. These examples help to reveal that the problem with such transactions does not reside in the agent’s diminished range of choice. So instead of obsessing about “voluntariness,” the presence of medical exigency should direct the researcher and the IRB to assess the fairness of the arrangement with the C-S. Is the deal exploitative? More precisely, given the interests and aspirations of both parties (and the poor bargaining position of one), is there a fair division of the benefits and burdens of cooperation?
The classic problem with research on medically vulnerable patients is an apparently ineliminable therapeutic misconception affecting the majority of these subjects.11 The patients know there are no satisfactory standard treatments and that, based on preclinical research, scientists are testing a drug that might be safe and effective. Despite warnings to the contrary, these subjects characteristically enter trials on the chance they will benefit from access to a drug that works. But Phase I clinical trials are not supposed to be about efficacy: They are designed to assess pharmacokinetics and safety. The research subject is vulnerable—so the story goes—because he or she is driven by a false but persistent hope for a cure and, accordingly, is likely to enter the study out of an unreasonable expectation of success.
Consider, for example, a fairly common protocol used in Phase I cancer research. Successive cohorts receive escalated dosages, reaching a theoretically therapeutic range toward the end of the trial. There might be six cohorts with three patients each. The first begins to receive dosage D1 at time T1. After an interval, at time T2, a second cohort begins receiving higher dosage D2. Patients at D1 continue to receive the drug only until their tumors progress by some predetermined degree or serious adverse reactions to the drug begin to appear. Assuming no adverse reactions stop the study, successive cohorts continue to enter at increasing dosages until, at the end of the last interval, six cohorts have received escalated doses for fixed intervals and the study ends. Although evidence of therapeutic efficacy might appear, researchers are not supposed to be looking for it. If it seems the drug can be taken at theoretically therapeutic levels without serious adverse reactions, Phase II and Phase III trials will be run to establish efficacy and optimum dosage.
Now even if the drug is, in reality, both safe and effective, it is often unlikely that a medically exigent research subject can benefit from it. First, patients in the early cohorts may receive theoretically subtherapeutic dosages. While researchers might have some reason to believe the drug is safe and effective, they do not have any expectation that efficacy can appear at those low dosages. When tumors progress, as they are expected to, those patients are removed from the study. Accordingly, these subjects run the risk of an adverse reaction without a compensating theoretical chance of benefit. And second, even if efficacy were to appear, the trial can end, leaving in the lurch patients who may be improving. There is commonly no guarantee that the drug will be made available, beyond the end of the trial, to research subjects who might be benefiting from it.
Given the improbability of benefit, consent procedures in Phase I trials often emphasize that there can be no promise of improvement. (Importantly, promises of improvement are rare in medicine generally.) But notwithstanding the caveats in the consent forms, it is evident that hope for remission or cure motivates the majority of Phase I subjects. One solution might be to beef up the disclaimers in Phase I consents. C-Ss could be solemnly warned that, even if the drug works, they might not get a dose large enough to do any good and, even if they did get such a dose and, accordingly, began to recover, they still would not be allowed to continue on it after the trial ended.
G-9
But these admonitions are unnecessary. Instead I suggest that clinical trials on medically vulnerable patients, in addition to being structured as scientifically sound, also be designed to maximize the likelihood of subject benefit. Patients should be assured they will have a chance of benefiting from participation if it turns out that the drug is safe and effective.
Consider, for example, a redesign of the Phase I trial described above. Once again, the first cohort enters at time T1 at dosage D1. As before, a second cohort enters at T2 and D2. Assuming that, at T3, no serious adverse reactions have appeared for the subjects at D2, a third cohort then enters at D3 and those whose tumors have progressed in the first cohort may have their dosages raised to D2. In general, any subject whose tumor has progressed may advance to the next higher dosage, but only if and when no serious adverse reactions have occurred with the subjects who have just completed an interval at that dosage.
Under this design, subjects enter onto the study with the guarantee that there are only five ways in which they will come off it. Either (#1) they choose to leave the study, or (#2) they seriously fail to comply with the protocol, or (#3) significant adverse reactions are seen in response to the drug, or (#4) they die, or (#5) they are cured. While C-Ss should be assured that #5 is unlikely, the study design takes seriously the medically exigent patient’s overriding interest in maximizing the possibility of therapeutic benefit.
But it also turns out that this revised design improves the scientific output of the study. In the first place, while it generates the same dose-related toxicity data that the initial version did, the revised study is better at revealing cumulative toxicity. This is because patients can stay on the revised protocol longer, well after their tumors progress. And because it can become evident sooner that the intervention is unsafe, the research effort can be halted sooner, reducing wasted research funds. Second, there would be fewer dropouts under this arrangement, and participation might be more attractive. Third, in the event that tumor growth is slowed, stopped, or reversed, the revised Phase I trial can evolve gradually into an early Phase II trial, accelerating the demonstration of efficacy. Finally, it should be added that this design may be especially appropriate for biologic approaches to cancer: angiogenesis inhibitors, for example, as opposed to cytotoxic agents. Adverse reactions are less of a concern with these therapies, and it is not as critical to determine the maximum tolerated dose.
The redesigned study effects a fairer distribution of the benefits and burdens of cooperation. It is a less exploitative arrangement. Under this maximum therapeutic benefit standard, the primary concern would still be the scientific validity of the research design. But, having satisfied that requirement, the patient’s powerful interest in improvement would have to appear prominently on the researcher’s radar screen. It must be explicitly acknowledged that medical exigency can justify a departure from the norm separating research and therapy. The conjoining of these two different purposes is justified when 1) illness is severe and 2) no safe, effective, and otherwise satisfactory treatments are available. It becomes reasonable to swim with the crocodiles. While there would still be ineliminable risks associated with receiving an unproven treatment—and no basis for any promise of improvement—the researcher could truthfully say that the study is designed to give each subject the maximum likelihood of benefit if the drug turns out to be safe and effective. To be sure, that is still far less than these patients want, but it is also far more than most of them now receive.
Allocational Vulnerability
If the internal benefit of research is a safe and effective therapy, the external benefits are the various other compensations research subjects receive. The patient in a state of medical exigency may be desperate for the internal benefit of research: a cure with a return to health. But those in a state of allocational disadvantage are seriously lacking in other socially distributed goods: money, housing, medical care, childcare, burial benefits, opportunities to benefit the community, and so on. The question for the investigator is, “Is the C-S seriously lacking in important social goods that will be provided as a consequence of his or her participation in research?” (On occasion, it may also be pertinent to ask whether the C-S is seriously burdened with social evils that will be relieved as a consequence of participation. This issue is especially pertinent for research on prisoners.)
G-10
Now, broadly, if Job-Seeker is destitute and hungry, and Business-Owner offers him a good job at a decent wage, and Job-Seeker accepts (notwithstanding that it is the only acceptable option), we wouldn’t concern ourselves with the voluntariness of the acceptance so long as the terms of the arrangement were fair. But if, on the other hand, Business-Owner is offering sub-subsistence compensation, and the work is dangerous, and there are no workers’ compensation benefits for the injuries sustained, we are likely to invalidate the agreement. We will do this, not because Job-Seeker had no other choice, but because the bargain was unconscionably exploitative. As with medical exigency, the vulnerability is to be found in Job-Seeker’s precarious position: economic in this instance. But this allocational disadvantage should direct our attention to the substance of the bargain: Is it fair to the party in the weaker position? The minimum wage, job safety regulations, and workers’ compensation benefits are all broadly supported means of reducing such exploitation.
In biomedical research, the vulnerabilities associated with allocational disadvantage can arise in many ways. The researcher needs to ask whether the deprivation has lead to acceptance of an exploitative offer. For persons lacking access to health care, participation in a clinical trial may provide essential services they have gone without. Prisoners, having lost their liberty, reside in an environment that is carefully designed to shut off opportunities: They may have no other chance to be of service to their communities. Children, whose discretionary economic resources can be scant, may be eager to endure sacrifice for the sake of a toy store gift certificate. Soldiers might seek out exemption from combat duty. Psychology students may lack the credits required for a degree. While allocations are often the result of impersonal socio-economic forces, the basis for ethical concern is compounded when someone with juridic authority over the C-S is distributing the goods in question. Prisons and the military, for example, may function in this way.
While it is easy to identify the allocational disadvantages in some cases, it is often harder to discern the difference between just and unjust compensation packages. Of the six types of vulnerability, allocational disadvantage is probably the most problematic. We are often inclined to honor the view that, if a bargain is satisfactory to both parties, third parties should not interfere. But participation as a subject in medical research can impose risks and burdens that properly attract community attention. While we do not want to see people treated unfairly, we are not very confident applying the concept of the just price.
At a minimum, I suggest we consider the standards we routinely apply to other comparable remunerative activities. Although the point has been urged before, it is hard to grasp why research subjects should not normally be entitled to medical treatment for the injuries they suffer and why they should be asked to subsidize the research enterprise in that unusually burdensome way. Surely if we extended broad community standards into this aspect of research, we would begin by securing a right to some version of workers’ compensation.
Infrastructural Vulnerability
Although IRBs, researchers, and subjects often take them for granted, there are many protections and resources that contribute importantly to the safety of the research subject. When a consent form asks subjects to call a listed telephone number if they have a question or complaint, those phrases presuppose access to a telephone system. When a protocol requires the long-term use of frozen biological agents, that provision presupposes a reliable supply of electricity. When an investigational drug regimen has to be skillfully administered, the researchers may be assuming the availability of skilled health care professionals and a responsible independent local review mechanism. At the structural level, essential political, legal, regulative, institutional, and economic resources may be missing, leaving the subject open to heightened risk. The question for the researcher is, “Does the political, organizational, economic, and social context of the research setting possess the integrity and resources needed to manage the study?”
G-11
Although egregious failings are likely to be more common in international research—particularly in undeveloped areas—it should not be assumed that U.S. citizens will always enjoy the protections most of us take for granted. Increasingly we hear of ethically flawed research at well-known universities where investigators are plainly confused about the ethical dimensions of their work and the review and monitoring committees are untrained, underfunded, and understaffed. Where procedures permit the participation of IRB members with conflicts of interest, the disinterested review of protocols may be an illusion.
Clearly the possibility of infrastructural vulnerability calls for attention to the contexts within which the research will be done. To some extent, national or international certifying bodies may be able to carry out the fieldwork for such inquiry: It may not be feasible for American research institutions to assess the resources in communities on the other side of the planet. Perhaps single or multiple project assurances can be secured from international partners: Pertinent inquiries could be directed to them.
Recommendations and Concluding Reflections
I have reconnoitered the terrain of vulnerability in research subjects, offering what we believe to be a more productive, a more nuanced account of the topic. I have tried to provide criteria for six discrete types, describing how each can impair the connection between consent and permissibility, I have alluded to some of the issues researchers might address in undertaking to accommodate the special needs of the vulnerable.
In the light of that discussion, the primary recommendation of this paper is that the traditional focus on discrete vulnerable subpopulations must now give way to something like the analytical framework proposed above. It is not now possible to develop subparts for every allegedly vulnerable group, and, even if it were, the absence of clear criteria for admission can only result in the politicization of our mechanisms for the protection of human subjects. What is needed is clear thinking about the species of human precariousness and the ethical response each calls for in the context of clinical research. The development of subparts could follow, but only if they are informed by a defensible analytical framework.
In the course of discussion, a number of more specific recommendations have been made. While more needs to be said about all of these, two suggestions are worth underlining. First, clinical trials should take far more seriously the needs of medically vulnerable research subjects. While good scientific design is a sine qua non, researchers should also be required to consider how they might provide maximum therapeutic benefit for patients who have run out of options. And, second, we need to consider the fair entitlements of research subjects who are disadvantaged in economic and other ways. It is a worry that we may be tolerating unfair arrangements in the context of clinical research that we would not find acceptable elsewhere.
Although the point has not been developed, it should be clear that members of a population may exhibit several types of vulnerability. Indeed research subjects can illustrate all six. For example, an eight-year-old girl in a third-world country could display cognitive limitations, could be under the authority of her parents or village elders, could be exceedingly deferential to any adults who are respected by her parents, could suffer from a serious medical condition for which there are no available treatments, could be lacking in general medical attention that would be provided in the course of the study, and could live in an environment in which resources critical to the success of the study were not reliably available. Instead of developing a discrete subpart for children (and assuming that when those regulations were satisfied, research on a child could then proceed), the analytic focus recommended here would highlight six problematics, each requiring further inquiry and, potentially, the implementation of compensating mechanisms.
While it still might make sense to develop standards and regulations for recurring subpopulations, these could no doubt be improved by concerted attention to something like the taxonomy of vulnerabilities that is set out here. It is possible to envision the eventual development of a master matrix, the columns of which
G-12
would be subpopulations and the rows of which would be the pertinent vulnerabilities, each cell detailing the compensating measures that might address them. Initially, such a resource could be developed from a review of ideas already recorded in approved protocols and on internet-based bulletin boards, such as MCWIRB. It would take funds and a concerted organizational effort to bring forth such a tool, crafting it as a living consensus document, continually improved by broadly submitted commentary and authoritative updates by well-respected advisory boards. And yet the availability of web-based and hardcopy versions of the matrix could be the most effective means of helping researchers and IRB members to measure up to the highest ethical standards in their work. Having served on an IRB, I can attest to the potential usefulness of such a resource.
Finally, it seems that the sensitive understanding of vulnerability—the many precariousnesses that afflict the human condition—exposes a certain universality in these themes even while grounding a broader case for kindness and sensitivity. None of us is without some cognitive limitation. Everyone is subject to juridic authority, not all of which is wisely benevolent. Socialization itself entails patterns of deference. All of us face an eventual and too real prospect of medical exigency. And no one is immune from extreme need and the harms that can flow from deficits in the systems we count on to provide us with essential services and protections. Nor are researchers the only ones who need to learn how to engage the vulnerable with sensitivity and honor.
The topic surely has an importance extending beyond the boundaries of research ethics.
Notes
1 Katz, J., ed., Experimentation with Human Beings (New York: Russell Sage Foundation, 1972), pp. 633, 1007–10.
2 Katz, J., ed., Op. cit., pp. 9–43.
3 Pence, G., Classic Cases in Medical Ethics, second ed. (New York: McGraw-Hill, Inc., 1995), pp. 225–52.
4 The National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, Belmont Report: Ethical Principles and Guidelines for Research Involving Human Subjects. Federal Register Document 79-12065, April 18, 1979.
5 Advisory Committee on Human Radiation Experiments, Final Report (Washington, D.C.: U.S. Government Printing Office, 1995).
6 See Austin, J.L., How To Do Things With Words (Cambridge: Harvard University Press, 1975); and Fingarette, H., Confucius: The Secular As Sacred (Prospect Heights, Illinois: Waveland Press, 1998 [reissue]).
7 Hill, T. E., Jr., “Servility and Self-Respect,” The Monist 57:99, January 1973.
8 Kipnis, K., “Full Consent and the Legitimacy of Experimentation on Prisoners” in Sargent, L.T., ed., Consent: Concept, Capacity, Conditions, and Constraints (Wiesbaden: Franz Steiner Verlag GmbH, 1979), pp. 181–188.
9 Advisory Committee on Human Radiation Experiments, Op. cit., p. 823.
10 Grady, D., “For Experimental Treatments, ‘Somebody Has to Be First,’” New York Times, June 25, 2000, section 15, p. 11, col. 3.
11 Brody, B., “Research on the Vulnerable Sick” in Kahn, J.P., Mastroianni, A.C., and Sugarman, J., eds., Beyond Consent: Seeking Justice in Research (New York: Oxford University Press, 1998), pp. 32–46.
G-13

REFLECTIONS ON THE ORGANIZATIONAL LOCUS OF THE OFFICE FOR PROTECTION FROM RESEARCH RISKS
Commissioned Paper Charles R. McCarthy
H-1
Introduction
Part 1 of this paper is written under the assumption that any decision concerning the optimal organizational site within the U.S. government for the oversight of human subjects research by the Office for Protection from Research Risks (OPRR) should be informed and guided by the historical origins of OPRR; the legislative mandate under which OPRR currently operates; Department of Health and Human Services (DHHS)/Food and Drug Administration (FDA) regulations for the protection of human subjects; compliance issues and regulatory experience; and the Common Rule and OPRR’s interface with regulatory activities of other federal departments and agencies. Treatment of these issues will constitute the background sections of Part 1 of the paper. The final portions of Part 1 will provide findings and recommendations.
Part 2 of the paper addresses similar organizational considerations pertaining to OPRR’s responsibilities for assuring the humane care and use of laboratory animals. The organizational location of that responsibility will be considered in the light of the historical background of oversight responsibilities for humane care and use of laboratory animals, with special emphasis on two major noncompliance cases; OPRR’s legislative mandate regarding laboratory animals; OPRR’s relationship to the U.S. Department of Agriculture (USDA) and the Animal Welfare Act; and animal welfare compliance issues and experiences. As will be seen, the nature of OPRR’s responsibility for laboratory animals, although superficially similar to its responsibilities for human subjects protections, is substantively different from them. The oversight functions pertaining to the care and use of laboratory animals strongly suggests that it be separated from oversight of human research subjects and placed in a different organizational context. Optimal organizational location of the responsibility for laboratory animals will be discussed in the final portion of Part 2 of this paper under findings and recommendations. Appendix I of the paper will comment on recommendations raised by John C. Fletcher, Ph.D.
The author of this paper served as Director, OPRR, for 14 years, from 1978 until 1992. Prior to 1978, he collaborated for 8 years with OPRR (and its predecessor office, the Institutional Relations Branch (IRB) of the Division of Research Grants (DRG), National Institutes of Health (NIH)). Consequently, virtually all of his 23 years as a federal employee were spent in dealing with policies, issues, and organizational questions related to the protection of human research subjects and the humane care and use of laboratory animals. Much of the information found in the paper is publicly documented. However, some of the information is derived from the memory of the author. To a considerable extent, this paper manifests his reflections on a public career devoted, in large measure, to providing protections for the rights and well-being of research subjects and promoting the humane care and use of laboratory animals. Reference is made to some of the individuals who made decisions that affected OPRR. No effort has been made to evaluate all the reasons why those decisions were made or to evaluate the overall performance of these individuals. Some of their decisions, in the author’s opinion, had negative consequences for OPRR, but no criticism of their overall performance is intended or inferred.
Part 1: The Historical Origins of OPRR’s Responsibilities for Human Subjects
OPRR came into existence officially in 1972. However, it had existed in another form since 1964. To understand the relationship of OPRR to the institutions that are subject to the regulations administered by OPRR, it is useful to look at the functions of the office that was the predecessor to OPRR—the IRB of the DRG/NIH.
In the final year of World War II, the NIH annual research budget was less than $80 million. Even by the standards of U.S. government agencies in that period of history, NIH was a small agency. After World War II and throughout the next two decades, NIH budgets increased precipitously. For most of the decade of the 1950s, a major portion of the NIH budget was consumed by its intramural clinical research program. The NIH Clinical Center (subsequently named the Warren Grant Magnuson Clinical Center) opened its doors to research subjects in 1953. At that time it was a 500-bed, state-of-the-art research facility.1
H-3
In its early days, the NIH Clinical Center housed the largest and most respected clinical research program in the world. From the time it opened its doors in 1953, the Clinical Center operated under a policy for the protection of “normal” volunteers involved in research.2 Normal volunteers were recruited for many studies in order to establish baseline data against which to measure data pertaining to disease or to serve as normal controls in clinical trials. Whenever normal volunteers were to be involved in research, the Clinical Center policy required prior review and approval of proposed research designs by a disinterested committee of scientists called a Clinical Research Committee (CRC). The policy required that informed consent be obtained from normal volunteer subjects each time they were invited to serve as subjects of research.
The Clinical Center policy also called for CRC review of research that involved unusual hazards, but few research projects were identified as involving such hazards. For practical purposes, the policy affected only normal volunteers.
Potential research subjects whose disease or condition was under study were referred to the Clinical Center by their personal physicians. Typically such patient/subjects had already exhausted standard treatments for their disease or condition. In many cases their best prospects lay in research. They came to the Clinical Center in hopes of finding in research a cure or amelioration of their disease or condition not found in the standard practice of medicine. These patient/subjects saw little, if any, difference between “innovative therapy” by a physician (ministrations that exceeded the boundaries of the standard practice of medicine that were administered with the intent of providing a therapeutic benefit to the patient) and “research” (a systematic study designed to produce generalizable knowledge about disease or underlying biological functions, primarily intended for the benefit of society). Patient/subjects also came because the Clinical Center enjoyed the reputation of providing better quality care than most hospitals at no financial cost to the patient/subject.
Research investigators at the NIH usually regarded persons referred to NIH by their physicians as “patients,” rather than “research subjects.” Research was commonly referred to as “treatment” or “patient therapy.”3 Given that environment, it is not surprising that the NIH had no policy of protections for patient/subjects involved in research. The amount of information given to these “patients” was left to the discretion of research investigators who were viewed and who viewed themselves primarily as physicians.
In 1966 Dr. Jack Masur, Director of the Clinical Center, appointed a committee headed by Dr. Nathaniel Berlin to update the Clinical Center policy. Masur was responding, in part, to the U.S. Public Health Service (PHS) policy issued in February of 1966 by Surgeon General Stewart. Although technically not bound by the PHS policy, the revised Clinical Center policy adopted some, but by no means all, of the provisions of the PHS policy. CRCs were created in the clinical units of each categorical Institute within the NIH that conducted intramural research.4 Controversial research projects could be referred to the CRC of the Clinical Center Medical Board (the governing body of the Clinical Center). Patient consent was required only to the extent that the investigator was expected to make a note in each “patient’s” chart that verbal consent had been obtained.
Following World War II, the NIH annual budget increased substantially each year until 1969. After a brief hiatus in 1970–1971 it has continued to grow steadily until the present time. The budget expanded from $80 million in 1944 to more than $1 billion in 1969. Currently the budget has climbed to nearly $14 billion, and the prospects for further growth seem almost limitless. By the year 1964, the expansion of the NIH intramural research program had slowed, but extramural research—that is research funded by the NIH but conducted in institutions throughout the U.S. and in many other countries—continued to grow at an impressive rate. DRG conducted, on behalf of the Institutes that comprised the NIH, peer review for scientific merit of research proposals submitted to the NIH by institutions outside NIH.
Biomedical research funds are, legally speaking, awarded to research institutions, not to the principal investigators (PIs) who conduct research. Some of these awards raised technical or ethical problems not governed by
H-4
general policies. They required special attention. A process gradually developed within DRG for handling problems not covered by general policy. Such matters as, for example, research cost overruns, ownership of research equipment when a PI moved from one institution to another, or the provision of supplementary funds for promising research, were handled on a case-by-case basis. The IRB/DRG/NIH was created to deal with and settle such problems on an ad hoc basis. From the outset, IRB/DRG/NIH dealt with extramural research institutions by means of negotiation. Its decisions took into account not only the interests of the taxpayers and the policies of the NIH but the organizational structure, traditions, and policies of the research institutions where the research was conducted. The talents and preferences of the investigators and the rights and welfare of research subjects were also considered, wherever appropriate.
In this way, the IRB/DRG/NIH had already begun to provide some protections for human research subjects before the publication of the first policy for the protection of human subjects. From the time of its inception, negotiation characterized and comprised most of the work of the IRB/DRG/NIH office.
Prior to 1966, the NIH intramural research program lacked a comprehensive policy for the protection of human research subjects, and the NIH extramural research program provided no protections of any kind for research subjects. The events that brought into existence the extramural Policy for the Protection of Human Subjects in 1966 are already well documented elsewhere. They are treated here only in summary fashion.5 In summary, the 1966 PHS policy pertaining to subjects of extramural research was occasioned by findings of serious abuses of the rights and well-being of research subjects involved in biomedical research. Hearings conducted by Senator Estes Kefauver in 1958–19596 demonstrated that most drugs were tested on patients who were unaware that they were research subjects. The dramatic televised account of the thalidomide tragedy that culminated in the birth of hundreds of deformed infants in Europe and Canada focused public attention on the regulation of investigational drugs; experimental transplantation of a sheep’s heart into a cardiac patient without independent review and without informed consent;7 whole-body radiation experiments in Ohio and their cover-up by Senator Robert Taft;8 the introduction of live cancer cells into elderly, indigent charity patients without their consent by investigators at the Sloan-Kettering Cancer Foundation and Jewish Chronic Diseases Hospital;9 and the Willowbrook study involving deliberate introduction of hepatitis into severely retarded children.10 This made NIH officials aware that if research was to continue to enjoy public confidence and if it was to continue to be funded with public dollars, then a policy for the protection of research subjects must be conceived and implemented.11 After several years of deliberation on the part of NIH officials, Dr. James Shannon, Director, NIH, recommended that Surgeon General Stewart issue a comprehensive policy for the protection of human subjects on behalf of the U.S. PHS—the health agencies within the Department of Health Education and Welfare (DHEW)—of which NIH is the largest.
On February 8, 1966, Surgeon General Stewart issued Policy and Procedure Order 129,12 the first comprehensive extramural federal policy for the protection of human subjects. Responsibility for implementing the policy was assigned to the IRB/DRG/NIH. That tiny office undertook the task of implementing the policy in a manner consistent with the way it had always done business—that is to say, it negotiated assurances of compliance with the PHS policy with each of the awardee institutions.
The “assurance” negotiations enabled each institution to create its own internal policy for the protection of human subjects that both complied with the very general terms of the PHS policy and allowed the institution to develop compliance mechanisms and policies consistent with the organizational structure, traditions, and preferences of the institution. The negotiations also enabled federal staff to explain to institutional officials why the requirements for prior review and approval by an institutional committee (later designated an “Institutional Review Board”), and why requirements for eliciting informed consent from subjects were included in the policy. It also enabled the NIH, acting through the IRB/DRG/NIH, to teach institutions that their obligation to
H-5
respect the rights and welfare of human subjects is or should be as important as their obligation to conduct sound scientific studies.
From the outset, the IRB/DRG/NIH, unlike most federal regulatory agents, used education as the primary tool of promoting compliance with the new policy. Although that office had authority to withhold awarded funds from an institution found to be noncompliant with the policy, it never actually used that power (though it sometimes threatened to do so).
For more than ten years after the Policy for the Protection of Human Subjects was issued in 1966, the only sanction imposed on any research institution was the discontinuance of the Tuskegee Syphilis Study (housed at that time in the Centers for Disease Control and Prevention (CDC), one of the PHS agencies). That action was taken by the Assistant Secretary for Health outside of ordinary channels of policy oversight.
No doubt the IRB/DRG/NIH is open to criticism for relying solely on education, persuasion, negotiation, and occasional threats to bring about compliance with the 1966 policy. Nevertheless, IRB/DRG/NIH can be applauded for recognizing that biomedical research institutions and investigators subject to the policy are, by profession, dedicated to improving the quality of life of fellow human beings. As a consequence, with rare exceptions, researchers are anxious to respect the rights and welfare of research subjects. The IRB/DRG/NIH believed that the best, most efficient, and least costly method of promoting compliance with the policy was to raise the consciousness of investigators and administrators concerning their moral obligations to research subjects. The policy required minimally acceptable ethical standards. Assurance negotiations and education promoted a higher level of compliance than that literally required by the policy. This view has governed compliance efforts from the inception of the policy. It accounts, in part, for the fact that most institutions voluntarily apply federal standards to all research conducted in their institutions, not just to research that is funded by the federal government. Education and persuasion were then and remain today the most effective tools of policy implementation.
The February 1966 PHS Policy for the Protection of Human Subjects underwent minor revisions in the summer of 1966, and it was further clarified in 1967 and 1969. The 1969 clarification made it clear that the policy extended to behavioral and social science research as well as to biomedical research.
In 1971 the policy was extended to all research studies involving human subjects conducted or supported by any agency within the DHEW.13 Consistent with the educational approach described above, the DHEW policy—called the “Yellow Book” because of the color of the pamphlet in which it was published—set forth policy requirements that included: 1) institutional assurances of compliance; 2) risk-benefit analysis; 3) review by committee; and 4) subjects’ informed consent. Of greater importance, it included a running commentary, in a column parallel to the policy requirements, presenting reasons why these requirements were necessary to safeguard the rights and welfare of human research subjects. The commentary, written primarily by Donald S. Chalkley, Ph.D., Director, IRB/DRG/NIH, came to be regarded as a classical defense of subjects’ rights and well-being.
In 1971 the news media published accounts of the infamous Tuskegee Syphilis Study conducted by PHS scientists in which approximately 400 syphilitic African-American males were systematically denied treatment for their illness over a period of more than three decades. Details of that tragic and scandalous study are published elsewhere.14 One of the consequences of the Tuskegee episode was a speech delivered at the University of Virginia by Robert Q. Marston, Director, NIH, calling for additional protections for vulnerable research subjects.15 Following that speech in 1972, Marston upgraded the IRB/DRG/NIH. He changed the name from the Institutional Relations Branch to OPRR and incorporated it into the Office of the Director, NIH. He increased OPRR staff and ordered it to report to Dr. Ronald Lamont-Havers, Associate Director for Extramural Research. OPRR Director, Donald S. Chalkley, was subsequently promoted to the Senior Executive Service. The fact that
H-6
OPRR reported to the Deputy Director for Extramural Research, who was ultimately responsible for all research awards, placed OPRR in a position of potential conflict with its own supervisor. So long as Dr. Lamont-Havers served in that position, the system worked well. As will be seen, conflict arose some four years later.
Dr. Marston also created a task force under the direction of Dr. Lamont-Havers to consider how best to implement the recommendations outlined in his speech at the University of Virginia. The task force was expanded to include representatives of all of the PHS agencies. It incorporated into itself a committee chaired by Dr. Charles Lowe of the Institute for Child Health and Human Development that was already addressing the ethical questions of fetal research. The task force was organized into subcommittees that developed position papers dealing with research involving human fetuses, research involving children, research involving prisoners, and research involving physically, mentally, and socially handicapped persons.
These position papers, in various stages of completion, were eventually submitted to the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission or Commission). The Commission incorporated much of the work of the task force into its final reports.
NIH was not the only component of the DHEW that responded to the Tuskegee Study. The Assistant Secretary for Health, Dr. Monty DuVal, created an investigative task force, chaired by Professor Jay Katz of Yale University, to review the Tuskegee Study and to make recommendations for action. The study was terminated within a matter of days. The U.S. Congress, particularly the Senate Health Subcommittee, chaired by newly elected Senator Edward Kennedy (D. MA), held a series of hearings that continued periodically for more than two years. The Senate hearings were among the earliest congressional hearings to be televised. Coming as they did, after the civil rights debates of the 1960s, the hearings evoked public criticism of injustices toward African Americans. As a consequence of the television coverage and the resulting widespread public knowledge of abuses carried out under the Tuskegee Study, the hearings had a substantial impact.
The Kennedy hearings touched on many health issues besides the Tuskegee trial and the rights of human subjects, but they dealt primarily with research ethics and the regulation of research involving human subjects. One of the topics Senator Kennedy scheduled for hearings concerned research involving whole-body radiation conducted on military veterans in Cincinnati. However, Robert Taft (R. OH) accused Kennedy of “meddling” in the affairs of the State of Ohio. The powerful Senior Senator succeeded in quashing the hearings. Nevertheless, Kennedy was able to amend the appropriations of the Department of Defense (which at that time included the Department of Veterans Affairs) to require informed consent for all research conducted by that department.16 Hearings similar to those conducted in the Senate were held in the House of Representatives by the House Health Subcommittee chaired by Representative Paul Rogers (D. FL). Numerous bills and amendments to pending bills were introduced in both the Senate and the House of Representatives. Virtually all of the proposed bills called for promulgation of regulations for the protection of human subjects. However, the proposed legislation in the House of Representatives manifested a very different approach to the regulation of research than did the Senate bills.
Until it became apparent that issuance of regulations was inevitable, NIH had steadfastly opposed the issuance of regulations for the protection of human subjects. Donald Fredrickson, Scientific Director of the National Heart, Lung and Blood Institute (subsequently the Director, NIH) was fond of repeating in staff meetings, “NIH is not a regulatory agency.” By this he meant that, in his judgment, the fewer administrative encumbrances that scientists faced, the better would be their scientific production. Although his view of the utility of regulations changed after he became the NIH director,17 he always referred to the regulations for the protection of human subjects as “the policy.” The view that regulations could stifle research was shared by most intramural scientists of the time.
The Senate bill introduced by Senator Kennedy called for creation of a permanent federal regulatory commission for the protection of human subjects that would be patterned after the federal Securities and Exchange
H-7
Commission that regulates each transaction that takes place in the U.S. stock market. The proposed commission was to be a separate regulatory agency with broad investigative powers. It could bring criminal charges against those who violated its regulations, and it could assess punitive damages on persons and institutions that failed to protect research subjects. It would have authority to regulate research involving human subjects funded by the federal government and research conducted in the private sector, including research funded by foundations, pharmaceutical companies, medical device manufacturers, and private individuals.
The House bill sponsored by Mr. Rogers borrowed concepts from S.J. Res. 75 introduced by Senator Walter Mondale (D. MN). It called for the creation of a National (Advisory) Commission for the Protection of Human Subjects of Biomedical and Behavioral Research to make recommendations to the Secretary, DHEW, concerning the protection of human subjects, particularly vulnerable subjects such as prisoners, children, fetuses, and the cognitively impaired. Much of its mandate derived from the Marston speech at the University of Virginia.
Senator Kennedy made it known to DHEW that if the department were to issue regulations for the protection of human subjects, he would support the House bill proposed by Mr. Rogers. The department, which had steadfastly opposed the issuance of regulations up until that time, quickly formed a drafting committee to produce regulations that would, it was hoped, enlist the support of Senator Kennedy for the Rogers bill.
The PHS Drafting Committee was given only a few weeks to produce a new set of regulations. The committee, inexperienced in writing regulations and pressed for time, elected to transform into regulatory form the provisions in the 1971 Policy for the Protection of Human Subjects (Yellow Book) issued by DHEW. However, the resulting regulations lacked the commentary found in the Yellow Book that instructed Institutional Review Boards on how to interpret the rules. Because of the time pressure imposed by Senator Kennedy, the customary DHEW clearance points for the issuance of regulations were either bypassed or given extremely brief deadlines. The result was a set of flawed regulations that did not extend to intramural research, that lacked requirements for recordkeeping, and that allowed broad exceptions to requirements for informed consent. On May 30, 1974, DHEW promulgated Regulations for the Protection of Human Subjects, at Title 45 Part 46 of the Code of Federal Regulations. Although the new regulations were little different in content from the DHEW Yellow Book, and although they lacked the educational commentary of the Yellow Book, they enjoyed the force of law. Senator Kennedy expressed himself as satisfied that DHEW was serious about protecting human subjects, and he agreed to back the Rogers bill.
I. The Legislative Mandate Under Which OPRR Currently Operates
Soon after Senator Kennedy lent his support to the Rogers bill, it was passed by both houses of Congress and enacted into PL 93-348, the National Research Act, signed into law on July 12, 1974. Title II of that act created the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research. By the time the National Commission completed its work in 1978, it had issued 17 major reports that included approximately 125 recommendations to the Secretary, DHEW. Many similar recommendations had been submitted to the Commission by the PHS Task Force and were supported by Donald S. Chalkley, Director, OPRR, and by Dr. Ronald Lamont-Havers.
One of the reasons that the National Commission exercised such a profound effect on regulations for the protection of human research subjects was the so-called forcing clause in the act that required the Secretary, DHEW, to accept the Commission’s recommendations or publish in the Federal Register reasons for not accepting them. Rather than go on record as opposing an ethics commission that had studied the issue for four years, DHEW Secretaries (Matthews, Califano, and Harris) accepted all of the Commission’s recommendations.
Among the many provisions in the National Research Act was a section that amended the Public Health Service Act. That section has now been updated and is currently incorporated in Sec. 491 of the Health Research Extension Act of 1985. The law requires the Secretary to issue regulations requiring Institutional
H-8
Review Board review and approval of all research involving human subjects (including intramural research) prior to funding (Sec. 491(a)). Additionally, however, Sec 491(b) requires that:
The Secretary shall establish a program within the Department of Health and Human Services under which requests for clarification and guidance with respect to ethical issues raised in connection with biomedical or behavioral research involving human subjects are responded to promptly and appropriately.
That section is incorporated in the act because Mr. Rogers, its primary sponsor, developed information in hearings before his subcommittee that supported the contention that the PHS policy, in existence since 1966, had been successful in part because of the educational efforts of the IRB (subsequently OPRR). The legislative history makes it abundantly clear that the law intends the department, through the OPRR, to promote a sound understanding of the ethics of research in all institutions that receive DHEW funding.
Section 491(c) of the act calls for prompt actions to enforce the regulations. It is interesting to note that the wording presumes that instances of noncompliance will be reported to DHEW. While this has not always been the case, very often educational efforts have emboldened whistle blowers to identify noncompliance with the regulations.
The author knows of no other federal regulatory mandate that includes a requirement for a program of guidance and education to accompany its regulatory effort. Beginning in 1978, OPRR subsidized a series of regional education programs for the protection of human research subjects. They were conducted in every segment of the country. Costs to participants were nominal. The growing number of Ph.D. level ethicists from universities across the country provided willing faculty leadership. In turn, the program provided visibility for these promising young scholars and high quality content to the educational programs. Coupled with intensive bioethics programs at the Kennedy Institute of Ethics at Georgetown University and efforts of a rapidly maturing community of bioethics scholars in America, the program enjoyed enormous success. One measure of its success was the number of telephone calls that poured into OPRR seeking guidance on difficult or controversial ethical issues.
At one point, in the mid-1980s, the number of daily incoming calls to professional staff in OPRR, largely from PIs and Institutional Review Board chairpersons, approached 200 per day during the academic year. The negotiation of Assurances of Compliance continued to be a means by which research institutions were periodically required to review and update their internal policies and procedures for the protection of human subjects. The negotiation associated with the assurance process continues to have some educational currency for research administrators who are expected to issue policies for their institutions and who are held personally responsible for the rights and well-being of research subjects in their institutions.
Nevertheless, in the opinion of this author, the process of negotiating assurances of compliance has become routinized. Institutions tend to reissue their policies with little reflection and little upgrading, and OPRR no longer travels to each institution in an effort to blend federal laws and regulations with institutional traditions and history. The transactions now take place via mail, telephone, and electronic communication. Thus the assurances of compliance have become a heavy administrative burden for OPRR. Worse, the assurance process has lost much of its original educational purpose. It needs to be replaced with a simple certification and by intense educational efforts that take a new form.
Although OPRR’s regional educational programs have continued to the present time, the federal subsidy began to shrink in the Reagan administration, and it largely disappeared in the Bush administration. It has not been restored by the Clinton administration, despite the fact that it has put more public effort and money into uncovering radiation research injustices that occurred in the years prior to the existence of regulations than it has into protecting subjects in the present time. Institutions are now required to underwrite the educational
H-9
efforts initiated by OPRR, which lacks funds to fully support the program. The number of programs has dwindled to about four regional programs per year.
In 1978 Dr. Lamont-Havers was upgraded to Deputy Director and was replaced by Dr. Thomas Malone. Dr. Malone continued to give the same level of support for OPRR and for the protection of research subjects begun four years earlier by Dr. Marston. Dr. Malone headed the search committee that selected Dr. McCarthy to succeed Dr. Chalkley, who retired in 1978.
When Dr. Malone was appointed Deputy Director, NIH, he continued to ask OPRR to report to him. However, when Dr. Malone was replaced by Dr. William Raub as Deputy Director, Raub ordered OPRR to report to the new Associate Director for Extramural Research, Dr. Kathryn Bick.18 The legal advisor to the PHS advised Dr. Raub at the time that to return to the previous arrangement in which OPRR reported directly to the Deputy Director for Extramural Research was to risk a conflict of interest. The reasoning of the Office of General Counsel was clear. Since OPRR was to exercise oversight authority over research projects that bore the stamp of approval of its immediate supervisor—the Deputy Director for Extramural Research—OPRR was placed in a position where it might have to overrule or criticize actions taken by its boss.
Dr. Bick had previously been employed as Deputy Director of the Neurology Institute (NINDS) which funded several animal studies that were discontinued by OPRR for their lack of compliance with the PHS Policy on Humane Care and Use of Laboratory Animals.19 The Neurology Institute had been severely criticized in the public media for funding these studies.
Shortly after Dr. Bick was named Deputy Director for Extramural Research, she froze personnel hiring in OPRR, cut its travel budget, and dramatically reduced its education budget. Her deputy was Dr. George Galasso, who succeeded her as Acting Deputy Director for Extramural Research. Dr. Galasso continued Dr. Bick’s policies of constraint of OPRR.
Dr. Bick also initiated a policy that required institutions that are subject to the regulations to underwrite the educational efforts initiated by OPRR. Consequently, the OPRR educational effort was overshadowed by the appearance of conflict of interest.
OPRR, a regulatory office, was forced (by lack of funds to fulfill its own legislative mandate) to invite regulated institutions to subsidize its programs of education. Support of such a program can cost the regulated institution upwards of $10,000. To refuse to host a program is perceived to be a risk of offending a regulatory office with power to interdict research monies flowing from the government to the awardee institution.
The Deputy Director for Extramural Research is the line supervisor of the Director, OPRR. Turning a deaf ear to OPRR’s appeals to the contrary, Dr. Bick ordered OPRR to carry out its educational mandate by asking regulated institutions to provide funds for its programs. Even though OPRR’s intentions were benign, the appearance of coercion was present. Dr. Bick also prohibited OPRR personnel from participating in programs operated by two Boston-based nonprofit organizations, Public Responsibility in Medicine and Research (PRIM&R) and Applied Research Ethics National Association (ARENA). PRIM&R has grown into a national organization whose national meetings dealing with the ethical and regulatory aspects of research involving human subjects are attended by more than 700 people. ARENA members are mostly Institutional Review Board administrators, Institutional Review Board members, and Institutional Review Board staff persons who exchange practical information on efficient methods for protecting human research subjects in institutions throughout the country. PRIM&R and ARENA address issues of interest, not only to institutions whose research is funded by federal agencies, but to institutions regulated by the FDA as well.
The policy of requiring awardee institutions to subsidize education programs was initiated by Dr. Bick and continued by her successors, Dr. Galasso, Acting, and Dr. Diggs. The potential conflict of interest has cast a shadow of suspicion on the educational efforts of OPRR, an office whose success demands impartiality and whose legislative mandate requires an education outreach. This situation should be changed.
H-10
As a part of its educational outreach, OPRR has worked closely with PRIM&R and ARENA. Educational efforts in the private sector, particularly those of PRIM&R and ARENA have partially supplied for the decline of OPRR-sponsored programs. Nevertheless, because the OPRR programs are regional and low cost and because they are official, they reach persons who do not attend the national meetings of PRIM&R and ARENA.
When OPRR educational programs were flourishing in the early and mid-1980s, the number of noncom-pliance cases reported to OPRR dwindled. Conversely, as OPRR educational programs have declined, numbers of noncompliance cases have risen dramatically. (The number of backlogged cases was said by an OPRR official to be about 150 about a year ago.) Although a direct correlation between preventive educational efforts and reduction in cases of alleged non-compliance cannot be demonstrated, it is reasonable to hypothesize that improved education efforts reduce noncompliance. Education efforts are far less costly than compliance investigations. Therefore, in the opinion of this author, a decrease in educational funding has contributed to an increase in compliance costs.
Only about half of the cases of alleged noncompliance actually demonstrate noncompliance. Only a small fraction of those cases where noncompliance is demonstrated involve direct physical harms to subjects, but all noncompliance involves an erosion of the rights of subjects. Education therefore prevents both harms to the welfare of subjects and damage to their rights.
II. DHHS/FDA Regulations for the Protection of Human Subjects
The National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research completed its tasks and was disbanded in 1978. Responsibility for implementing the Commission’s recommendations was delegated by the Secretary, DHEW, to the Director, OPRR. OPRR organized a Human Subjects Regulations Drafting Committee that included representatives of all of the relevant agencies within the DHEW, including the Office of the Secretary. Mr. Richard Riseberg, Office of General Counsel, played a key role on the committee. The committee scrapped the 1974 version of Regulations for the Protection of Human Subjects and rewrote them in the light of 1) the recommendations made by the National Commission; 2) public comments on the Commission’s reports and on proposed rulemaking; and 3) public hearings on proposed rulemaking.
A major step forward occurred when FDA, with encouragement from the Secretary’s office and leadership from Dr. John Petricianni of FDA’s Bioresearch Monitoring Program allowed the Drafting Committee to redraft FDA regulations for Clinical Investigations and Informed Consent (21 CFR50 &56) so that the FDA regulations reflected the recommendations of the National Commission and were, in nearly all respects, congruent with the DHHS regulations. The DHHS regulations differ from those of the FDA in three ways: 1) DHHS regulations allow a waiver of informed consent under certain limited circumstances, whereas FDA regulations allowed no such limited waiver;20 2) the FDA regulations do not require regulated institutions to negotiate assurances of compliance prior to IRB review and approval of research involving human subjects, whereas DHHS regulations do require negotiation of assurances (thus placing FDA in the position of having to approve IRBs after they complete their work, rather than before); and 3) FDA regulations require inclusion of a statement in all consent documents that informs subjects that FDA personnel may review their records. In all other respects the DHHS regulations that pertain to federally funded research and FDA regulations that apply to research carried out in the private sector are identical.
Both the DHHS Regulations for the Protection of Human Subjects and the FDA Regulations for Clinical Research and Informed Consent were signed by DHHS Secretary Harris on January 19, 1981, one day before the Reagan administration replaced the Carter administration.
The regulatory significance of the melding of FDA and DHHS regulations is difficult to overstate. It has had a salutary effect on research ethics that far exceeds that of the Common Rule. Hundreds of institutions that had previously been required to follow two sets of regulations were now able to follow a single set of rules. The
H-11
consequence has been that institutions can operate under a single internal policy for the protection of human subjects. This made it both feasible and attractive to extend the same protections to all human research subjects, irrespective of the source of funding.
Furthermore, it was now practical for FDA to join OPRR in educational efforts. Joint OPRR/FDA educational programs could now reach out, not only to universities and clinics that conduct federally supported research, but to research foundations, pharmaceutical houses, device manufacturers, small businesses, and research data banks. Finally, the DHHS/FDA congruent regulations allow the FDA and OPRR to share compliance information and to cooperate in investigations of alleged noncompliance.
Because the FDA budget for education programs was virtually nil, the issuance of congruent regulations and the resulting partnership in education placed further strains on the education budget of OPRR. Nevertheless, the partnership has proved to be a valuable and workable, if financially strapped, arrangement.
An unknown fraction of research activities involving human subjects remains unregulated. Research studies are not covered by either DHHS regulations or FDA regulations if the research is conducted by private sector institutions that 1) enjoy no federal support and 2) are not covered by either DHHS or FDA regulations because they involve no drugs, biologics, or medical devices. Failure to regulate such research constitutes a double standard that sends a message that the government has less concern for subjects of research conducted by unconventional sources than it does for other subjects.21 The publication of the DHHS/FDA congruent regulations, updated in the light of the findings and recommendations of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, propelled OPRR into the role as the lead agency within the federal government for the protection of human research subjects.
Because responsibility for implementing FDA regulations is spread across the three major FDA Centers—Drugs, Biologics, and Medical Devices—there is no central office within the FDA that exercises direct line authority over research protections, although Dr. Stuart Nightingale has exercised strong leadership in this area for many years. Because FDA has no central authority within its organization, it cannot exercise leadership across other federal agencies. That leadership has been centered on OPRR and is taken for granted by most federal agencies since the close of the National Commission’s deliberations in 1978. Nevertheless, OPRR has never been given legal authority, personnel, the prominence, or the funding to play that role properly. The role has always been an add-on responsibility for which no personnel or funding has been provided.
III. Compliance Issues22
One of the many advances accomplished by the promulgation of the new Regulations for the Protection of Human Subjects (45 CFR 46) in 1981 was a clarification of responsibilities of institutions and research investigators. Because of ambiguities and lacunae—particularly with respect to reporting and record keeping requirements—in the 1974 version of the regulations, it was difficult (between 1974 and 1981) to demonstrate whether a given research activity fell within or outside of the regulations. Without adequate records, it was often impossible to develop clear findings of noncompliance, and consequently it was difficult to evaluate allegations of noncompliance and to impose sanctions on institutions or investigators who were noncompliant. These shortcoming were corrected in the 1981 version of the rules.
One of the difficulties faced by OPRR was the unwillingness of the NIH intramural program to comply with the 1981 version of regulations for the protection of human subjects. Although the Clinical Center was technically out of compliance during the period 1974–1981, it followed a policy very similar to the DHEW policy. When the DHEW regulations were updated in 1981, OPRR was informed by the Director of the Clinical Center that the intramural program would not negotiate an assurance of compliance with the new regulations but would continue to follow its own internal rules. The Director, OPRR, turned to the Director, NIH, for
H-12
backing, but was bluntly told to “leave the Clinical Center alone.” Clearly this was a case of an abuse of authority and an open conflict of interest.
Nevertheless, the Director, OPRR, notified the Director of the Clinical Center, Dr. Mortimer Lipset, that he would inform the public media that all of the Clinical Center studies, including a National Institute of Mental Health sleep study in which a subject unexpectedly died, were being conducted out of compliance with federal rules. Within 24 hours, the Clinical Center initiated the process of negotiating the required assurance. As it happened, the death of the subject in the National Institute of Mental Health sleep study was not caused by the research, but by an unreported health condition of the subject herself. Negligence in screening subjects (the young woman who died had a condition that would have excluded her from the study) and negligence in using and monitoring faulty equipment contributed to the subject’s death.
With the assistance of the DHHS Secretary’s Office of General Counsel, the 1981 version of the regulations coupled with the assurances of compliance signed by senior executives in the research institutions made it possible, in most cases, to determine whether research was conducted in accordance with the rules. For example, the 1981 regulations required records of all research protocols, records of all decisions made by IRBs, and retention of informed consent documents. These requirements simplified compliance evaluations. No longer could an institution plead the excuse that records were unavailable to determine whether a violation had occurred, because lack of careful record keeping was itself a violation of the regulations. As it turned out, careful record keeping exonerated many studies where alleged violations were claimed.
The education program of OPRR stressed that violation of the rights of subjects would not be tolerated and that whistle blowers would be, so far as possible, protected. (No whistle blower was ever publicly identified during the years 1981–1992, except in cases where the whistle blower chose to identify himself/herself.) Furthermore, OPRR taught administrators that if they identified noncompliance in their own institutions and notified OPRR, they would be allowed to correct the situation without automatically triggering a federal investigation. Of course, a full report of the institution’s findings and corrective action(s) would be forwarded to OPRR for review. Follow-up reports were also periodically required. If OPRR found that the investigation had been thorough and the institution’s corrective action had been appropriate, the case was closed. For many institutions that meant that adverse publicity about the institution was avoided. The system worked remarkably well.
In complex cases, institutions often invited OPRR to join with the institution in carrying out an investigation. This cooperation was fostered by the education programs that made it clear that OPRR and institutions both had a stake in assuring compliance with the regulations.23 A few examples may be illustrative:
| A. |
Martin Cline, M.D., was an investigator at UCLA. He submitted a protocol to the UCLA medical Institutional Review Board, which deliberated for nearly a year but never approved Dr. Cline’s research. The protocol called for administering recombinant genetic material to thalassemia patients. The Board consulted with a number of experts, but was never satisfied that the animal data supported an attempt to carry out the procedure in humans. Dr. Cline went to Italy and carried out the research in the clinic of a colleague. He then went to Israel and was able to obtain Institutional Review Board approval of a falsified version of his recombinant DNA protocol. Cline involved several patients from Israel in his study, which was in reality his UCLA protocol. A whistle blower notified OPRR, which conducted a thorough investigation. UCLA readily supplied Institutional Review Board information that confirmed Dr. Cline’s noncompliance. UCLA removed Dr. Cline from his role as a department head, and NIH declared him ineligible to compete for awards to carry out human subjects research. |
H-13
| B. |
Robert P. Gale, M.D., also from UCLA, wished to conduct rescue research in leukemia patients. These patient/subjects, all of whom were considered terminal patients, were, according to Gale’s protocol, to donate their own bone marrow, which was treated to make it disease free and stored. Then, drugs would be administered to kill all of the patient/subject’s remaining marrow. The patient/subject’s stored allogeneic marrow would then be reinserted in hopes of “rescuing” him or her from death due to loss of bone marrow. Dr. Gale failed to obtain approval from the UCLA Institutional Review Board, so he falsified IRB approval documents and falsified consent documents to indicate that they had been Institutional Review Board-approved. All of the patients died. A nurse recognized the consent document that Dr. Gale was using as bearing a number that was approved for a different research project. She checked with the Institutional Review Board and found the consent document was never approved for Dr. Gale’s protocol. After a careful investigation by a disinterested faculty committee, Dr. Gale was notified by the academic Senate that he would never be promoted. He was given a “Scarlet Letter” punishment by OPRR—that is, a description of his infractions would accompany all of his future award applications. He was never given another federal award. |
| C. |
In rare cases, institutions failed to cooperate with OPRR investigations. One such case involved Dr. Mark Straus of Boston University who was accused by the Eastern Cooperative Oncology Group of falsifying data in a cancer research project funded by the National Cancer Institute. The university fired Dr. Straus for unspecified reasons. Then the university claimed that because Straus was no longer employed by it, it had no responsibility to cooperate with the OPRR investigation. The university failed to sequester the data in question, so that the accused and others had the opportunity to destroy or alter records after the investigation began. The President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research also held hearings in Boston concerning this case. Witnesses were not sworn, and cross examination did not take place. Inaccurate information was delivered to the commission, and unsupported accusations were made by witnesses against one another. The hearing led to inaccurate press reports. The President’s Commission hearings made it more difficult for OPRR to discover the truth in the matter and to have Dr. Straus acknowledge that truth. It took nearly three years to locate all of the laboratory reports and patient records from other hospitals to reconstruct patient records and demonstrate that, indeed, Dr. Straus and no one else had falsified research data to such an extent that he had placed subjects at significant risk. Dr. Straus was debarred for four years from eligibility to compete for federal research support. Approximately $7 million was recovered by NIH from a $19 million grant. The investigation cost more than $3 million.24 |
| D. |
On at least one occasion, the Director, NIH, appeared to be taking a step toward interfering with an OPRR investigation. In the same case, a member of Congress urged punishment for the accused before all of the evidence was evaluated. Dr. Robert Gallo, a prominent NIH intramural scientist, was credited by many with discovering HIV. (Crediting Gallo with this finding was disputed by French scientists who claimed Gallo stole their findings.) |
The case proceeded as follows: Dr. Gallo developed material in his laboratory that stimulated immune responses in laboratory animals. He forwarded the material to a colleague, Dr. Zagury, in Paris, France. Dr. Zagury modified the material and injected it into terminally ill human patient/subjects with advanced AIDS. Evaluation by French officials showed that the treatment hastened the death of several of Zagury’s patient/ subjects. Dr. Zagury also used the material to develop a “vaccine for AIDS” that was injected into a number of citizens of Zaire. Some of Dr. Zagury’s Zairian laboratory workers and Dr. Zagury himself were also injected with the material. A brief summary of this research project was published in Nature magazine. Drs. Zagury and Gallo were identified as primary and secondary investigators. Alert NIH employees called the article to the
H-14
attention of OPRR. They indicated that the preparation had been clearly labeled “for use in laboratory animals only.” OPRR investigated and found the facts to be as described above. Dr. Gallo did not deny the facts, but he pleaded that the regulations do not apply to him because he is a bench scientist who had no direct contact with human subjects. Nevertheless, Dr. Gallo was severely reprimanded for collaborating with a clinician in research involving human subjects that was conducted in violation of the regulations. As a result of this case, records of all shipments out of NIH intramural laboratories have been monitored. The French government administered sanctions to Dr. Zagury. Dr. Zagury was also declared ineligible to compete for future NIH awards.
While the Gallo/Zagury investigation was under way, newly appointed Dr. Bernadine Healy, Director, NIH, sent a strongly worded memorandum to the Director, OPRR, directing him to give her a full accounting of the status of the Gallo investigation. She sent a similar memorandum to the Office of Research Integrity (ORI), which was examining the French claims that Dr. Gallo had “stolen” the credit for discovering the HIV virus from French scientists.
The Director, OPRR, responded to Dr. Healy by memorandum stating that briefing her could appear to be a conflict of interest because the investigation concerned alleged misconduct by one of her most prestigious employees. The Director, OPRR politely declined to provide the briefing. The Director of ORI gave Dr. Healy the requested briefing. Subsequently Dr. Healy was severely criticized in a congressional hearing by Rep. John Dingell (D. MI) for interfering with the investigation carried out by ORI.
In the meantime, Mr. Dingell, Chairman of the House Energy and Commerce Subcommittee on Oversight and Investigations, directed his own investigative staff to interrogate OPRR on the status of its investigation of Dr. Gallo. OPRR provided congressional staff only with information that had already appeared in the public media. However, the legal implications of denying investigative material to a congressional oversight committee were not clear. The Office of General Counsel had advised OPRR to surrender all of the relevant information. Mr. Dingell chose not to make an issue of OPRR’s failure to provide him with investigative information, but he publicly criticized OPRR for the slowness of its investigation. His own staff began a parallel investigation. The Dingell staff traveled to Paris but were rebuffed by the French government. The French, on the other hand, prompted by interventions from the U.S. Department of State and the NIH Fogarty International Center, provided information to OPRR about Dr. Zagury through their Health Attaché in the French Embassy. Under pressure from the French government, Zagury, accompanied by his assistants, traveled at his own expense to NIH and provided significant information. Because of political turmoil and violence in Zaire and tensions between Zaire and the U.S. government at that time, complete records from that country were impossible to obtain. Nevertheless OPRR was able to get enough information to complete its report, take action, and close the case.
In an exit interview several years after the Gallo/Zagury case, Dr. Healy acknowledged that she regarded OPRR’s failure to brief her as an act of defiance that infuriated her. Only after she was criticized by Mr. Dingell for interfering with the ORI investigation did she come to believe that OPRR’s action was in the public interest.
These cases illustrate different kinds of situations that can face OPRR. The Cline case required an astute whistle blower to bring it to the attention of OPRR. No amount of oversight would have enabled OPRR to uncover secret noncompliant activity by a U.S. investigator in Italy and in Israel. It was necessary for a well-informed scientist to recognize the situation and to report it. Once reported, it was necessary for OPRR to have access to competent scientists to evaluate the protocol as proposed and as actually conducted. This case teaches us that OPRR must not only have persons competent in clinical research on its staff, but it must have the ability to consult with experts in order to base regulatory decisions on a clear understanding of the evidence, including the scientific evidence. At the present time, OPRR has no permanent physician with clinical research background on its staff. It relies on two part-time volunteers for assistance in this area.
H-15
The Gale case also required a whistle blower. In this case the whistle blower was an alert nurse. No oversight of the situation would have uncovered the fraud without help from inside the institution. OPRR’s limited resources were, in this case, greatly enhanced by the full cooperation of the UCLA administration and a disinterested faculty committee determined to learn what actually happened and to take appropriate steps.
The Straus Case illustrates how difficult it is for OPRR to function without the assistance of the regulated institution. Future regulations may need to address the obligation of the institution to assist the government in evaluating compliance. Straus was extremely clever. His case cost OPRR—with invaluable assistance from the NIH Division of Management Survey and Review (usually involved with investigation of fiscal mismanagement or fraud)—hundreds of hours of precious staff time.
The Gallo/Zagury case illustrates the fact that at times OPRR must have high political backing. The case was resolved only because the Department of State and the NIH Fogarty International Center had relationships of trust with the Health Attaché in the French Embassy. On the surface, neither the Director, NIH, nor Congressman Dingell and his staff actually did anything wrong. Yet OPRR felt that signals as to how the case should be adjudicated were being given by powerful political forces—the Director, NIH, to whom OPRR must turn for personnel, budget, and cooperation, and a powerful chairman of a congressional investigative committee. Part of the challenge of finding the proper organizational locus for OPRR is to give OPRR the political backing it needs to withstand pressure from highly placed leaders in the Congress or other agencies in the executive branch, including the White House itself. OPRR would not survive very long if it were a separate agency. OPRR must also be protected against interference by its own supervisors.
IV. Development of the Common Rule25
In December, 1981, the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research recommended the following: 1) All federal agencies should adopt the regulations of DHHS (45 CFR 46); 2) the Secretary, DHHS, should establish an office to coordinate and monitor government-wide implementation of the regulations; and 3) each federal agency should apply one set of rules consistently to research conducted or supported by the federal government.26 The Secretary, DHHS, through the Assistant Secretary for Health, designated OPRR as the “lead” office to develop a common set of regulations across the government. However, OPRR was dealing with reduced budgets and severe downsizing restrictions. Requests for personnel and salaries to carry out the task were quickly denied by NIH, which refused to forward the requests to the Office of the Secretary. Since most offices within the department were facing downsizing and OMB placed each department and agency under personnel and budget ceilings, it is unlikely that OPRR’s request would have been approved even if it had gone forward to the Secretary.
OPRR approached each agency in the federal system with a request for compliance with the recommendation of the President’s Commission. Most agencies sent an employee to the organizational meeting, but they delivered messages that stated—in effect—that they had no locus for human subjects protections, that they had no budget for such protections, and that they too were downsizing and operating under an Office of Management and Budget directive that no office or function could be added in a federal agency unless an equivalent function was discontinued.
Nevertheless, OPRR was able to obtain some backing from the OMB on grounds that what was being proposed was a simplification of regulatory structure. With prodding from OMB and nagging from OPRR (which had no authority to require action), the agencies finally agreed to review DHHS regulations. The response was disheartening. Each agency agreed to promulgate DHHS regulations, so long as it was able to add clauses of exception or additional protections to the DHHS rules. Literally dozens of exceptions were proposed. Had action been taken at that point, there would have been no Common Rule. The Department of Education
H-16
(DOE), for example, agreed to follow the Common Rule on condition that it could add an additional subsection dealing with protection for the rights and welfare of handicapped persons. The Department of Agriculture and the Environmental Protection Agency sought exceptions for pesticide research and food testing research. The Department of Justice sought an exception for research conducted in federal prisons. On the other hand, OMB said that no variation of any kind from DHHS rules would be allowed.
OPRR was able to persuade most agencies to drop their request for modification, but DOE was adamant. Neither DHHS nor any other agency would accept the DOE proposals. DOE refused to drop its demands. A DOE political appointee, Madeline Will, who enjoyed the friendship of the President refused to yield. OMB would not proceed without DOE. After nearly three years of standoff, there was a change in personnel at DOE, and progress toward producing a Common Rule began again. The turnover in leadership that marked the change from the Reagan administration to the Bush administration, returned the project to its starting point. No one in the Bush administration felt obligated to honor commitments made during the Reagan administration. OPRR had no authority to force the issue, but it turned to the Office of Science and Technology Policy (OSTP) headed by the President’s Science Advisor for assistance.
Armed with support from both OSTP and OMB, where a change in personnel had reduced rigidity, drafting progress was made. However, the legal advisor to the President refused to approve the final draft because, in his opinion, the requirement that each IRB include both men and women constituted a quota. The Bush administration was on record as opposing all quotas and considered them to be illegal. OPRR then turned to the DHHS Office of General Counsel for assistance. After several months and many meetings, a rewording of the IRB membership clause won approval from the White House. Armed with new wording in the regulation and strong support from OSTP, OPRR once again initiated a clearance process in each of the affected departments and agencies. Finally on June 18, 1991, 16 departments and agencies simultaneously published the Common Rule.
Given the difficulty of the getting so many departments and agencies to agree on the rule, serious questions concerning any further changes in the rule are raised. Unless the process is altered, the rule is fixed for perpetuity.
V. Findings
Note that the findings and recommendations below relate not only to the optimal organizational locus of OPRR but to its relations to 1) other federal components with ethics responsibilities; 2) staffing; 3) OPRR’s responsibilities; and 4) OPRR’s functions. The author believes these items cannot be separated.
| 1. |
The historical functioning of the IRB/DRG/NIH and the OPRR suggests that when the office responsible for the rights and welfare of human subjects is conducting a significant level of educational effort, it achieves the highest level of compliance. When funding for its educational function is decreased, it tends to have many more compliance problems. Therefore, a constant and reliable funding mechanism for a major educational outreach should be included in the mission of this unit wherever it is finally located. |
| 2. |
The legislative authority under which OPRR operates is delegated to it by the Secretary, DHHS. That legislation requires regulation, education, and compliance. These functions should be retained in any new organizational configuration. They should be carefully distinguished, but they must also be balanced and coordinated. |
| 3. |
The present setting of OPRR constitutes an apparent conflict of interest. As noted above, on a number of occasions in the past this appearance manifested itself as a reality. Potential conflict surfaces as a concern each time OPRR forwards its proposed budget request to the agency that it regulates. OPRR should not regulate the agency within which it is located and to whom it looks for funding, personnel, promotions, and |
H-17
staff honors and bonuses. Furthermore, OPRR often has difficulty in enforcing rules on research conducted or supported by the CDC and other components of PHS. This is true, in part, because OPRR is regarded as being a small part of a sister agency, rather than a representative of the Secretary, DHHS, over all research within the department.
| 4. |
OPRR must be in a position to obtain technical assistance in a wide variety of disciplines. This is important for educational, compliance, and legal issues. OPRR must therefore be in a position to seek advice and assistance from intramural scientists, science administrators, and general counsel. Such advice will be promptly provided if the request comes from the Office of the Secretary, DHHS. |
| 5. |
OPRR must, on occasion, work closely with other offices that have ethical responsibilities toward research. Research integrity—that is, the function of preventing fraud, plagiarism, theft of intellectual property, overpayment or double funding, “kiting,” etc., and investigating and punishing such unethical behavior when it occurs, must proceed hand-in-hand with protections for human subjects of research. |
| 6. |
Responsibility for protection of human subjects should be established in a law that establishes the “lead responsibility” for the Common Rule. To be effective, the office that exercises “lead responsibility” must have the full support of a cabinet-level officer. The law must require that the unit exercising lead responsibility report regularly to Congress concerning implementation of the Common Rule. |
VI. Recommendations
In the light of all that has been said above, the following recommendations are offered:
Recommendation 1. There be established by law within the Office of the Secretary, DHHS, an Office of Research Ethics (ORE). The ORE shall be directed by an Assistant Secretary, DHHS, who shall be a member of the Senior Executive Service. (Not a political appointee.) The Director of the ORE shall answer directly to the Secretary, DHHS.
Recommendation 2. The ORE shall have at least two divisions. Division 1 shall be called the Human Subject Protections Division (formerly OPRR); Division 2 shall be called the Scientific Integrity Division.
Recommendation 3. The Human Subject Protections Division of the ORE shall have at least two branches: an Education Branch and a Compliance Branch.
Recommendation 4. The Director, ORE, shall prepare and submit to Congress once each year a report of all the major educational and compliance activities of the ORE for the year. Additionally, reports of all completed inquiries and investigations shall be forwarded to the Congress. The Congress, on the other hand, shall make it unlawful for any person to interfere with ongoing inquiries and investigations of unethical activities or noncompliance with laws, regulations, or policies. Should any inquiry or investigation extend beyond one year, the Director, ORE, shall explain to the Congress why the inquiry or the investigation has not been completed.
Recommendation 5. Included in the annual report of the Director, ORE, shall be an evaluation of the performance of each of the agencies within the DHHS with respect to compliance with laws, policies, and regulations setting forth ethical standards of conduct for research.
Recommendation 6. Included in the annual report of the Director, ORE, shall be an evaluation of the performance of each of the departments and agencies that function under the Common Rule. The report shall address both educational efforts and compliance efforts of these departments and agencies in areas related to protection of the rights and welfare of human subjects.
H-18
Recommendation 7. Included in the annual report of the Director, ORE, shall be an estimate of the personnel and budgetary needs of the Office, including each of its components.
PART 2: The Historical Background of OPRR’s Responsibilities for Humane Care and Use of Laboratory Animals
In 1963, NIH contracted with the Institute of Laboratory Animal Research27 (ILAR) of the National Academy of Sciences to prepare guidance for awardee institutions concerning the care, housing, and husbandry that should be provided for vertebrate animals involved in research.
NIH had three motivations in issuing its contract to ILAR: a) recognition of a moral obligation to house and care for living, sentient nonhuman animals involved in research in a humane and respectful manner; b) recognition that obtaining reliable scientific results based on research involving animals requires that research animals be maintained in a contented and healthy state; and c) recognition that public support of research involving animal subjects is contingent upon the animals being treated in a humane manner.
ILAR produced the first edition of the Guide for the Care and Use of Laboratory Animals in 1963. This edition was so titled because it emphasized the housing and care that should be provided for laboratory animals. The Guide was updated in 1965, 1968, 1972, 1978, and 1985. The most recent version of the Guide was published in 1996. Although the current version of the Guide provides more information than previous editions concerning the care and housing of laboratory animals, much of the new information included in the Guide deals with so-called performance or outcome standards for treating laboratory animals.
Each edition of the Guide published after 1966 includes recommendations that meet and exceed the standards set forth in the Animal Welfare Act passed in 1966 and amended in 1970, 1976, and 1985.28 ILAR has attempted to include in the Guide the best information available, from both research studies and hands-on experience, concerning the care and use of laboratory animals.
The Guide for the Care and Use of Laboratory Animals29 has been translated into many languages, and it is recognized throughout the world as providing an excellent foundation on which to erect a laboratory animal care and use program.30 The PHS Policy on Humane Care and Use of Laboratory Animals issued in 1979 required institutions that receive research awards from any of the PHS agencies to provide assurances to OPRR’s Division of Animal Welfare (DAW) that the institution will comply with the recommendations set forth in the Guide. Prior to 1979, awardee institutions were encouraged to follow the Guide, but Assurances of Compliance were not required, and little more than a token effort to require compliance was made.
From 1963 until 1979, the primary influence exerted by OPRR on awardee institutions came by way of education and persuasion of staff veterinarians in the institutions. OPRR encouraged the hiring of Diplomates of the American College of Laboratory Animal Medicine—veterinarians with advanced training and experience who are recognized as experts—to direct programs in the awardee institutions. Furthermore, it encouraged, but did not require, institutions to seek accreditation from AAALAC.31 The 1979 PHS policy was inadequate in many ways. Assurances provided little detail beyond a statement that the institution intended to comply with the recommendations in the Guide. Assurances did not make it clear which senior institutional official would be held responsible for compliance with the policy. (Because no institutional official was designated, compliance was often left to the discretion of department heads or laboratory chiefs. Thus in the same institution, the quality of care for animals often ranged from very poor to excellent.) Furthermore, assurances did not require prior review and approval of protocols, and they required minimal recordkeeping. As a consequence, although the 1979 assurances probably contributed in a small way to the improvement of care and use of animals, their impact was small.
H-19
It was apparent that the quality of the animal programs in most institutions depended primarily on the institutional veterinarians and their staffs. If the veterinarians were well trained, given adequate resources, and were allowed to exercise authority over the housing, care, and use of the animals, the programs were usually compliant and strong. On the other hand, if institutional veterinarians lacked training, resources, or administrative support, their programs were usually weak.
Many veterinarians complained that they were cast in the role of “research cops” who recognized obligations stemming from their veterinary oath, rather than the PHS policy and the Animal Welfare Act to see that animals were properly cared for and humanely used in research. Unfortunately, in many cases, veterinarians lacked authority to insist that research investigators use animals properly. In a typical research institution, there was tension rather than cooperation between research investigators who used animals for their research and veterinarians who recognized an obligation to care for animals and to see that their use in research involved as little pain or distress as possible to the animal. In virtually all of the older institutions and many newer ones there was no central vivarium. Animals were housed in convenient locations for research investigators. Typically either department heads or individual research investigators were responsible for the animals involved in their research. In most cases, such persons were not trained to care for the animals. Staff veterinarians were available for consultation, but many investigators failed to consult with their staff veterinarian because correction of the problem was charged against the award money assigned to the researcher. Thus, investigators were often loath to consult with staff veterinarians.
In the period between 1979, when the PHS policy was revised, until 1981, OPRR was preoccupied with responding to the recommendations of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research. OPRR staff energy was devoted primarily to efforts to incorporate the recommendations of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research into the DHEW Regulations for the Protection of Human Subjects.
The sole veterinarian on the OPRR staff retired, and hiring freezes prevented recruiting a replacement veterinarian. OPRR’s program for animal welfare was maintained but not improved during this period. After the Regulations for the Protection of Human Subjects were promulgated in 1981, OPRR began to devote more resources and efforts to improve its oversight of awardee programs involving laboratory animals.
As soon as OPRR focused renewed attention on enforcement of the 1979 Policy, the policy’s shortcomings began to come to light. The policy required Animal Welfare Assurances of Compliance to be negotiated by awardee institutions, but was unclear as to the level of detail required in an assurance document. Consequently, assurance documents were often brief and vague. The policy required animals to be maintained in a manner consistent with the recommendations in the Guide, but failed to require either a plan for accomplishing that goal or evaluation of whether the goal was achieved. The policy did not require prior review and approval of protocols by an Institutional Animal Care and Use Committee. For that reason, some studies involved more animals than necessary to obtain sound scientific data. Others failed to use a sufficient number of animals to achieve reliable scientific results. Inhumane procedures were sometimes carried out in the name of science. The policy was virtually useless in preventing these abuses. The policy required little record keeping, and it made no provision for voluntary reporting of problems associated with the care or the use of laboratory animals. OPRR recognized early on that not only was the policy seriously flawed, it was also extraordinarily difficult to enforce.
In 1982 OPRR began to gather information necessary to revise and upgrade the PHS policy. Until then, the policy had been backed by the authority of the Assistant Secretary for Health, who made compliance with the policy a condition of receiving an award to carry out research involving laboratory animals. Issuance of the policy was not required by law, and Congress paid little attention to laboratory animals and the policies that governed their care and use.
H-20
In late summer, 1982, Mr. Alex Pacheco, then a student at George Washington University and a leader in a newly formed organization called People for the Ethical Treatment of Animals (PETA), took a summer job in the Silver Spring, Maryland, Laboratory of Dr. Edward Taub. While Dr. Taub was away from his laboratory on vacation, Mr. Pacheco arranged to have several veterinarians visit the laboratory, which housed approximately 15 deafferented primates (the motor and sensory nerves of one arm of each animal had been severed). Dr. Taub was studying regeneration of damaged nerves. Mr. Pacheco took a series of colored photographs of the laboratory and the condition of the animals. Then he arranged for a state police raid on the facility under the Prevention of Cruelty to Animals law of the state of Maryland. The visiting veterinarians, the colored photographs, and the police report all indicated that the animals were housed in a filthy, fetid environment that constituted cruelty to the animals.
Dr. Taub claimed that his laboratory was clean and well run when he left on vacation. He claimed that Mr. Pacheco had trashed the laboratory, failed to clean cages, neglected the animals, and subjected the laboratory to false reports of animal cruelty. Mr. Pacheco, for his part, claimed that he merely documented the deplorable state of the laboratory and the condition of the animals. Initially the matter was handled in the courts of the state of Maryland. Dr. Taub was convicted on six counts of animal cruelty, but a court of appeals set aside the conviction on the grounds that since the laboratory was subject to the PHS policy, the issue was a federal matter. The court remanded custody of the animals to NIH. OPRR was directed to investigate.
OPRR was never able to determine with a high level of confidence whether Dr. Taub operated an abominable laboratory, Mr. Pacheco had trashed a well-run laboratory in Taub’s absence, or neglect by Taub and trashing by Pacheco combined to create a dreadful situation.
Taub claimed that he had been “set up” by PETA in such a way that he appeared to be in serious noncom-pliance with the PHS policy. Some of the facts in the case made such a defense plausible. The prosecuting attorney for the state of Maryland subsequently took an administrative position with PETA. Furthermore, the state temporarily housed the animals, in violation of a number of city ordinances, in the basement of a Rockville house owned by Ingrid Newkirke, President of PETA, and the animals were stolen from the Newkirke residence—only to be returned with no questions asked. These facts provide some circumstantial evidence to support Dr. Taub’s contention that PETA had indeed “set up” Dr. Taub.
Dr. Taub acknowledged to OPRR that his records were intact. The records showed that the animals had not received routine veterinary care for a period of years. Because the animals were deafferented, they required more specialized care than most other primates. Absence of veterinary care for a period of years constituted a serious violation of the PHS policy. Taub’s defense that he personally had provided care for the animals was considered inadequate.
Dr. Taub’s grant was suspended until such time as his laboratory could be brought into compliance and he was able to demonstrate that he could meet all the standards set forth in the Guide. Taub appealed the decision, but lost the appeal. Taub’s laboratory was never restored, and the animals remained, by court order, in the custody of NIH (despite a series of lawsuits brought by PETA) for many years until all died or were euthanized. Custody suits brought by PETA were taken all the way to the Supreme Court, which confirmed decisions of the lower court that PETA had no legal standing on which to base its claim to custody of the animals. The case of the Silver Spring Monkeys, as it was called in the media, lasted for a period of approximately ten years.
In 1983 another case made national headlines. A group that identified themselves as the Animal Liberation Front (ALF) broke into the University of Pennsylvania Head Injury Clinic in Philadelphia. Equipment was smashed and files were scattered. Most important, approximately 60 hours of audio/videotapes were stolen. The tapes had been used as a tool by research investigators to capture visual images of research animals; data concerning heartbeat, blood pressure, and brain wave activity; and investigator’s verbal observations concerning the animals involved in the research study of head injuries.
H-21
The protocol called for sedated baboons to be injured in a machine that simulated the whiplash motion that often inflicts damage to the neck and spine of humans involved in rear-end auto crashes. The nature of the injuries to the animals were to be studied, and the animals’ unassisted recovery from injury was to be compared with the recovery of animals that received a variety of treatment modalities. The protocol was controversial because it required the infliction of a severe injury on the baboons. Each animal ultimately would be examined in terminal surgery.
The ALF gave the stolen audio/videotapes to PETA. PETA edited the tapes, added a voice over commentary, and circulated the edited tape entitled Unnecessary Fuss32 to schools, newspapers, Congress, television networks, and dozens of television stations. Congress and members of the general public were shocked at the cruelty to and disregard for the research animals presented on the tape. PETA then petitioned the PHS to close the laboratory and to punish the investigators, Drs. Langfit and Genarelli, for violation of the PHS policy. OPRR refused to act on the basis of evidence contained in an edited tape. The University of Pennsylvania claimed that Unnecessary Fuss was a caricature of the actual proceedings that had taken place in the laboratory. PETA refused for more than a year to turn over the evidence it had to the OPRR. In the spring of 1984, PETA sent the unedited tapes to the USDA, which in turn sent them to OPRR.
OPRR asked 18 veterinarians, mostly Diplomates of the American College of Laboratory Animal Medicine, who were, for the most part, employed by various Institutes within NIH, to review the tapes and report on their findings concerning violations of the PHS policy or the Animal Welfare Act. In the meantime, OPRR conducted several site visits to the Head Injury Laboratory. On the last of those site visits, Dr. Generelli performed a surgical procedure in the presence of the visitors that he claimed was typical of those involved in the study. OPRR was astonished to learn that aseptic technique was sloppy, that smoking was allowed in the operating theater (improper on many grounds, and a dangerous procedure where oxygen tanks are stored and used), and that the depth of induced anesthetic coma in the animals was questionable. OPRR also learned that most of the animals were not seen by an attending veterinarian either prior to or after suffering whiplash.
OPRR discovered that the Unnecessary Fuss presented the case history of only 1 of approximately 150 animals that had received whiplash. By clever editing and inaccurate voice over comments, the viewer was led to believe that the inhumane treatment depicted on the film was repeated over and over and over again. In actual fact, one baboon was badly treated, and the film showed that single mistreatment over and over again while the commentator narrated that the mistreatment was repeated on a long series of different animals. In all, OPRR identified about 25 errors in the description of what was taking place. Typical was the statement accompanying film showing an accidental water spill that acid had been carelessly poured on a baboon.
Despite the fact that Unnecessary Fuss grossly overstated the deficiencies in the Head Injury Clinic, OPRR found many extraordinarily serious violations of the Guide for Care and Use of Laboratory Animals. Veterinary and post-trauma nursing care for the animals were inadequate, survival surgical techniques were not carried out in the required aseptic manner, the operating theater was not properly cleaned, the holding facility lacked the required number of air changes per hour and other features required of a holding facility, and occupational health safeguards were not enforced. Furthermore, OPRR found deficiencies in the procedures for care of animals in many other laboratories operated under the auspices of the university. The university was put on probation by OPRR. The Head Injury Clinic was closed. The chief veterinarian was fired, the administration of animal facilities was consolidated, new training programs for investigators and staff were initiated, and quarterly progress reports to OPRR were required.
Although OPRR dealt with a small number of additional cases of violation of the 1979 PHS Policy for the Humane Care and Use of Laboratory Animals, the case of the Silver Spring Monkeys and the University of Pennsylvania Head Injury case were the two events that caught the attention of the public and Congress, illustrated the serious weaknesses in the 1979 policy, and focused the attention of the Assistant Secretary for Health and the Director, NIH, on the importance of upgrading the PHS policy.
H-22
I. OPRR’s Legislative Mandate
OPRR took three major steps to upgrade the Policy for the Humane Care and Use of Laboratory Animals. First, it convened a committee drawn from across the PHS to provide advice. Second, it persuaded Congress (particularly Congressman Doug Walgren) to postpone legislation long enough for the new policy to be promulgated and tested. Third, it initiated a series of educational workshops that were presented in every region of the country. The proposed policy was discussed and comments elicited at all of these events.
The revised PHS policy was promulgated in May of 1985. Promulgation of the policy was coordinated with the publication of the 1985 version of the Guide for Care and Use of Laboratory Animals edited and published by ILAR.
The new policy included many new provisions. The most important new requirements were 1) requiring each assured institution to identify, both by name and office, the institutional official who was to be held responsible for assuring that the institution’s entire laboratory animal program would meet or exceed the recommendations in the Guide; 2) establishing an Institutional Animal Care and Use Committee (IACUC) in each awardee institution; 3) requiring semi-annual inspection of all animal holding facilities followed by a report to OPRR of all deficiencies in facilities, staffing, or training and steps taken to remedy the deficiencies; 4) requiring an occupational health program including standard operating procedures for all persons who had contact with laboratory animals (this program would protect both human and animals); 5) requiring prospective and ongoing protocol review by the IACUC and periodic reporting to OPRR with a special proviso for immediate reporting of serious problems; 6) the beginning of a system of evaluation that allowed a program to be evaluated, at least in part, on performance standards—that is, judging the worth of a program by the health and well-being of the animals rather than engineering standards that specify requirements for cage sizes, facility cleanliness, heating and air conditioning systems, and the like.
OPRR had found that institutions could be in compliance with the technical requirements of the Guide and nevertheless have an unhealthy colony of laboratory animals. It had also found the converse proposition was sometimes true. The 1985 version of the Guide and the concurrent education program stressed evaluation of the health and comfort of the animals in addition to requirements for good husbandry practices that included caging, housing, and sanitation.
On November 20, Congress enacted the Health Research Extension Act of 1985 (PL 99-158), that required the Secretary, DHHS, acting through the Director, NIH, to promulgate the very Guidelines for the Care and Use of Laboratory Animals that were issued in May of 1985 and that had been tested over a six-month period. The law, in essence, provided congressional sanction for a policy that had already been promulgated, implemented, and evaluated. Most of the provisions in the policy were born of experience of noncompliance with the 1979 policy and the experience of the NIH intramural animal research programs that provided ready and immediate feedback to OPRR.
The policy relied almost entirely on hands-on experience rather than the literature that was beginning to come from the bioethics movement in the United States dealing with the moral status of animals. The policy represented an act of trust that IACUCs would, over time, develop standards by which to judge prospective protocols involving animal subjects. That act of trust has been fully justified. IACUCs have examined virtually every procedure employed by investigators and have evaluated virtually every system, method, and technique for caring for animals.
The revised policy—assisted no doubt by strident, though often illegal and inaccurate criticisms of the animal activists—jump-started the improvement of programs for the care and use of laboratory animals from a system that was, at best, mediocre, to one in which Americans may legitimately take pride.
Within a few months after the PHS Policy for the Humane Care and Use of Laboratory Animals was backed by law, OPRR found it necessary to close the facilities of Columbia University’s school of Physicians and
H-23
Surgeons and the animal facilities at the City of Hope University in southern California. Neither institution had made an acceptable effort to come into compliance with the new policy.
As a result of their suspension, the two institutions rebuilt their animal research programs and came into compliance in a matter of a few months. Not only were facilities improved, but staff were increased, training was initiated, and a proper chain of command was established. The drastic actions of closing entire programs (at Columbia it was estimated that $90 million of research was suspended for a period of more than four months) served as a warning to the entire research community that the policy, which enjoyed the support of the scientific community, would be fairly but rigorously enforced. Although there have been many other minor cases of noncompliance, the history of implementation of the PHS policy has been, since the Columbia case, characterized as a partnership between the DAW and the research community rather than a regulator/regulatee relationship.
About a month after the PHS policy was bolstered by the enactment of the Health Research Extension Act, the Congress incorporated amendments to the Animal Welfare Act in the Food Security Act of 1985 (PL 99-158). The new law was detailed, complex, and specific. Careful interpretation was necessary to make it internally consistent. Among other provisions in the act were controversial provisions that called for exercise for dogs and psychological well-being of primates. It also called for harmonization with the PHS policy through consultation with the Secretary, DHHS.
Initially USDA minimized the USDA/DHHS harmonization clause, and it published proposed rulemaking in 1987. A storm of criticism greeted the proposed rules that relied exclusively on engineering standards. After a second unpopular proposal of regulations, OMB convened a meeting involving the Acting Secretary of Agriculture and the Acting Director of NIH.
Although both of the senior officials were present, negotiations were carried on by OPRR and the Director of the Animal Plant Health Inspection System (APHIS) within the USDA. The historic outcome of that meeting was an agreement to incorporate in many places in the USDA regulations performance standards in addition to engineering standards. Although engineering standards would be used, the seriousness of a violation of such a standard would be judged in terms of whether it negatively affected the health and well-being of the animals.
The USDA regulations produced in 1991 met with instant approval and endorsement from Congress and the research community. They were criticized by animal activists who claimed they were too vague, unenforceable, and filled with loopholes. The regulations were challenged in court by a group known as the Animal Legal Defense Fund. That group won its case—that the regulations did not adequately implement the law—in the lower court, but on appeal was found to have no standing to sue. The matter has recently been referred to the Supreme Court.
II. OPRR’s Relationship to the USDA
From 1970–1980 relationships between USDA officials with responsibility for implementing the Animal Welfare Act and OPRR staff were cool and distant. Rivalry and suspicion and a very different approach to regulations characterized the relationship. Clearly, the USDA approach was established by its own Office of General Counsel, which sought to produce rules that could be enforced in court proceedings. Thus, emphasis on issues that could be clearly measured, weighed, or documented characterized the USDA rules. In the years 1980–1985 the OPRR and APHIS began to cooperate in their efforts to promote sound practices of care and use for laboratory animals. However, until the 1985 amendments to the Animal Welfare Act, the USDA’s authority was confined to holding facilities for animals. It had no jurisdiction over the use of laboratory animals in research. USDA inspectors had been trained to check lists of engineering standards, including such items as cage sizes, the expiration dates on feed bags, sanitation, air flow, clean water dispensers, thermostats, pest control, lighting, bedding, and cage washing. They had little training or expertise in evaluating the health and
H-24
comfort of the animals. Because USDA exercised no jurisdiction over rats and mice, (about 90 percent of all the animals used in research), inspectors never visited laboratories that used no other species.
Because there were so many items on the USDA checklist, virtually every institution failed to meet some USDA standards. On Monday mornings, for example, most cages are littered in most laboratories. Inspectors visiting a holding facility on a Monday almost always found sanitation to be wanting because the cages had not been cleaned since Friday. If a bulb burned out, a cage washer needed repair, or a crack formed in a wall or a ceiling (that could possibly harbor vermin), even though it was sprayed weekly with hot water and disinfectant, the institution could fail inspection. Under the new regulations all of these items would be evaluated, but the primary evaluation is directed to the health of the animals. If the animals exhibit normal behavior and eating habits, have good coats, are neither too thin or too fat, have been checked periodically by a veterinarian, are socialized to other animals and to their human caretakers, then mechanical failures and floor cracks are not judged to be as serious as they would be if the animals were in poor health.
In other words, the engineering standards are viewed in the light of outcome or performance standards and judged accordingly. Performance standards require better trained inspectors who are qualified to evaluate animals. OPRR staff from DAW have worked harmoniously with USDA inspectors to teach them how to evaluate facilities using performance standards. A survey of IACUCs conducted by the Scientists Center for Animal Welfare and a survey of the opinions of USDA inspectors have indicated that performance standards have greatly improved the care and use of animals.
Since 1990 the cooperation between OPRR’s DAW and the USDA has been outstanding. Both agencies have profited, and the quality of both care and use of animals has, by every measure, risen dramatically.
In testing policy interpretations and in perfecting approaches to making reasonable performance standards, DAW works closely and harmoniously with the NIH Office for Animal Care and Use and with the administrators, veterinarians, research investigators, technicians, and caretakers at NIH facilities. Many of these individuals are called upon to assist in the training of USDA inspectors and in OPRR educational programs and site visits. Credit is due to Dr. John Miller, Director of DAW and to his successor Dr. Nelson Garnett for improving relationships with USDA, improving relationships with the NIH intramural program, and as a consequence improving the oversight of the care and use of laboratory animals in awardee institutions. Recent meetings in Boston of more than 500 members of IACUCs indicate that these bodies have become highly sophisticated in evaluating the protocols that come before them. These bodies have been remarkably successful in developing procedures for inspecting facilities, maintaining high performance standards, and improving protocols proposing to involve animals in research. IACUCs have had dramatic success in putting practice the three Rs of animal research: reduction, refinement, and replacement.
III. Findings
| 1. |
The statutory authority of DAW is delegated through the Director, NIH, and, by law, is implemented by guidelines (policy) rather than by regulations. The law encourages flexibility. It confers a certain amount of discretionary authority on OPRR’s DAW. Such discretion needs the kind of ongoing reality check provided by the intramural animal welfare program at NIH. |
DAW should be so situated that performance standards can be tested and perfected. DAW can afford to spend time on these matters because the USDA—although it has changed dramatically—still emphasizes engineering standards, allowing DAW to emphasize performance standards.
| 2. |
The history of DAW is one of trust relationships. While that also characterized the human subjects approach of the 1980s, the trust relationship relative to the human subjects research community has seriously eroded in the 1990s. This may be an appropriate time to separate the two divisions, so that the trust relationship of DAW can be maintained, while a new trust relationship of the Human Subjects Division is built. |
H-25
| 3. |
Because many failures to comply with the PHS policy require changes in architecture, plumbing, heating, air conditioning, or building maintenance, compliance with the PHS policy cannot occur overnight. DAW works for months or even years to improve facilities and bring them to full compliance. (It took several years for Congress to appropriate the money for a new primate facility at NIH. In the meantime, DAW worked closely with NIH to bring the old facility as close to compliance as possible.) For this reason, DAW’s operating procedures differ markedly from those of human subjects, even though they are superficially similar. Separation should not hurt either division. |
| 4. |
Since 1985 the Director of OPRR has given less and less time to animal concerns, because the policy has been so widely acclaimed and functioned so well. DAW can easily stand on its own feet. |
| 5. |
Although DAW, like the rest of OPRR, has policy oversight responsibility of NIH programs as does the Division of Human Subjects, it has little history of actual conflict of interest. The potential for conflict of interest may be offset by different means than is the case for protection of human subjects. |
IV. Recommendations
| 1. |
Because the PHS Act requires the PHS Policy for Humane Care and Use of Animals to be promulgated by the Secretary, DHHS, through the Director, NIH, there are legal barriers to moving the DAW to the department level as was recommended for the Division of Human Subjects in OPRR. It is therefore recommended that DAW remain within the NIH. |
| 2. |
Because the PHS policy increasingly emphasizes performance standards, the DAW needs to be closely associated with the NIH intramural program where performance standards and environmental enrichment efforts for laboratory animals are routinely tested and evaluated prior to being recommended for general use. DAW should therefore remain within the NIH. Although other DHHS agencies have small intramural animal programs, they fund little extramural animal research. Therefore, the need to be at a secretarial level to exercise oversight of those programs is far less for DAW than for the Human Subjects Division. DAW should remain within NIH. |
| 3. |
Because the expertise of staff from many components of NIH is needed for training of USDA personnel, DAW should remain within the NIH and in close cooperation with such staff persons. |
| 4. |
DAW should not remain under the Deputy Director for Extramural Research because that raises the same potential conflict of interest problems that were addressed above with respect to human subjects. Rather, DAW should be answerable only to the Director, NIH. Like the OPRR, DAW should be required to report annually to the Congress. |
| 5. |
The DAW budget should not fluctuate at the whim of its current supervisor. Rather DAW should have a stable budget that is adequate to provide for both site visits, education, and administrative responsibilities. |
Notes
1 McCarthy, Charles R., Encyclopedia of Bioethics, Warren T. Reich, ed., in Chief, Macmillan Library Reference, USA, Simon, Shuster, and Macmillan, New York, 1995. “Research Policy” Vol. 4, pp. 2285–2287.
2 Group Considerations of Clinical Research Procedures Deviating from Accepted Medical Practice and Involving Unusual Hazard, Policy Statement Issued by NIH Clinical Center, 1953.
3 Even today, IRBs often find words like “treatment” and “therapy” used in consent documents for research studies that offer little chance of direct benefit to the subjects. The author reviewed many such documents while serving on the Human Subjects Subcommittee of the Recombinant DNA Advisory Committee.
H-26
4 NIH’s Clinical Center Policy issued July 1966. The policy was updated in 1976 and 1977. In the Carter administration an effort was made to reduce the number of federal agencies. Since, in technical government parlance, a committee qualifies as an agency (though such vocabulary is seldom used), all of the CRCs were combined into a single CRC of which each of the institute committees was called a subpanel. Similar groupings of committees and subcommittees occurred throughout the government. In this way it could be reported to the public that the number of federal agencies was dramatically reduced.
5 Frankel, Mark, Public Policymaking for Biomedical Research: The Case of Human Experimentation, George Washington University Press, May 9, 1976. Frankel has done a masterful job of presenting the mosaic of scientific, political, ethical, and public policy issues that coalesced to form the initial PHS Policy for the Protection of Human Subjects.
6 U.S. Senate Committee of the Judiciary, Subcommittee on Anti-trust and Monopoly, 86th Congress. Hearings were held in 1959–1960, but the resulting amendments to the Food, Drug and Cosmetic Act were not enacted into law until 1962. When the bill came to the Senate floor, Senator Jacob Javits, R. NY, introduced an amendment from the floor requiring informed consent in the testing of drugs. The FDA struggled for several years trying to find a way to implement the informed consent requirements. It was not until Institutional Review Boards were developed in universities and pharmaceutical houses that an instrument was found to require informed consent.
7 Cooley, D.Z., Hallman, G.L., Bloodwill, et al. “Human Heart Transplant: Experience with Twelve Cases,” Am Journal of Cardiology, 22:804–810, 1977. See also Fox, Renee C., and Swazey, Judith P., The Courage to Fail, A Social View of Organ Transplantation and Dialysis, University of Chicago Press, 1974, pp. 149–211.
8 Frankel, p. 124.
9 Langer, Elinor, “Human Experimentation: Cancer Studies at Sloan-Kettering Stir Public Debate on Medical Ethics,” Science 143:551–553, 1964. See also Frankel, p. 68.
10 Beecher, Henry K., “Ethics and Clinical Research,” New England Journal of Medicine 274:1364–1360, 1966.
11 Frankel, p. 153. Officials at the NIH, including its Director, Dr. James P. Shannon, were in close touch with Congress about issues of abuse of research subjects. NIH came to believe that Congress would hold NIH responsible for misconduct in NIH-supported research, even though the NIH of that time had no control over the conduct of extramural research. Shannon realized that not only were the subjects of research vulnerable to research abuses, but also that the entire PHS, including NIH, was vulnerable to negative criticism occasioned by such conduct.
12 Frankel, p. 153.
13 The Institutional Guide to DHEW Policy on Protection of Human Subjects, HEW Publication No. 72-102, Dec. 1, 1971.
14 Jones, J.H., Bad Blood, 2nd edition, New York Free Press, 1993.
15 Marston, Robert Q., “Medical Science, the Clinical Trial, and Society,” presented at the dedication ceremonies for the McLeod Nursing Building and Jordan Medical Education Building, University of Virginia, 10, 1972. The speech was written by Mr. Storm Whaley, Public Affairs Officer, NIH, who taught ethics at the University of Arkansas prior to coming to Washington, DC.
16 Frankel, p.183. It should be noted here that in each year after the disclosure of the Tuskegee debacle, the U.S. government has paid compensation to the survivors of the study and to their heirs. In FY 1995 $2.8 million and in FY 1996 $1.88 million was paid in compensation. Personal communication to the author from OPRR. President Clinton made a public apology on behalf of the nation to the survivors and heirs of the Tuskegee Study in 1997. Although care and compensation have been provided to the victims of the Tuskegee Study and—following disclosures by the President’s Advisory Committee on Human Radiation Experiments—to some survivors of radiation experiments no general policy for care and compensation for injured research subjects has ever been implemented.
17 In 1979 Dr. Fredrickson was asked to present a paper on the occasion of Professor Tristram Engelhardt’s installation at the Kennedy Institute of Ethics, Georgetown University, Washington, DC. A draft of the talk was prepared by OPRR. In the talk, Fredrickson committed himself and NIH to improvement of the protections afforded to human research subjects. He provided in the talk some historical instances of abuse and pledged that, to the best of his ability, such abuses would not recur. From that point forward, Dr. Fredrickson championed the work of OPRR. Note: This is a personal recollection of the author.
18 Dr. Bick was subsequently promoted to Deputy Director for Extramural Research, although her responsibilities remained unchanged and her supervision of OPRR was not affected by the promotion.
19 One of these cases involved the work of Dr. Edward Taub in the case that the media referred to as the Silver Spring Monkey case. NINDS received bad publicity for its funding of Taub’s research.
H-27
20 See 45 CFR 46.116 (d). Recently the FDA added a waiver of informed consent for emergency research in circumstances where it is not feasible to obtain the consent of a subject or a subject’s authorized representative within the available “window of opportunity” to carry out the research.
21 Sites where unregulated research is conducted include colleges and universities not receiving federal funds that conduct research in the behavioral and social sciences; in vitro fertilization clinics; some physician’s offices; some dental offices; some psychiatric offices; some legal service clinics; some corporate and industrial health safety and fitness programs; and weight loss and diet clinics (often posing high risks to obese subjects who suffer from cardiac problems, diabetes, and circulatory problems). In a letter to the author, Dr. Ellis, Director, OPRR, states that OPRR receives many complaints about unregulated research both by mail and by telephone. These complaints include both physical and psychological harms, breaches of privacy and confidentiality, and affronts to dignity. It should be noted, however, that devising a set of regulations to cover such research is far from a simple matter. Such research cannot be controlled by interdicting federal funds (DHHS regulations), and it involves no articles involved in interstate commerce (FDA regulations). To control such research with criminal penalties is likely to drive it underground. Since many research infractions are minor offenses, if they were criminalized the cost of enforcement would be disproportionately high.
22 All of the cases cited below are filed at OPRR. Since the author was involved with them and therefore remembers their general outlines, and since the cases are cited only for the purpose of identifying strengths and weaknesses in the present organizational location of OPRR, he has not obtained copies of the reports and re-read them. He is prepared to do so if NBAC determines that such additional research is necessary.
23 One such example was the widely publicized Baby Fae case in which a baboon’s heart was transplanted into a newborn infant suffering from a degenerative heart condition. Loma Linda invited OPRR to conduct an investigation, even though the research was not federally funded.
24 Everyone involved in the case agreed that the data had been falsified. By reconstructing the records it was determined that alterations in data always occurred on Thursday afternoons, the only period of time when Dr. Straus was alone in the clinic. When confronted with this information, Straus initiated plea bargaining through his lawyer.
25 The Model Federal Policy for the Protection of Human Subjects is better known as the Common Rule. It remains the only successful “cross-cutting” (across all federal departments and agencies) in the federal government. It was published in 56 Federal Register June 18, 1991 at 28002–28032. For discussions of various aspects of the development of the Common Rule, see Porter, Joan P., and Dustera, Alicia K., “Lessons from Two Federal Initiatives: Protecting Human Research Subjects, and Handling Misconduct in Science,” Academic Medicine 68(9):551–555, 1993. See also Porter, Joan P., “Federal Policy for the Protection of Human Subjects,” IRB 13(5):8–9, 1991. Finally, see Porter, Joan P., Development of a Federal Policy for the Protection of Human Subjects of Research, Food Drug and Cosmetics Law Journal 45(6):623–629, 1990. Dr. Porter was a senior employee in the OPRR during the entire period of the development of the Common Rule. She worked tirelessly to get clearances from all of the affected departments and agencies as well as the OMB, the Department of State, and the OSTP in the White House.
26 President’s Commission for the Study of Medicine and Biomedical and Behavioral Research, “Implementing Human Research Regulations,” U.S. Government Printing Office, 1981.
27 The Institute of Laboratory Animal Research (ILAR) operates within the National Research Council of the National Academy of Sciences. Founded in 1953, ILAR, formerly called The Institute of Laboratory Animal Research, has produced reports, publications, educational events, and studies aimed at improving the preservation, shipping, care, and use of laboratory animals, farm animals, and animals in their natural habitat. In 1997, ILAR’s title was changed to the Institute of Laboratory Animal Research.
28 The Animal Welfare Act of 1966 (P.L. 89-544) was amended in: (a) 1970 by P.L. 91-579, (b) 1976 by P.L. 94-279; and 1985 by P.L. 99-198.
29 The current Guide for Care and Use of Animals was published under the auspices of the Institute of Laboratory Animal Resources, by the National Academy Press, Washington, DC, 1996. Funding was provided by NIH and the USDA.
30 The 1995 issue of the Guide has been or is in process of being translated into Spanish, Portuguese, French, German, Japanese, Russian, Korean, and Chinese.
31 The Association for Assessment and Accreditation of Laboratory Care (AAALAC), International, formerly known as the American Association for the Accreditation of Laboratory Animal Care. This association provides on-site visitation, evaluation, and assessment of the quality of the laboratory animal care and use programs of its member institutions in the United States, Canada, and Europe. AAALAC uses the Guide for the Care and Use of Laboratory Animals as its standard for evaluating the quality of laboratory animal programs.
32 The title Unnecessary Fuss was derived from a statement by Dr. Weingarden, then Director, NIH, to the effect that the ALF and PETA had raised an “unnecessary fuss” over research involving animals, particularly research conducted at the Head Injury Clinic at the University of Pennsylvania.
H-28
PROTECTIONISM IN RESEARCH INVOLVING HUMAN SUBJECTS
Commissioned Paper Jonathan D. Moreno University of Virginia

I-1
“We can never rest comfortably in the belief that the soil from which our satisfactions sprout is not watered with the blood of martyrs. But a troubled conscience compels us, the undeserving beneficiaries, to ask: Who is to be martyred? in the service of what cause? and by whose choice?” Hans Jonas1
In the ethics of human subjects research, protectionism is the doctrine that human beings should be protected from the risks of participation in research. Evidently, unless one believes that scientific progress always trumps the interests of human subjects, protectionism per se is hardly a controversial view. Controversy enters mainly when several nuanced interpretations of the doctrine are distinguished with an eye toward its application to actual research projects.
There are alternative perspectives to protectionism, from the standpoint of subjects and of investigators. From the subjects’ point of view, a philosophy that calls for ease of access to clinical research emerged in the 1980s, and I will allude to it later in this paper. But enthusiasm for research participation as an alternative “back door” to medical care has waned in recent years.
From the standpoint of investigators an alternative to protectionism is reliance on their moral virtue, thus emphasizing the high degree of discretion over the management of human subjects that has traditionally been accorded scientists. On closer inspection, however, a position that favors investigator discretion is not an alternative to protectionism but a particular version of it, one that places the onus for protecting subjects on the researcher.
In this paper I shall analyze the historical origins of protectionism as a philosophical position in the ethics of human subjects research. I shall also distinguish three versions of protectionism that have emerged in this history: moderate, strong, and weak versions, framed in terms of how much discretion investigators should be allowed concerning the management of human subjects. Weak protectionism entails reliance on the discretion of the investigator with modest constraints understood as guidelines. Moderate protectionism makes room for investigator discretion but within a framework of rules. Strong protectionism involves greatly reduced investigator discretion in a context of direct intervention by a third party, perhaps in the form of monitoring of actual research activities.
There are several critical issues for a protectionist policy in human subjects research. The first is the relationship between the interests of the subject and those of science and “future patients.” The second is whether and in what manner the conduct of the investigator may be monitored or controlled by third parties. A corollary of these issues is the question of special arrangements for subject populations that are vulnerable by virtue of age, medical condition, or social status. All of these topics will be recurrent themes in this paper.
Individuality and Society
No endeavor presents more strikingly the tension between individual and social interests than does medical research involving human subjects. Although the origins of the contemporary idea of individuality with its associated rights and interests are complex, largely Western, and relatively recent, the originality of the idea of individuality should not be exaggerated. What seems to have emerged since the Enlightenment is not so much the notion of the individual, which was surely available to ancient thinkers who meditated on the meaning of human subjectivity, as it is the inferences (moral and otherwise) drawn from that notion. Eastern and traditional cultures, too, are hardly ignorant of the idea of individuality, though again they may attribute different implications to it.
I-3
The fundamental ideas behind our contemporary understanding of society are arguably more continuous with the ancient world than that of individuality. From the Greeks we inherited the ideal of social solidarity and the conception of social roles that entail role-related duties, as well as a general sense of public responsibility. Enlightenment thinkers, the founders of our Western political framework, reached back to classical sources for their inspiration but also developed medieval notions of consent as the basis for governmental legitimacy. It is perhaps in that ambiguity, deep in the Enlightenment tradition, that we may locate the tension between individual and societal interests.
Yet in another sense individual and society are complementary rather than conflicting ideas. Few thinkers (apart from the most extreme libertarians and “objectivists” on the one hand and radical collectivists and “pan-psychists” on the other), have found it acceptable to treat matters concerning human nature as reducible to one or the other. Most have presupposed an anthropology of “social individuals,” with the true battleground mainly a matter of line-drawing. Even our current preoccupation with genetics tends to accept this presupposition, couched in terms of genomic background and phenomic expression.
It is also useful to recall that the same period that gave rise to modern experimental method also refined ideas about personal dignity that we today take for granted. That scientific progress is finally in the service of improving opportunities for human beings to express their best and most humane selves is today hardly questionable. In that sense scientific activity that undermined human dignity would be a cultural contradiction. It is this sensibility that underlies the nearly universal condemnation of the use of human beings as mere means to scientific ends.
Traditional medical ethics embodies a resolution of the tension between individual and societal interests. Hippocratic tradition favors care for the individual patient but also emphasizes the continuous learning or “practice” that must take place, clearly with an eye toward benefiting future patients and thereby society in general. Experimentation in an emergency is authorized though care must be taken to avoid engendering more harm than good. Presumably the learning that takes place through experimental results can be applied to future practice and passed on to one’s apprentices in the fraternity. All this is in the spirit of the Hippocratic tradition and survives in modern medical values.
Clearly the modern experimental environment creates vast new complications for application of the Hippocratic “harm” principle, but the principle itself rests on a presumption of protection of the immediate patient. Vulnerable persons, exemplified as slaves in the versions of the Oath that antiquity has bequeathed to us, must be specifically included in this protectionist attitude. How, then, to effect the resolution called for by the Hippocratic tradition in the modern experimental environment? Because protectionism is a doctrine that is rooted in experience, an understanding and justification of the ways it has been implemented require an historic approach.
From Havana to Nuremberg
Concerns about the involvement of human beings in research are at least a century old. Many institutionalized children were subjects in vaccine experiments in the nineteenth century, in Europe and the United States, and by the 1890s anti-vivesectionists were calling for laws to protect children. At the turn of the century the Prussian state imposed research rules and Congress considered banning medical experiments for certain populations, such as pregnant women, in the District of Columbia. In the ensuing decades there were occasional well-publicized scandals, mostly involving child subjects, and the first attempt to test a polio vaccine was stopped after the American Public Health Association censured the program.2 Prior to World War II, however, medical researchers were largely inoculated against regulation by the nearly legendary status of the self-experimentation by members of U.S. Army physician Walter Reed’s Yellow Fever
I-4
Commission in Cuba. One of the commissioners, Dr. Jesse Lezear, died after subjecting himself to the mosquito’s bite, helping to confirm the hypothesis of the disease’s spread. A less celebrated but equally notable element of the Reed story is his use of an early written contract for the Spanish workers who were among the commission’s other subjects, which itself appears to have followed a controversy involving yellow fever research subjects.3 For some reason Reed himself was widely thought to have been one of the volunteer subjects, perhaps due to his untimely death only a few years later that resulted from a colleague’s error. This misconception added to the legend and to the model of medical researchers as of exceptional moral character, even to the point of martyrdom. The Reed mythology became a singular reference point and justification for the self-regulation of medical science. During the 1960s, when physician researchers were coming under new levels of scrutiny, the distinguished physician-scientist Walsh McDermott referred to the Reed story to demonstrate the social importance of medical research, with the high moral standing that went with it.4 An occasion for the significant revision of this picture became available at the end of the Second World War, when 23 Nazi doctors and medical bureaucrats were tried for crimes associated with vicious medical experiments on concentration camp prisoners. The defendants were selected from about 350 candidates. Although only 1,750 victims were named in the indictment, they were a handful of the thousands of prisoners used in a wide variety of vicious experiments, many in connection with the Nazi war effort. Some involved the treatment of battlefield injuries or in preventing the noxious effects of high altitude flight. Others, such as the sterilization experiments, were undertaken in the service of Nazi racial ideology, and still another category had to do with developing efficient methods of killing.
A strong defense mounted by the defendants’ lawyers pointed to the fact that the Allies, too, had engaged in medical experiments in the service of the war effort. As the prosecution’s attempt to demonstrate that there were clear international rules governing human experimentation faltered, the judges decided to create their own set of rules, known to posterity as the Nuremberg Code, the first line of which is “The voluntary consent of the human subject is absolutely essential.” Although the court seemed to believe that protections were needed, it is not clear how intrusive they wished these protections to be in the operations of medical science. The judges declined, for example, to identify persons with mental disorders as in need of special provisions, although urged to do so by their medical expert. The very requirement of voluntary consent for all undermined the relevance of their code to experiments involving persons with diminished or limited competence, and the extreme circumstances that gave rise to the trial itself seemed quite distant from normal medical research.5
Discovering Informed Consent
Unlike the medical profession as a whole, in 1947 the new Atomic Energy Commission apparently took note of the Nazi doctors’ trial and attempted to impose what it termed “informed consent” on its contractors as a condition for receiving radioisotopes for research purposes. It also established—or attempted to establish—a requirement of potential benefit for the subject. Both of these conditions were to apply to nonclassified research. This relatively protectionist attitude may not have been adopted with a great deal of appreciation of its implications. In any case, the AEC’s position met with resistance among some of its physician contractors, but not its physician advisors. The AEC’s early protectionist stance finally did not become institutionalized, and the letters setting out the requirements seem to have soon been forgotten. (The potential benefit requirement seems itself to have been incompatible with all the trace-level radiation research the AEC sponsored shortly thereafter.) Similarly, in the early 1950s the Department of Defense adopted the Nuremberg Code, along with written and signed consent, as its policy for defensive research on atomic, biological, and chemical weapons, but a 1975 Army Inspector General report pronounced that initiative a failure.6 Thus by the early 1950s although there were gestures in the direction of a protectionist attitude toward human subjects, even these expressions were in a fairly abstract philosophical vein rather than in a robust set
I-5
of institutionalized policies and procedures. An example is the Army’s failure to implement a compensation program for prisoners injured in malaria or hepatitis studies when it was contemplated in the late 1940s.7 The essential feature of the weak form of protectionism that prevailed at that time was its nearly wholesale reliance on the judgment and virtue of the individual researcher. Deliberations on the World Medical Association’s Helsinki Declaration of 1964 (Helsinki I) began in 1953. Informed consent was a far less prominent feature of the first Helsinki Declaration than of the Nuremberg Code. Further, Helsinki introduced the notion of surrogate consent, permitting research when individuals are no longer competent to consent themselves. These moves place a substantial burden on the self-control of the individual researcher, a point to which I shall return later.8 To be sure, until the middle and later 1960s, and with the significant exception of the Nazi experience, to many there did not seem to be good reason for worries about human protections. The development of penicillin, the conquest of polio, and the emergence of new medical devices and procedures apparently unmarked by inappropriate conduct, all bolstered the public prestige of biomedical research. Nevertheless, there were some inklings of a continuing, albeit low-intensity, concern about the concentrated power of medical researchers even in the 1950s, exemplified perhaps in the gradual disappearance from professional discussions of the term “human experiment” and its replacement with the more detached and comforting “research.” On the whole, then, the world of clinical studies from the late 1940s up through the mid-1960s was one in which a weak form of protectionism prevailed, one defined by the placement of responsibility upon the individual researcher. Written informed consent (through forms generally labeled “permits,” “releases,” or “waivers”), though apparently well established in surgery and radiology, was not a common practice in clinical research and in any case cannot be said to provide more than a modicum of increased protection to human subjects. For example, whether a medical intervention was an “experiment” or not, and therefore whether it fell into a specific moral category that required an enhanced consent process, was a judgment largely left up to the researcher. Partly that judgment depended on whether the individual was a sick patient or a healthy volunteer. The former were as likely as not to be judged as wholly under the supervision of the treating doctor even when the intervention was quite novel and unlikely to be of direct benefit. Therefore an individual might be asked to consent to surgery but not be informed beyond some generalities about its experimental aspect.
There were, however, some important exceptions. For example, the Atomic Energy Commission established a set of conditions for the distribution of radioisotopes to be used with human subjects, including the creation of local committees to review proposals for radiation-related projects. Early Institutional Review Boards (IRBs) were established in several hospitals (including early ones at Beth Israel in Boston and the City of Hope in California), in order to provide prior group review for a variety of clinical studies. Another exception seems to have been the Clinical Center of the National Institutes of Health in Bethesda, Maryland, which opened in 1953. A government-supported research hospital, the Clinical Center appears to have been one of a handful of hospitals that required prospective review of clinical research proposals by a group of colleagues.
As advanced as the Clinical Center might have been in this respect, the prior group review process it established seems, at least at first, to have been confined to healthy, normal volunteers. The moral equivalence of at least some sick patients who would probably not be helped by study participation to normal subjects who would not be benefited (with the possible exception of vaccine studies) was apparently not appreciated in policy. These subtleties were largely lost in a period in which medical discretion and societal benefit weighed heavily.
I-6
In Search of the Best Approach
Prior group review is essential to the transition beyond weak protectionism and was not common before the 1970s. Yet decades earlier there was a keen awareness of the psychological vulnerability inherent in the patient role, a vulnerability that could have argued for independent review of a research project. An extensive psychological literature, founded mainly on psychoanalytic theory, propounded a skeptical view of the underlying motivations of experiment volunteers as early as 1954. That year, Louis Lasagna and John M. Von Felsinger reported in Science on the results of Rorschack studies and psychological interviews of 56 healthy young male volunteers in drug research. The authors concluded that the subjects exhibited “an unusually high incidence of severe psychological maladjustment.” “There is little question,” they wrote, “that most of the subjects...would qualify as deviant, regardless of the diagnostic label affixed to them by examining psychiatrists or clinical psychologists.” The authors theorized that this group may not have been representative of the population from which it was drawn (college students), and that they might have been attracted to the study for various reasons having to do with their deviance, beyond financial reward.9 I describe this study at length not to endorse its psychology or its conclusions, nor to imply that neurotic tendencies are either typical of research volunteers or a priori disqualifying conditions for decisionmaking capacity. The point is, rather, that thought was being given as early as 1954 to the question of the recruitment of subjects who may be vulnerable despite their healthy and normal appearance. The article was published in a major scientific journal. It would have been natural to ask further questions about the vulnerability of potential research subjects who are known to be seriously ill. Yet despite this psychological theorizing, which could be viewed as quite damning to the moral basis of the human research enterprise, protectionism was at best a weak force for years to come.
Historians of research ethics generally date the increasing vigor of protectionist sentiment among high-level research administrators, as well as the general public, to the series of events that began with the Thalidomide tragedy and continued with scandals such as the Brooklyn Jewish Chronic Disease Hospital Case and, later, the Willowbrook hepatitis research. These cases cast doubt on the wisdom of leaving judgments about research participation to the researchers’ discretion. The Jewish Chronic Disease Hospital Case, in which elderly debilitated patients were injected with cancer cells, apparently without their knowledge or consent, was one of those that attracted the attention and concern of the NIH director, James S. Shannon. Shannon’s intervention, and the resistance from within his own staff, was an important and revealing moment in the history of human subjects protections.
In late 1963 Shannon appointed his associate chief for program development, Robert B. Livingston, as chair of a committee to review the standards for consent and requirements of NIH-funded centers concerning their procedures. The Livingston Committee affirmed the risks to public confidence in research that would result from more cases like that of the Jewish Chronic Disease Hospital. Nonetheless, in its 1964 report to Shannon the committee declined to recommend a code of standards for acceptable research at the NIH, on the grounds that such measures would “inhibit, delay, or distort the carrying out of clinical research....” Deferring to investigator discretion, the Livingston Committee concluded that NIH was “not in a position to shape the educational foundations of medical ethics....”10 Disappointed but undeterred by the response of his committee, Shannon and Surgeon General Luther Terry proposed to the National Advisory Health Council (NAHC) that the NIH should take responsibility for formal controls on investigators. The NAHC essentially endorsed this view and resolved that human subjects research should only by supported by the Public Health Service if “the judgment of the investigator is subject to prior review by his institutional associates to assure an independent determination of the protection of the rights and welfare of the individual or individuals involved, of the appropriateness of the methods used to secure informed consent, and of the risks and potential medical benefits of the investigation.”11 The following year
I-7
Surgeon General Terry issued the first federal policy statement that required PHS-grantee research institutions to establish what were subsequently called Research Ethics Committees.12 The seemingly innocent endorsement of “prior review by institutional associates” was the most significant single departure from the weakly protectionist tradition to a process that finally yielded the moderately protectionist system we have today.
The surgeon general’s policy was, however, hardly typical of contemporary attitudes, and the practice it sought to implement is one we are still trying to effect. To appreciate the weakness of the form of protectionism that prevailed through the 1960s, it is useful to recall the dominant role that prison research once had in drug development in the United States. By 1974 the Pharmaceutical Manufacturers Association estimated that about 70 percent of approved drugs had been through prison research. Pharmaceutical companies literally built research clinics on prison grounds. Although in retrospect we may think of modern limits on prison research as a triumph of protectionism (on the grounds that prisoners cannot give free consent), at the time it was a confluence of political and cultural forces that had little to do with actual abuses (though there certainly were some), and was resisted by prison advocates. Perhaps the most important public event that signaled the inevitable end of widespread prison research was the 1973 publication of “Experiments Behind Bars” by Jessica Mitford in the Atlantic Monthly.13
Within the medical profession itself, then, weak protectionism remained the presumptive moral position well into the 1970s, if not later. Neither of the most important formal statements of research ethics, the Nuremberg Code and the Helsinki Declaration, had nearly as much effect on the profession as a 1966 New England Journal of Medicine paper by Harvard anesthesiologist Dr. Henry Beecher. The importance of timing is evident in the fact that Beecher had been calling attention to research ethics abuses since at least 1959, when he published a paper entitled “Experimentation in Man,”14 but his 1966 publication “Ethics and Clinical Research”15 attracted far more attention. One important distinguishing feature of the latter work was Beecher’s allusion to nearly two dozen cases of studies alleged to be unethical that had appeared in the published literature. By “naming names” Beecher had dramatically raised the stakes.
It would, however, be an error to conclude that Beecher himself favored external review of clinical trials that would remove them from medical discretion. To the contrary, Beecher was one among a large number of commentators who favored (and in some instances continue to favor) reliance primarily upon the virtue of the investigator. Although he strongly defended the subject’s right to voluntary consent, he argued in his 1959 paper that “an understanding of the various aspects of the problem” being studied was the best protection for the human subject, and was quite critical of the Nuremberg Code’s dictum that the subjects themselves should have sufficient knowledge of the experiment before agreeing to participate.
Beecher’s attitude toward the Code’s provisions was hardly limited to philosophical musings. In 1961 the Army attached a new provision to its standard research contract, rules that were essentially a restatement of the Nuremberg Code. Along with other members of Harvard Medical School’s Administrative Board, Beecher protested and persuaded the Army Surgeon General to insert into Harvard’s research contracts that its Article 51 were “guidelines” rather than “rigid rules.”16 Beecher’s attitude was shared by many other distinguished commentators on research practices through the 1960s and 1970s. In 1967 Walsh McDermott expressed grave doubt that the “irreconcilable conflict” between the “individual good” and the “social good” to be derived from medical research could be resolved, and certainly not by “institutional forms” and “group effort”—apparently references to ethics codes and peer review. McDermott’s comments were by way of introduction to a colloquium at the annual meetings of the American College of Physicians on “The Changing Mores of Biomedical Research.” In his remarks McDermott alluded to the growing contribution of research to the control of disease, beginning with Walter Reed’s yellow fever studies. Thus, he continued, “medicine has given to society the case for its rights in the continuation of clinical investigation,” and “playing God” is an unavoidable responsibility, presumably one to be shouldered by clinical investigators.17
I-8
Another distinguished scientist who made no secret of his skepticism toward the notion that the investigator’s discretion could be supplemented by third parties was Louis Lasagna. In 1971 Lasagna wondered “how many of medicines greatest advances might have been delayed or prevented by the rigid application of some currently proposed principles to research at large.”18 Rather, “for the ethical, experienced investigator no laws are needed and for the unscrupulous incompetent no laws will help....”19 When the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research proposed a moratorium on prison research in 1977, Lasagna editorialized that the recommendations “illustrate beautifully how well-intentioned desires to protect prisoners can lead otherwise intelligent people to destroy properly performed research that scrupulously involves informed consent and full explanation and avoid coercion to the satisfaction of all but the most tunnel-visioned doctrinaire.”20 It is perhaps worth noting that both Beecher and Lasagna had good reason to reflect on the problem of research ethics, stemming from some work they did together. Between 1952 and 1954 Louis Lasagna had been a research assistant in an Army-sponsored project, directed by Beecher, in which hallucinogens were administered to healthy volunteers without their full knowledge or consent. Recalling the episode for the President’s Advisory Committee on Human Radiation Experiments in 1994 interview, Lasagna reflected “not with pride” on the study.21
Tuskegee Changes All
Among those who developed an interest in research ethics during the 1960s was Princeton theologian Paul Ramsey. Although Ramsey is today remembered as one who took a relatively hard line on research protections, and he did in fact significantly advance the intellectual respectability of a protectionist stance, in retrospect his position seems remarkably modest. In his landmark 1970 work, The Patient as Person, Ramsey declared that “No man is good enough to experiment upon another without his consent.”22 In order to avoid the morally untenable treatment of the person as a mere means, the human subject must be a partner in the research enterprise. However, Ramsey was prepared to accept unconsented treatment in an emergency, including experimental treatment that might save life or limb. He also acceded to the view that children who cannot be helped by standard treatment may be experimental subjects if the research is related to their treatment and if the parent consents.
By 1970 the notion that consent was ethically required was well-established in principle (including surrogate consent for children and incompetents), however poorly executed in practice. Ramsey’s contribution was in calling attention to the problem of nonbeneficial research participation, a decision that required at a minimum the human subject’s active participation. As though to underline the point, only two years after Ramsey’s book was published the Tuskegee Syphilis Study scandal broke into the open, a case in which the subjects were clearly not informed participants in the research. The subsequent federal review panel appointed to review the study, the Tuskegee Syphilis Study Ad Hoc Panel, concluded that penicillin therapy should have been made available to the participants by 1953. The panel also recommended that Congress create a federal panel to regulate federally sponsored research on human subjects, a recommendation that foreshadowed and helped define the later transition from weak to moderate protectionism.
A casualty of the syphilis study was the attitude exemplified in the 1967 essay of Walsh McDermott and the 1969 paper by Louis Lasagna. In the years immediately following Beecher’s 1966 article it was still possible to argue that scientists should take responsibility to make what McDermott regarded as appropriately paternalistic decisions for the public good, decisions that recognize that societal interests sometimes take precedence over those of the individual. Although there clearly are instances in which this general proposition is unobjectionable, following the syphilis study such an argument became much harder to endorse in the case of human experiments.
I-9
As the implications of the Tuskegee revelations became apparent, philosopher Alan Donagan published an essay on informed consent in 1977 that symbolized the altered attitude. In Donagan’s essay the invigorated informed consent requirement is taken as nearly a self-evident moral obligation in clinical medicine. In his discussion of informed consent in experimentation, Donagan explicitly compared the arguments of a Nazi defense attorney with those of McDermott and Lasagna, concluding that they are both versions of a familiar and (one infers), a rather primitive form of utilitarianism. Donagan concluded that, by the lights of the medical profession itself, the utilitarian attitudes instanced in the Nazi experiments and the Brooklyn Jewish Chronic Diseases Hospital case, cannot be justified. Perhaps still more telling about the evolution of the moral consensus concerning research ethics is the mere fact that Donagan, a highly respected moral philosopher and not an easily marginalized “zealot,” could associate the arguments of Nazis with those of some of America’s most highly regarded physicians. Donagan’s essay underlined a leap in the evolution of protectionism through the Tuskegee experience, especially on the question of the balance between the subject’s interests and those of science and the public, and on the subsequent discretion to be granted the lone investigator.23
Social Science Research
Less scholarly and regulatory attention has been given to protecting subjects in social science research than in clinical trials, and it might well be said that the emphases of this paper reflect that deficit. Nevertheless, there have been some spectacular instances in which social science research issues erupted into public debate, though the regulatory response has, again, been modest. Perhaps the most intense reaction in this area was generated by Stanley Milgram’s research on obedience to authority.24 Milgram purported to show that normal subjects could be induced to cause pain to others, or to think that they were, simply by being asked to do so by an individual perceived to be in authority, in this case an experimenter. Although there were criticisms of Milgram’s methodology, much of the reaction focused on the harm the study design may have caused the deceived subjects. Also in the early 1970s Philip G. Zimbardo conducted a study of male volunteers’ reactions to a mock prison environment in which some of them were assigned roles as prisoners, others as guards.25 The experiment elicited such strong reactions from the participants, including abuse of the “prisoners” by the “guards,” that Zimbardo halted the study. Milgram’s study design is more typical than Zimbardo’s, in which deception was not an element. Still, both of these cases raise important questions about the relationship between consent and risk.
Deception is an important element of much social psychological research, and is still largely permissible within the framework of a broad consent process. The Ethics Code of the American Psychological Association (APA) requires psychologists to attend to the potential participant’s capacity to consent, and to provide sufficient information about the nature of the research. The code bars excessive financial or other inducements, and mandates an explanation of the voluntary nature of research participation. The APA code permits deception only if its use is justified by prospective scientific benefits and alternatives are not feasible. The deception may not pertain to experiences that would affect prospective subjects’ willingness to participate.26 A new subsection, currently under consideration, would allow participants to withdraw their data once debriefed.27 Although many of the elements of the APA code reflect the standard protectionist model, in context the code also exhibits familiar tensions between scientific progress and individual interests. The mere fact that deception is permitted, albeit carefully hedged with protections, exemplifies the view that research may often justificably violate the usual moral rule that prohibits lying, and to do so in a highly sophisticated and systematic fashion.
There have also been well publicized cases of important social science research that appear to go beyond deception to outright invasions of privacy. In the course of preparing his landmark (and sympathetic) study Tearoom Trade,28 about homosexual behavior among men of high social standing in a large Midwestern city,
I-10
sociologist Laud Humphreys observed men entering a public rest room in a city park, confirmed that they engaged in anonymous homosexual acts, recorded their license tag numbers, and obtained their names from a contact in the bureau of motor vehicles. He was then able to confirm their identity and status in the community. About a year later Humphreys disguised himself and interviewed them in their homes about their personal lives.
Defenders of such research practices argue that they are acceptable so long as the researcher does not disclose the identities of the sometimes unwitting participants. A similar argument may be made for survey research that seeks information concerning intimate and sometimes illegal behavior. Yet one may question whether even knowing participation in potentially embarrassing or, at an extreme, surveys that pose some personal risk to the subjects should be required to undergo more intensive review than is currently the case. Under the Common Rule “survey procedures” are generally considered exempt from protections unless the individual subjects could be identified and “disclosure of the subjects’ responses outside the research could reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects’ financial standing, employability, or reputation (emphasis added).”29 As they will generally not be trained as lawyers, one wonders how much assurance either the subject or investigator can have that information about criminal behavior will not be subject to subpoena by a court of law.
Injuries to Research Subjects
One dimension of Beecher’s attitude toward protectionism expressed a much stronger position than he was prepared to take with regard to investigator discretion. In 1969 Beecher urged that, because damage to subjects may occur even if all appropriate precautions have been taken, “It is unreasonable to expect that the society which profits actually or potentially should not share in the responsibility for what was done.”30 Writing in Science in 1970, a year after Beecher, legal scholar Clark Havighurst argued that societal responsibility would help ensure that unjustifiable risks would not be undertaken if a system would “not only compensate the unlucky subject but also place the burden on those best able to evaluate and control the risks attending the experiment.” Though Beecher and Havighurst both advocated a compensation scheme, Havighurst seemed more inclined to design it in such a way that researchers and research agencies shoulder the burden and not simply society at large. In 1973, the Commission on Medical Malpractice recommended that some party—the researcher, the research institution, the research sponsor, or the federal government—should be required to insure research subjects against injuries.31 Today, however, researchers are only required to disclose on consent forms whether or not they will provide compensation for research risks.32 With a few exceptions, such as veterans of the armed forces who may be eligible for compensation for injuries sustained as part of a Veterans Administration study, there is normally no insurance provided against injuries incurred in the course of a study. Instead, it is standard for consent forms to include language to the effect that emergency care will be provided, but that the sponsoring institutions have made no provisions to compensate for research-related injuries. Some consent forms go further. In the words of one: “[the research institution] will not provide you with financial compensation or reimbursement for the cost of care provided to treat a research-related injury or for other expenses arising from a research-related injury. The institution or group providing medical treatment will charge your insurance carrier, you, or any other party responsible for your treatment costs.” Although this waiver would presumably not apply to injuries flowing from a successful malpractice claim, not all “adverse events” that result in injury to the research subject can be attributed to malpractice. Under those conditions the failure of any involved entity to take financial responsibility for persons who have answered the call to contribute to scientific progress and the public good is hardly the act of a grateful society. In this area the reality of our practice grievously fails to match even the rhetoric of our protectionist philosophy.
I-11
Classified Research
Elsewhere I have explored in detail the history of debates about the ethical problems entailed by research undertaken on sensitive matters related to national security.33 Much of this discussion took place immediately prior to and during the cold war, and relevant documents have only recently become available to scholars. The upshot of this complex story is that government officials did engage in detailed debates about the rules that should apply, and that policies were in fact articulated, though often they were inadequately implemented. Although direct physical injuries to those involved have been difficult to confirm, the experience has indisputably left behind a legacy of distrust that continues to trouble many Americans and depresses the morale of many in the armed forces.
In response to a 1995 recommendation by the Advisory Committee on Human Radiation Experiments (ACHRE), the Clinton administration issued an executive memorandum requiring that all classified research meet the requirement of informed consent and prior group review. Obviously all involved would have to receive appropriate security clearances, including the subjects themselves. Any IRB member who disagreed with the majority concerning a classified study would have the right to appeal to the head of the sponsoring agency or the President’s science advisor. The 17 agencies that have signed onto the Common Rule are now developing an amendment to the regulations that would regularize the requirements set forth in the President’s memorandum.
Protectionism Today: An Assessment
On the account I have presented, protectionism is the view that a duty is owed those who participate as subjects in medical research. The underlying problem is how to resolve the tension between individual interests and scientific progress, where the latter is justified in terms of benefits to future individuals. Weak protectionism is the view that this problem is best resolved through the judgment of virtuous scientists. Moderate protection-ism accepts the importance of personal virtue but does not find it sufficient. Strong protectionism is disinclined to rely on the virtue of scientific investigators for purposes of subject protection to any substantial degree.
The Common Rule largely relies on a moderately protectionist approach to subject protection. In so doing, it deploys two principle techniques to constrain investigator discretion: informed consent and prior group review. More strongly protectionist approaches, such as monitoring procedures, would gradually impose more direct controls over the actual consent process and the study activities themselves. Data safety and monitoring boards provide some precedent for such intervention, but their primary rationale is as compensation for the methodological necessity of double-blind study design.
In many respects our contemporary system of human subjects protections is a triumph of moderate protec-tionism. Consider for example the position exemplified in a recent essay on ethics in psychiatric research, in which the authors state that “the justification for research on human subjects is that society’s benefit from the research sufficiently exceeds the risks to study participants.” But then the authors continue, “potential risks and benefits must be effectively communicated so that potential subjects can make informed decisions about participation.”34 The current battleground, then, is not whether the subjects should in theory be full participants, or whether prior review of experiment proposals should be required, but whether, or to what extent, subjects can take an active role in the clinical trials process. The extent to which such active participation is possible may help to forestall the introduction of more strongly protectionist requirements.
The tone for the current debate was established by the late 1970s and embodied in the work of the National Commission. With the storm of the syphilis study at their backs, the members of the National Commission could go further in specifying protections for research subjects than would have been possible only a few years before. The National Commission made three critical contributions to the protectionist movement:
I-12
the establishment of principles underlying human subjects protections; the identification of populations that needed to be singled out for special protections (fetuses, prisoners, children, and the mentally infirm); and the distinction between research and medical practice. The distinction between research and practice is especially important because it goes to the question that I have argued is critical in the emergence of stronger forms of protectionism: the scope of the physician-investigator’s discretion. One National Commission recommendation that would have substantially modified the scope of discretion for some investigators was that of the “consent auditor” who, “where appropriate,” would be charged by the IRB to observe and verify the adequacy of the consent process for persons institutionalized as mentally infirm.35 Nonetheless, the story I have to tell is not one of an inexorable march toward a stronger form of protectionism, even in the past 20 years. Although the tendency since the advent of the Nuremberg Code—greatly strengthened in the United States by the “Belmont Report”—has been to limit the scope of investigator discretion, there have been countervailing forces. One of these has been the Declaration of Helsinki, which uses the concepts of therapeutic and nontherapeutic research, defining the former as “Medical Research Combined with Professional Care.” According to Helsinki IV (1989), “If the physician considers it essential not to obtain informed consent, the specific reasons for this proposal should be stated in the experimental protocol for transmission to the independent committee.” Thus Helsinki continues to contemplate a relatively permissive attitude toward investigator discretion, as it has since the first version in 1954. Notably, Henry Beecher preferred Helsinki to Nuremberg precisely because the former is a “set of guides” while the latter “presents a set of legalistic demands.”36 Another force counteracting the tendency to limit investigator discretion has been movements on behalf of greater access to clinical trials. The most pronounced expression of this effort has occurred among AIDS activists, who successfully insisted upon the creation of alternative pathways for anti-AIDS drugs in the late 1980s. In the face of a disease that resisted treatment and struck down people just entering the prime of life, the determination to find solutions was understandable. The slogan of ACT-UP (AIDS Coalition to Unleash Power) that “A Drug Trial is Health Care Too,” was a political expression of confidence in the power of science. As well, the slogan betrayed assumptions about the benefits of research participation and the self-discipline of the medical research community, as well as relying on the very protections it sought to undermine. It should be said that activist organizations have largely revised their attitude toward alternative pathways of access to nonvalidated medications.
Other developments at the federal level in the 1980s and 1990s have been more consistent with the trend toward strengthened protections. The President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research made recommendations on the evaluation and monitoring of IRB performance,37 and also endorsed the proposition that research-related injuries should be compensated.38 Among the recommendations of the Advisory Committee on Human Radiation Experiments in 1995 were several that addressed improved human subject protections. For example, the ACHRE urged that regulations be established to cover the conduct of research with institutionalized children and that guidelines be developed to cover research involving adults with questionable competence. The ACHRE also recommended steps to improve existing protections for military personnel concerning human subject research. Substantial improvements were urged in the federal oversight of research involving human subjects: that outcomes and performance should be evaluated beyond audits for cause and paperwork review; that sanctions for violations of human subjects protections be reviewed for their appropriateness in light of the seriousness with which the nation takes failures to respect the rights and welfare of human subjects; and human subjects protections be extended to nonfederally funded research. The ACHRE also recommended that a mechanism be created for compensating those injured in the course of participation as subjects of federally funded research.39 On May 17, 1997, the National Bioethics Advisory Commission (NBAC) unanimously adopted a resolution that “No person in the United States should be enrolled in research without the twin protections of informed
I-13
consent by an authorized person and independent review of the risks and benefits of the research.”40 That same month President Clinton stated that “[w]e must never allow our citizens to be unwitting guinea pigs in scientific experiments that put them at risk without their consent and full knowledge.”41
Federal Rules and Reports: In Pursuit of Protections
The contemporary presumption that protectionism is and ought to be the governing philosophy of modern human subjects research has been reflected in several federal reports on the efficacy of prevailing research rules in protecting human subjects, especially the adequacy of the IRB system. The IRB concept is predicated on the protectionist assumption that, contrary to the views of Beecher and other earlier commentators, physician authority concerning the appropriateness of research participation must be subject to the formal constraints of a third party, in this case, a committee of peers and laypersons. It may be useful to review the provenance of the IRB system.
Since the passage of the 1974 National Research Act (Public Law 94-348), universities and other research centers have been required to use what it called Institutional Review Boards to protect the rights and welfare of human subjects. Research institutions provide the Department of Health and Human Services with single- or multi-project assurances that their IRBs will apply the federal rules to all federally funded research conducted at the institution or by its employees; many assurances encompass all research with human subjects regardless of sponsorship.
The National Research Act also transferred oversight of research involving human subjects to a new organization within the National Institutes of Health, the Office for Protection from Research Risks (OPRR). In 1974 the Department of Health, Education, and Welfare (DHEW, now DHHS), also adopted regulations (45 CFR 46 under Section 491 of the Public Health Service Act) that made IRBs responsible for determining whether potential subjects are “at risk” in proposed research, and if so, whether the risks outweigh the possible benefits to them and the importance of the knowledge to be gained.
In 1991 a single set of regulatory protections governing human subjects research was adopted by sixteen federal departments and through an executive order, applied to the Central Intelligence Agency as well. These general provisions are known as the Common Rule, and are identical to the basic DHHS policy for the protection of research subjects, 45 CFR 46, subpart A. Subsequently, the Food and Drug Administration (FDA) changes in its informed consent and institutional review regulations to bring them into general conformity with the Common Rule.
However, in March 1996 the United States General Accounting Office (GAO) published “Scientific Research: Continued Vigilance Critical to Protecting Human Subjects.”42 Conceding a lack of systematic studies of government efforts to ensure compliance with human protections standards, the report found that the current activities generally work to prevent harm to research participants. Through interviews with individuals familiar with the system, the GAO report anticipated a number of themes that resurfaced in subsequent studies. It stated that the oversight system is “impaired by IRBs’ heavy workloads and competing demands, limited funds for on-site inspections, the complexity and volume of research under review, and reliance on researchers’ self-assurances that they are complying with requirements.” In the same spirit as the GAO report, in June 1998 the Department of Health and Human Services Inspector General (IG) published, “Institutional Review Boards: A Time for Reform.” The IG report was organized in four separate documents, one an “Overview and Recommendations,” and the others on different aspects of the current status of IRBs: “Their Role in Overseeing Approved Research,” “The Emergence of Independent Boards,” and “Promising Approaches.”43 The IG recommendations included several that would reform federal IRB requirements so that they would have more flexibility but also more accountability. To strengthen IRB
I-14
oversight the IG suggested mandating Data Safety Monitoring Boards (DSMBs) for multi-site trials. It would also require the FDA to inform IRBs about sanctions against investigators, and sponsors and investigators to inform them about prior IRB review of a research plan. The report recommended that IRBs increase their awareness of actual research practices by visiting study sites. Although the authors noted that such observations would represent a departure from the historic relationship between IRBs and investigators, in fact IRBs already have the authority to conduct active monitoring, though this is rarely done.
The report also recommended that both investigators and IRB members receive training in research ethics. To this end, it urged that the Public Health Service require that all its grantee institutions have a program to train investigators in human subject protections, similar to the current NIH requirement for trainees. Investigators should be required to sign a written attestation that they are familiar with and will uphold federal subject protection policies, and institutions should certify that there is a continuing education program for IRB members. There were also recommendations concerning conflicts of interest, workload pressures on IRBs, and strengthening the federal capacity to deal with IRB performance problems as they arise.
The Inspector General noted the increase in independent or private IRBs, which are created outside of organizations that conduct research in order to satisfy federal requirements for board review of clinical research proposals. Although these boards are more efficient than traditional research center-based IRBs, they are not the sort of local review bodies envisioned in previous understanding of human subjects protections. They are also alleged to contribute to conflict of interest concerns and worries about the potential for “IRB shopping,” in which sponsors go from one board to the next until they find one that approves their study.
The Inspector General concluded that the IRB system is in jeopardy because the local boards are overworked, they fail to oversee approved studies, their members lack sufficient training, and they face inherent conflicts of interest. These problems persist, the IG report continued, because the Office for Protection from Research Risks and its counterparts in other departments have neither the resources nor the independence to provide adequate guidance to IRBs, much less to monitor their activities. Two years after the 1998 report, in April 2000, the Inspector General expressed her concern that in the intervening years there had been “minimal progress in strengthening continuing protections for human subjects participating in research.” Some “promising steps” have been taken by NIH, however, including a new requirement that DSMBs share information with IRBs, new initiatives for IRB member and investigator education, and a website of bioethics resources.44 Although I am largely in agreement with the Inspector General’s continuing criticisms of the current system—especially with regard to the lack of fit between the current research environment and the decades-old IRB arrangement, the need for IRB member and investigator education, and increased study monitoring—the extent of the problem should not be exaggerated. It is worth recalling some of the conclusions of the only comprehensive empirical study of the IRB system, the 1998 report of the NIH Office of Extramural Research, which found that about 10 percent of IRBs review nearly 40 percent of the protocols, indicating that the large academic research centers are especially hard pressed. This result is somewhat reassuring insofar as it suggests that the problems are mostly manageable and found at institutions that have considerable stocks of human (if not financial) resources to deal with them.45 One population that the National Bioethics Advisory Commission itself singled out for special protection is that of persons with mental disorders that may affect decisionmaking capacity. In its December 1998 report the NBAC issued a number of recommendations concerning IRB approval of research proposals involving this population. The report recommended that IRBs reviewing such proposals have two members familiar with the concerns of persons with mental disorders in research, and that protocols should not include persons from this population in research if the research can be done with others. It would also have IRBs look for specific elements of protocols before granting approval to clinical studies with this population, for example, that the capacity assessment of potential subjects is conducted by a psychiatrist not involved in the research, and that investigators specify methods for minimizing risk and evaluate risks and benefits.
I-15
The NBAC report also recommended the creation by the DHHS Secretary of a Special Standing Panel (SSP) on research involving persons with mental disorders that may affect decisionmaking capacity. The SSP would review research that could not otherwise be approved with this population under the NBAC recommendations and promulgate guidelines for local IRBs that may reduce the need for SSP approval. The SSP thus has some characteristics that may apply to a national human subjects office, although the report did not address the broader role of such an entity.
Confidentiality
Considering that patient confidentiality is perhaps the most ancient and deeply held moral value in medicine, it may be surprising that modern protectionism, at least as expressed in the bioethical literature, has had relatively little to say about this topic. A classic paper by Siegler in 1982 depreciated confidentiality as a realistic attribute of modern medical institutions and may have served to dampen interest in the topic. In support of his suggestion that confidentiality may be a “decrepit” concept in practice, Siegler found that at least 75 individuals in one academic medical center had legitimate access to a patient’s chart.46 At the policy level, some protection of medical information is afforded by the 1974 Federal Privacy Act (P.L. 93-579), and the National Privacy Commission filed a report in 1976, but there is still no comprehensive federal legislation to protect medical information. The protection of sensitive information stemming from clinical research is to some degree covered by the Public Health Service Act. The Act “provides for ‘certificates of confidentiality’ which offer a legal basis for protection against civil, criminal, administrative, legislative, or other proceedings to force disclosure of personally identificable data.”47 However, the certificate system places a higher burden on the claim of confidentiality than is usually thought to be required in physician-patient relations.
Several factors have motivated a renewed concern about confidentiality protections, including utilization review as part of “gatekeeping” strategies in the proliferating managed care marketplace, the increasing use of electronic records, and the foreseen integration of genetic data into patient histories. Specifically with regard to clinical trials, the need to recruit larger numbers of subjects for more complex studies makes access to patient records an attractive opportunity to identify medically appropriate potential subjects. Individuals sought for studies that attempt to measure the prevalence of genetic alterations in a population may also feel themselves to be at risk if positive test results become known.
In spite of longstanding expressions of concern about the privacy of electronic records and genetic information in particular, it has been difficult to achieve agreement on confidentiality standards. The continuing confusion about medical records and confidentiality protections is reflected in the current debate about rules currently proposed by the Department of Health and Human Services. In 1996, Congress passed a law that required DHHS to issues rules protecting medical records that were transmitted through computers if Congress itself failed to pass legislation on medical privacy with a certain period. As the self-imposed deadline came and went last year with a new law, the rule-making process was triggered.
The proposed rules would give patients the right to view and amend their medical records, and require physicians and health care institutions to give notice of their intent to use medical information and track that which is disclosed. They would also make health plans and insurers responsible for monitoring the activities of outside contractors who have access to patient data. However, some critics charge that there would be no informed consent for access to records if they are being used for treatment, to obtain payment for health care services, or for what the proposed rules call “health care operations.” In some cases the rules would also enable health care providers to release medical information to policy, employers, government data banks, and researchers without consent.48 Apart from the limits of the currently proposed rules, a comprehensive approach to the problem of confidentiality of data gathered in the course of research probably cannot avoid confronting the problem posed by
I-16
the Common Rule’s narrow definition of research: “a systematic investigation designed to develop or contribute to generalizable knowledge.” Under this definition there are numerous “nonresearch” projects that systematically collect and utilize data from medical records, including program evaluations in public health and utilization review in health services management.49 Semantic niceties should not be allowed to circumvent the legitimate public policy goal of maintaining the confidentiality of medical information.
Summary and Recommendations
The current system of human subjects protections in the United States, formally embodied in the Common Rule, is expressive of a moderately protectionistic philosophy of research ethics. For example, I have asserted that the first critical issue in a system that regulates human subjects research is the relationship between the interests of the subject and those of science and “future patients.” The common rule permits legally competent individuals to consent to research participation even though it is not designed to benefit them, but the risks must fall within an acceptable range as determined by an IRB. A weakly protectionist philosophy could dispense with IRB approval, while a strongly protectionistic approach might not find informed consent for certain kinds of research acceptable, even with IRB approval (owing, perhaps, to institutional or other pressures that are substantial but may not rise to the level of coercion or manipulation).
The second critical issue that determines the level of protectionism in a human subjects research regulatory system is whether and in what manner the conduct of the investigator may be monitored or controlled by third parties. The current system in the United States is again moderately protectionistic in this respect because it requires prior review of protocols by an IRB and permits the IRB to engage in concurrent monitoring of the study itself. Thus it provides more protection than a system that places a greater burden on the virtue of the individual investigator, as advocated by Beecher and other early commentators. But the common rule currently provides less protection than a system that requires external assessment of the consent process. A step in this direction is exemplified in NBAC’s recommendation that an independent assessment should be sought for a potential subject’s capacity to consent to research protocols involving greater than minimal risk, in cases when that subject has a mental disorder that may affect decision making capacity.50 However, institutional resistance to the National Commission’s related proposal for consent auditing for those institutionalized as mentally infirm in 1978 suggests that more protectionist proposals have long been against the grain of our system and does not augur well for NBAC’s recommendation.
A system that attempts to balance scientific advancement with the interests of individuals (while holding the latter as ultimately constraining the former) is bound to require continuous reinterpretation and “tuning up.” The following recommendations are therefore made in an evolutionary spirit and presume that our society is, in its collective judgment, currently moving toward a more vigorously interventionist interpretation of what remains at bottom a moderately protectionist attitude toward the regulation of clinical trials. At the same time, they do not presuppose significant changes in the attitudes of the clinical research community, which can be relied upon to continue to resist, not wholly without merit, regulation that it perceives as creating bureaucratic obstacles rather than genuine protections.
Informed Consent
NBAC should reaffirm its 1997 resolution that “No person in the United States should be enrolled in research without the twin protections of informed consent by an authorized person and independent review of the risks and benefits of the research,”51 and should further resolve that this standard become federal law.
There is no good reason—moral, constitutional, or financial—to do without a federal law that guarantees these protections regardless of the source of funding or sponsorship. The Common Rule already serves as a
I-17
virtual common law standard and scientific researchers who work with human subjects would be foolish indeed to ignore informed consent, no matter who is supporting their projects. Specific provision should be made for a requirement of informed consent for classified research.
Financial Conflict of Interest
Investigators should be required to disclose to potential subjects any financial interests in the research.
The disclosure of financial interests that could reasonably be construed as presenting conflicts is a well-recognized duty in other professions. Considering the growing proportion of research that is privately funded and the commercial nature of much of this research, the exceptionalism traditionally granted to physicians with respect to financial disclosure is hard to justify. Possible delays in recruiting subjects for promising research and embarrassment on the part of investigators are not acceptable reasons for failure to bring this information to light. In fact, subjects themselves will likely find this information less interesting than IRBs, who will have to face the problem of determining whether certain financial arrangements should be modified.
Decisionmaking Capacity
Investigators should be required to explain to IRBs how they will assess decisionmaking capacity on a continuing basis for persons known to have a history of diminished capacity or are likely to lose capacity for a significant period during a study.
Capacity assessments should not be a windowless box within which investigators have unlimited discretion, particularly considering that important human rights are engaged when persons are exposed to circumstances (regardless of level of risk or theorized benefit) to which they might not otherwise agree. Research involving persons with questionable capacity to consent will increase as new experimental medications to treat neurologic and psychiatric disorders become available, and as new treatment for those who are gravely ill is developed. It is not an undue burden to ask investigators to document a procedure that, presumably, must already be part of their ethically conducted research.
Surrogate Consent
States should clarify the circumstances under which a legal authorized representative (LAR) may give permission for research involving a person who lacks decisionmaking capacity, and whether individuals may give advance authorization for such research if they should lose decisionmaking capacity.
Currently there is often uncertainty about who can function as a LAR under state law and about the scope of their decisionmaking authority. As a result, many clinicians are operating in legally and morally ambiguous territory. In particular, states should clarify whether a LAR has the authority to authorize important research that poses some risk to the subject without the prospect of direct benefit to that person. States should also consider whether individuals should be able to express their wishes concerning such research participation while they still have the capacity to express themselves.
Research Risks
The NBAC or another appropriate federal panel should design and recommend an indemnification system for persons injured in the course of participation as subjects in clinical trials.
Consent forms commonly warn that the sponsoring institution cannot be responsible for injuries incurred as a result of the study. Whatever their legal status, from a moral standpoint these warnings have a distinctly hollow ring, and leave the impression that our society places little value in the willingness to be part of the research enterprise. The recommendations of the 1973 Commission on Medical Malpractice should be revisited and a scheme for insuring persons against the risk of injuries sustained due to research participation should be devised.
I-18
Confidentiality
Federal research regulations should provide for clear and unambiguous limitations to access to medical records linked to individuals.
Those who agree to be subjects in medical research should not have to be concerned about the disposition of data with implications about their health status that may be obtained in the course of a study. Regulations should clearly prohibit unconsented access and release of medical records, including those accumulated in a research context, that can be associated with identified individuals. Activities that skirt the definition of research, such as “program evaluations” in public health and “quality assurance” in managed care, should be subject to scrutiny. Effective action in this area may require that the statutory definition of research, couched in terms of “generalizable knowledge,” be revisited.
IRB Activities
IRBs should be required to register with the Office for Protection from Research Risks, to compile annual data on the number of research proposals reviewed and the number approved, and the number of subjects in research that has been approved.
Many have commented on the peculiarity that more is known about Animal Care and Use Committee activities than is known about IRB activities. These modest requirements would help to correct that imbalance.
Education
All IRB members should receive initial and continuing education in the history and philosophy of human subjects research, in the current regulations governing such research, and in current issues in the field. Familiarity with federal human subjects protections should also be required of researchers who function as principle investigators.
Many observers have noted wide disparities in the familiarity of IRB members with the regulations they are responsible for interpreting and enforcing. Similarly, investigators should be aware of the rules that condition their work. Current initiatives to create accreditation programs for institutions and their research review system should serve as an impetus on the IRB side. Further measures may be required to help ensure investigator familiarity with the regulations, such as a signed attestation as part of the material submitted for IRB review.
Notes
1 Hans Jonas, quoted in Experimentation with Human Beings, Jay Katz, ed. (New York: Russell Sage Foundation, 1972), p. 735.
2 Susan E. Lederer and Michael A. Grodin, “Historical Overview: Pediatric Experimentation,” in Children as Research Subjects: Science, Ethics, and Law, Michael A. Grodin and Leonard H. Glantz, eds. (New York: Oxford University Press, 1994).
3 Susan E. Lederer, Subjected to Science: Experimentation in America before the Second World War (Baltimore: Johns Hopkins University Press, 1995).
4 Walsh McDermott, “Opening Comments on the Changing Mores of Biomedical Research,” Annals of Internal Medicine 67 (Supl. 7):39–42, 1967.
5 Jonathan D. Moreno, Undue Risk: Secret State Experiments on Humans (New York: W.H. Freeman, 1999).
6 Advisory Committee on Human Radiation Experiments, The Human Radiation Experiments (New York: Oxford University Press, 1996), p. 63.
7 Advisory Committee on Human Radiation Experiments, The Human Radiation Experiments (New York: Oxford University Press, 1996), p. 55–56.
8 Ruth R. Faden and Tom L. Beauchamp, A History and Theory of Informed Consent (New York: Oxford University Press, 1986).
9 Louis M. Lasagna and John M. Von Felsinger, quoted in Experimentation with Human Beings, Jay Katz, ed. (New York: Russell Sage Foundation, 1972), pp. 623–624.
I-19
10 Advisory Committee on Human Radiation Experiments, The Human Radiation Experiments (New York: Oxford University Press, 1996), pp. 99–100.
11 Dr. John S. Reisman, the Executive Secretary, NAHC, to Dr. James A. Shannon, 6 December 1965 (“Resolution of Council”).
12 William J. Curran, “Governmental Regulation of the Use of Human Subjects in Medical Research: The Approach of Two Federal Agencies,” in Experimentation with Human Subjects, Paul A. Freund, ed. (New York: George Braziller, 1970), pp. 402–454.
13 Jessica Mitford, “Experiments Behind Bars Doctors, Drug Companies, and Prisoners,” Altantic Monthly 23:64–73, January 1973.
14 Henry K. Beecher, “Experimentation in Man,” JAMA 169:461–478, 1959.
15 Henry K. Beecher, “Ethics and Clinical Research,” New England Journal of Medicine 274:1354–1360, 1966.
16 Advisory Committee on Human Radiation Experiments, The Human Radiation Experiments (New York: Oxford University Press, 1996), pp. 89–91.
17 Walsh McDermott, “Opening Comments on the Changing Mores of Biomedical Research,” Annals of Internal Medicine 67 (Supl. 7):39–42, 1967.
18 Louis Lasagna, “Some Ethical Problems in Clinical Investigation,” in Human Aspects of Biomedical Innovation, Everett Mendehlsohn, Judith P. Swazey and Irene Taviss, eds. (Cambridge, MA.: Harvard University Press, 1971), p. 105.
19 Ibid., p. 109.
20 Louis Lasagna, “Prisoner Subjects and Drug Testing,” Federation Proceedings 36(10):2349, 1977.
21 Louis Lasagna interview by Jon M. Harkness and Suzanne White-Junod (ACHRE), transcript of audio recording, 13 December 1994 (ACHRE Research Project Series, Interview Program File, Ethics Oral History Project), 5.
22 Paul Ramsey, The Patient as Person: Explorations in Medical Ethics (New Haven, CT: Yale University Press, 1970), pp. 5–7.
23 Alan Donagan, “Informed Consent in Therapy and Experimentation,” Journal of Medicine and Philosophy 2:318–329, 1977.
24 Stanley Milgram, Obedience to Authority (New York: Harper & Row, 1974).
25 Ruth Faden and Tom L. Beauchamp, A History and Theory of Informed Consent (New York: Oxford University Press, 1986), pp. 178–179.
26 American Psychological Association, “Ethical Principles of Psychologists and Code of Conduct,” American Psychologist 47(1597–1611), 1992.
27 Celia Fisher, personal communication, May 15, 2000.
28 Laud Humphreys, Tearoom Trade: Impersonal Sex in High Places (Aldine de Gruyter, 1975).
29 45 CFR 46.101(b)(2)(i).
30 Henry Beecher, “Human Studies,” Science 164:1256–1257, 1969. Clark Havighurst, “Compensating Persons Injured in Human Experimentation,” Science 169:153–154, 1970.
31 Medical Malpractice: Report of the Secretary’s Commission on Medical Malpractice (DHEW Pub. No. OS 73-88, 1973), p. 79.
32 45 CFR 46.116(a)(6) (1981).
33 Jonathan D. Moreno, Undue Risk: Secret State Experiments on Humans (New York: W.H. Freeman, 1999).
34 Jeffrey A. Lieberman, Scott Stroup, Eugene Laska et al. “Issues in Clinical Research Design: Principles, Practices, and
Controversies,” in Ethics in Psychiatric Research, Harold A. Pincus, Jeffrey A. Lieberman, and Sandy Ferris, eds. (Washington, DC: American Psychiatric Association, 1999), pp. 25–26.
35 National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, Report on Those Institutionalized as Mentally Infirm (Washington, D.C.: Department of Health Education and Welfare, 1978), pp. 8–11.
36 Sir William Refshauge, “The Place for International Standards in Conducting Research for Humans,” Bulletin of the World Health Organization 55, 133–35 (Supl. 1977) (Quoting Henry K. Beecher, “Research and the Individual,” Human Studies 279 (1970)).
37 President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research, Implementing Human Subject Regulations (Washington, D.C.: GPO, 1983).
I-20
38 President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research, Compensating for Research Injuries: The Ethical and Legal Implications of Programs to Redress Injured Subjects, Vol. I, Report (Washington, D.C.: GPO, June 1982).
39 Advisory Committee on Human Radiation Experiments, op. cit., pp. 527–528.
40 National Bioethics Advisory Commission, Full Commission Meeting, Arlington, Virginia, May 17, 1997.
41 William Jefferson Clinton, Morgan State University Commencement Address, May 18, 1997.
42 “Scientific Research: Continued Vigilance Critical to Protecting Human Subjects” (Letter Report, 03/08/96, GAO/HEHS-96-72).
43 Department of Health and Human Services, Inspector General, Institutional Review Boards: A Time for Reform (Washington, D.C.: Department of Health and Human Services, 1998).
44 Office of Inspector General, “Protecting Human Subjects: Status of Recommendations,” April 2000.
45 Jonathan D. Moreno, “IRBs Under the Microscope,” Kennedy Institute of Ethics Journal 8(3):329–337, 1998.
46 Mark Siegler, “Confidentiality in Medicine-A Decrepit Concept,” New England Journal of Medicine 307:1520, 1982.
47 Charles R. McCarthy and Joan P. Porter, “Confidentiality: The Protection of Personal Data in Epidemiological and Clinical Research Trials,” Law, Medicine and Health Care 19:240, 1991.
48 “Groups Warn of Breaches of Privacy Laws for Patients,” The Washington Post, April 16, 2000, p. A02.
49 Robert Amdur, Marjorie Speers, and Elizabeth Bankert, “IRB Triage of Projects That Involve Medical Record Review,” IRB.
50 National Bioethics Advisory Commission, “Assessing Potential Subjects’ Capacity to Decide about Participating in a Research Protocol”(Recommendation 8), Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity, December 1998.
51 National Bioethics Advisory Commission, Full Commission Meeting, Arlington, Virginia, May 17, 1997.
I-21

FEDERAL AGENCY SURVEY ON POLICIES AND PROCEDURES FOR THE PROTECTION OF HUMAN SUBJECTS IN RESEARCH
National Bioethics Advisory Commission Bethesda, Maryland
J-1
Introduction
In 1991, a single set of regulations, referred to as the Common Rule (The Federal Policy for the Protection of Human Subjects in Research), was published in the Federal Register and adopted independently by 16 federal departments and agencies (see Table 1).1 The adoption of this set of common regulations was a benchmark event in the United States for addressing concerns about the uniformity of the system of human subjects protection throughout the federal government. It represented the culmination of a 10-year effort to produce a single set of regulations, something that had been recommended by the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research in 1981.
In 1995, the final report of the Advisory Committee on Human Radiation Experiments expressed concerns about the adequacy and uniformity of the implementation of the regulations among the signatory agencies. In his 1995 Executive Order (12975) establishing the National Bioethics Advisory Commission (NBAC), President Clinton directed every federal department and agency that conducts, supports, or regulates research involving human subjects to report to NBAC regarding the protections afforded to human subjects by each department’s or agency’s existing policies and procedures. This analysis describes the final results of a data collection process conducted by NBAC staff and consultants over a three-year period to describe and assess federal policies and practices related to protecting human subjects in research. The data reported here reflect the status of agency activities as of February 2000. This report does not include descriptions of activities or changes in agency functions that have occurred since February 2000.2
Background on NBAC Data Collection Efforts
The initial responses by federal agencies to the President’s 1995 request were variable. To follow up on the initial data provided, NBAC staff and consultants undertook an examination of each department’s activities to protect human subjects in research and the structures, policies, and procedures in place for the review and oversight of human subjects protections. Each department head was contacted and invited to appoint a representative to discuss with NBAC all departmental activities, policies, and procedures involving the protection of human subjects in research.
A survey tool for “Phase I” of the data collection was developed as the basis for individual interviews that were conducted with each departmental representative. The survey was designed to assess both the department’s level of compliance with the Common Rule, and, for those departments that were in compliance, any difficulties they encountered in adhering to the regulations. In addition, several questions solicited suggestions from the departments for the improvement of federal protections for human subjects. Both the questions and methods of the survey were reviewed by outside experts.3 After several pilot interviews, survey questions were added to include commonly mentioned topics, such as ethical issues in international research. Each interview included follow-up questions and department-specific discussions, which served to illuminate those practices unique to each institution.
The survey questions, along with an information sheet, were mailed to each departmental representative. NBAC staff arranged to meet, in person, with representatives from every department, including those departments that responded to the survey in writing. Interview notes were supplemented by departmental charts, written policies and regulations, and other materials collected at the meeting. Several departments found it difficult to provide a department-wide response to the survey; in such cases, the agencies within the department that sponsored research with human subjects were interviewed separately.
The major purpose of the Phase I survey was to examine what structures—i.e., organizational units, personnel, and written policies and procedures—were in place to protect human research subjects, especially those related to the Common Rule.
J-3
Staff and consultants then embarked on “Phase II” of the survey, the purpose of which was to examine, among those organizations with structures in place, what processes were followed to protect human subjects, particularly related to the Common Rule. The respondents were asked to respond to open-ended questions about their perceptions of policies and procedures in their agency. Phase II of the study was never completed; it was thought that the data from Phase I deserved a fuller evaluation before continuing with lengthy interviews.
Based on the data collected in Phase I (and only somewhat in Phase II), staff and consultants characterized the status of agency compliance with the regulations (at the time of NBAC’s interview with each agency) of implementation of procedures and policies to protect human research subjects. Determinations of agency status were made based on staff/consultant interpretations of the data collected in the two phases of the survey. They are, in large part, based on staff assessments of what constitutes research, minimal risk, vulnerable subjects, and adequacy of structures and procedures in place. Out of these analyses a draft report was written and distributed to the federal agencies for comment. The report was also shared with NBAC.
On October 2, 1998, the Office of Science and Technology Policy sponsored a meeting with federal agency representatives, NBAC commissioners, and staff to discuss the draft report. Commissioners R. Alta Charo, James Childress, and Bette Kramer attended that meeting. In addition, an ongoing exchange of information occurred between NBAC staff and the agencies. At several meetings of the Human Subjects Research Subcommittee of the Committee on Science, National Science and Technology Council, NBAC staff briefed agency representatives on the status of NBAC’s work. NBAC staff also invited agencies to submit, on an ongoing basis, information about changes in their human subjects protections policies and procedures. Thus, data have accumulated over time.
On May 4, 1999, NBAC Chairman, Harold T. Shapiro sent a memorandum to the President summarizing general concerns about human subjects protections that had been raised by these initial reviews. Areas of concerns were the following:
Methods for Completing Data Collection and Analysis
In fall 1999, NBAC staff and consultants began to re-evaluate the data collected in Phase I of the study and concluded that these data provide a useful starting point for the Commission’s assessment. The partial data collected in Phase II, however, are not particularly informative. It was decided that because significant time had been passed since the initial data collection, and because the evolving work of NBAC had raised new issues and concerns about human subjects protections, it would be necessary to collect more timely and complete data from the same set of agencies.
J-4
On November 12, 1999, Dr. Shapiro sent a letter to each of the agency representatives informing them of NBAC’s intentions. The letter requested that the agencies provide NBAC with an update on any changes that have occurred in their human subjects protections policies and procedures since they last reviewed and approved the Phase I data. In December 1999, a questionnaire was sent to each department or agency head requesting a response by mid-February 2000 (see Appendix A). Sixteen agencies and their relevant subcomponents responded to the survey (see Table 2). As of October 1, 2000, the Department of Agriculture has not responded to this request. This report describes the results of the survey analysis.
Survey Results
The Size and Scope of Human Subjects Research Supported by the Federal Government
All 16 federal departments and agencies responding to the survey conduct or support research involving human subjects, although some components within departments reported that they do not sponsor or conduct humans subjects research (e.g., the Administration on Aging in the Department of Health and Human Services [DHHS]). Each agency’s human subjects research program is distinctive in terms of its size, scope, organization, and focus, all of which reflect the primary mission of the agency. The following examples illustrate the diverse types of research conducted and/or supported by the federal agencies:
J-5
In the questionnaire NBAC suggested that agencies use the definition of “human subject” as provided at Section 102(f) of 45 CFR 46: “A human subject means a living individual about whom an investigator (whether professional or student) conducting research obtains (1) data through intervention or interaction with the individual, or (2) identifiable private information…” This definition was uniformly applied in the agency responses. Only DOT indicated a divergence from this definition. DOT treats human cadavers as human subjects in safety research conducted by the agency.
Budget Data
Each agency was asked to provide estimates of total budgets, research budgets,4 and human subjects research budgets5 for Fiscal Year (FY) 1999 (see Table 3). Of the responding agencies, only the Central Intelligence Agency (CIA) was unable to provide budget data because it is classified.
These data were gathered to understand the relative resource allocations to research in each agency, that is 1) the proportion of the budget spent on research regardless of the type (e.g., human, animal, weapons),
J-6
2) the proportion of the research budget spent on research involving humans, and 3) the proportion of the human subjects research budget conducted within (intramural or in-house) the agency. Some departments provided detailed budget data by agency (e.g., DOJ provided budget data for its four divisions; see Table 2 for listing). Others aggregated all data into one overall figure.
The amount of funding devoted to research or human subjects research as a percent of total department funding varied enormously. For example, although the SSA had a budget of over $421 billion, it spent less than $30 million on human subjects research. Likewise, although DOD had the largest overall research budget (nearly $36 billion), only $37 million was allocated to human subjects research.
By far, DHHS is the largest federal sponsor of research involving human subjects, totaling nearly $9.3 billion in FY 1999, the largest portion of which is allocated to the National Institutes of Health (NIH) ($8.6 billion). NIH supports 82.8 percent of all federally funded human subjects research in the United States. Of note, NIH has a sophisticated system for assigning codes to research proposals involving human subjects, including exemption status, existence of assurances, and whether concerns about protections have been expressed by scientific or Institutional Review Boards (IRBs). This system provides relatively accurate real-time estimates of the amount of human subjects research currently supported.
In contrast to DHHS, some agencies dedicate relatively small amounts of their total budget to human subjects research. For example, DOJ spent less than one percent of the department’s total budget on human subjects research in FY 1999.
In addition, NBAC asked for the percent of human subjects research conducted by agency employees or other staff (e.g., students) on site. With this question, NBAC was trying to determine where responsibility for IRB review of research studies lies, i.e., with an agency IRB versus a grantee’s or contractor’s IRB. Some agencies do not themselves conduct human subjects research, rather they support research conducted by contractors or grantees or through cooperative agreements (see Table 4). Of the agencies that do conduct research (thereby requiring some level of review by the agency), most have some mechanism for review of protocols by an IRB or similarly constituted body (see Exhibit A for some examples), although there were a few exceptions (EPA and SSA). Almost all federal agencies that conduct human subjects research within their own facilities have intramural IRBs whose members include agency staff and at least one member who is not affiliated with the agency.
Exhibit A: Distinctive Mechanisms for Review of In-House Research
Environmental Protection Agency All human subject research studies supported by EPA must either be approved or be determined to be exempt research by the EPA Human Subjects Research Review Official before any contract, grant, cooperative agreement, or cooperative research and development agreement (CRADA), interagency agreement, or any formal agreement involving EPA support of such studies is awarded or entered into. All human research studies conducted by EPA also must be approved or determined to be exempt by the Review Official before work can start (EPA Order No. 1000.17 Change A1, July 30, 1999).
Indian Health Service The Indian Health Service (IHS) has a two-tier IRB system. IHS is divided into 13 Areas or regions; each Area has its own IHS IRB. Each Area IRB is the IRB of record for research conducted in that area in which IHS is involved in any way. There is also a Headquarters IRB that oversees the IRB system; it reviews all research in which IHS is involved, including all research reviewed by one or more Area IRBs, as well as research that takes place at the national level.
U.S. Coast Guard The infrastructure in place to monitor the human subject protections is a newly established Coast Guard Formal Review Board that reviews and approves all of the test procedures and documentation prior to every experiment.
Social Security Administration SSA’s extramural research is reviewed for compliance with the Common Rule’s informed consent guidelines, as well as the Privacy Act and SSA privacy rules. Project and contract officers conduct this review, with advice from SSA’s Privacy Officer in the Office of Disclosure Policy and the Office of General Counsel. For review of extramural biomedical or behavioral research, SSA relies on contractors’ or grantees’ IRBs and the existing DHHS Multiple Project Assurance (MPA) system. SSA’s intramural research, which includes neither biomedical nor behavioral research, receives a similar review throughout each project’s planning, conduct, and evaluation.
J-7
Table 4 displays the number of IRBs found at those agencies that conduct research and the number of protocols reviewed in FY 1999. The range of protocols reviewed was large. DOD, with 43 IRBs, reviewed more than 3,500 protocols in FY 1999 while the CIA’s IRB reviewed just 2. Although the VA has 101 IRBs of record, there is no centralized system to tabulate the number of protocols reviewed in FY 1999. It is notable that a few agencies that conduct human subjects research have no constituted IRB available to review such research.
Types of Sponsored Research
Of those agencies that sponsor human subjects research, most support more than one type (see Table 5). All agencies reported supporting social science/behavioral research. Nine of the 16 support clinical research. Some agencies predominately support one type of research. For example, DOT principally supports human factors research, and the Consumer Product Safety Commission (CPSC) primarily supports social science/behavioral research and consumer product testing. A few agencies receive funds from other agencies to conduct research. For example, in addition to conducting its own research, the Census Bureau is funded by Congress to conduct large population surveys, such as the Survey of Income and Program Participation, and also receives interagency transfers of funds to provide field collection, data processing, and analysis services for other federal agencies such as the Bureau of Labor Statistics, the Bureau of Justice Statistics, and ED.
Research with Vulnerable Populations
Agencies were asked to respond to whether research is sponsored or conducted that targets vulnerable populations (as specified at Section 111(a)(3)). The responses to that question can be seen in Table 6. The large number of agencies responding positively to this question was unexpected. Upon follow-up with several of the agencies, it became clear that they understood the question to mean, “are members of vulnerable populations ever subjects in your research, in contrast to the targeted population of the research?” For example, ED indicated that it might conduct studies that inadvertently include pregnant women, although their pregnant condition is inconsequential to the research. In contrast, some agencies do target these populations, for example, the Department of Housing and Urban Development (HUD) conducts studies in economically disadvantaged neighborhoods to assess needs.
Administrative Oversight of Human Subjects Research
NBAC asked a series of questions related to administrative roles and responsibilities related to oversight of human subjects research, ranging from decisionmaking regarding review to the size of the administrative unit(s) devoted to protections.
Determination of Need for Review
NBAC asked, “What are the policies and procedures of your agency for determining whether a particular activity constitutes human subjects research? Please describe agency procedures for making determinations for 1) research conducted by agency employees or other staff and 2) research conducted by grantees, contractors and other funded entities.” Each federal department structures its program of administrative oversight of human subjects research somewhat differently, despite the fact that all operate under the requirements of the Common Rule. Some departments conduct reviews of research documentation out of one central departmental office, while others rely on local review (e.g., within an agency division or by a contractor’s or grantee’s IRB); some provide detailed interpretive guidance on human subjects protections to subsidiary intramural research offices, contractors, and
J-8
grantees, while others simply reference the Common Rule; and some departments audit or review IRB performance routinely, while others conduct investigations only when problems emerge.
Many agencies have an officer, either full or part time with assigned duties in this area. Often, if the agency primarily or exclusively supports research conducted by grantees and contractors (i.e., not by federal employees), a high-ranking individual responsible for grants and contracts is charged with making decisions about which research involves human subjects and whether it is exempt. For example, the Director of Grants Policy and Oversight Staff at ED makes the final determinations regarding need for review. At EPA, such determinations are made by a Human Subjects Research Review Official. In some agencies, the Office of the General Counsel primarily is involved. For example, at DOJ, the OJP Office of the General Counsel works with the Human Subjects Protection Officer to make determinations of this kind. The Food and Drug Administration (FDA) Office of the Senior Advisor for Science in the Office of the Commissioner will be responsible for reviewing determinations of exemptions.
Technical officers at USAID, NASA, and the National Institute of Standards and Technology (NIST, in DOC), who might be involved in grantmaking or contracting activities, often make the determination in cooperation with legal counsel that human subjects are involved.
In general, agencies rely on the grantee or contracting institution to make the initial determination of whether human subjects are involved. For research conducted in-house, or intramurally, the process might be different. If an agency has one or more IRBs, often the Chair will make these determinations. This is the case at CIA, DOD, and VA, and components of DOJ.
As mentioned previously, a few agencies conduct research but have no IRB. In the cases of SSA and HUD, the agencies report that the research conducted qualifies for an exemption, therefore there is no need for an IRB. SSA recognizes, however, that the requirements for their DHHS MPA pre-empt that exemption.
Determination of Exempt Research
Agencies were asked about their policies and procedures for determining whether a human subjects research activity is exempt under Section 101 and were asked to estimate the percent of human subjects research determined to be exempt from the Common Rule. The responses to this question are summarized in Table 7. Many agencies reported that all or nearly all of their research is exempt. The most common exemption cited (14/16) was:
Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures or observation of public behavior, unless:
(i) information obtained is recorded in such a manner that human subjects can be identified, directly or through identifiers linked to the subjects; and (ii) any disclosure of the human subjects’ responses outside the research could reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects’ financial standing, employability, or reputation.
In general, agencies use the same mechanisms to determine exempt research as that used to determine whether human subjects are involved—that is the IRB Chair for in-house research and a combination of technical and legal staff for grantees and contractors. For example, the Chair of the CPSC Human Subjects Committee, in consultation with the Office of General Counsel, determines whether the proposed activity is exempt under Section 101.
Some agencies have customized administrative mechanisms for making these determinations to meet their statutory and mission-related requirements. For example, the Census Department considers all of its research to be exempt under Federal Policy 15 CFR 27.101(b)(3)(ii) which exempts survey procedures if “federal
J-9
statute(s) require(s) without exception that the confidentiality of the personally identifiable information will be maintained throughout the research and hereafter.” However, privacy and confidentiality issues that relate to human subjects are brought to the Census Bureau’s Policy Office. The Disclosure Review Board has primary responsibility for ensuring confidentiality in published reports and data products.
As mentioned above, SSA does not have an IRB, because it claims all research is exempt. This exemption took effect on April 4, 1983, as a result of a final DHHS rule published on March 4, 1983 (FR 9266). Research carried out under section 1110(b) of the Social Security Act, however, remains subject to the Common Rule’s informed consent requirements. The 1983 notice states that “In order to insure the continued protection of human subjects participating in such [otherwise exempt] research activity, the Department is adding a specific requirement of written, informed consent in any instance, not reviewed by an IRB, in which the Secretary determines that the research activity presents a danger to the physical, mental, or emotional well-being of a participant.” In the case of biomedical and behavioral research, in the 1983 Federal Register notice, DHHS makes clear the need for IRB review, but states such review would be “unnecessary and burdensome in the context of research under the Social Security Act and otherwise.” DHHS discusses, but rejects, several proposals for IRB review of research and demonstrations to support public benefit or service programs and concluded that “ethical and other problems raised by research in benefit programs will be addressed by the officials who are familiar with the programs and responsible for their successful operations under state and federal law.” SSA has reviewed the 1983 regulation with the Office for Protection from Research Risks (OPRR, now the Office for Human Research Protections [OHRP]) and has concluded that it continues to apply to SSA research and demonstrations. In 1999, SSA did not conduct any extramural human subjects research or demonstrations under section 1110(b).
The Health Resources and Services Administration (HRSA, in DHHS) reported that practically all of its research activity comprises program evaluation or evaluation of demonstration projects. All such evaluations are technically exempt under the public “benefit and service” criterion. However, HRSA Policy 96.05 requires such a claim of exemption to be approved by the HRSA Human Subjects Committee; otherwise IRB oversight is required.
Qualifications for IRBs
Clearly, local review is a key component of the oversight system. The Common Rule requires IRB review and approval prior to the granting of federal funding for research on human subjects. Agencies that conduct human subjects research and that are signatories to the Common Rule should have an IRB or IRB-like body to review its research. The systems by which IRBs are formed are relatively uniform across those agencies that have one—that is, they are formed and charged according to the requirements of the Common Rule. In general, a high-ranking official makes the determinations about IRB members. For example:
J-10
Many agencies have policy directives and manuals that supplement the Common Rule, specifying in greater detail the required composition of the IRB. For example, at the Bureau of Prisons (DOJ), a majority of the members must be from outside the Bureau and must include a prisoner representative. The members are appointed by the Bureau Director, who must give final approval to IRB decisions.
Sizes and Functions of Administrative Units
In overseeing human subjects research conducted in-house or supported extramurally, federal agencies assume the following responsibilities: 1) communication of practice guidelines to research institutions and IRBs based on the policies of the Common Rule; 2) establishment of a structure whereby research proposals involving human subjects are peer reviewed for scientific merit as well as for IRB approval and the adequacy of subject protections; 3) negotiation of assurances with research institutions that ensure that adequate protections will be in place for research subjects; 4) verification that institutions, their IRBs, and researchers are complying with the federal human subjects regulations; and 5) investigation of complaints of noncompliance and adverse outcomes for subjects of research.
The method, intensity, and frequency of research oversight and inspection activities may depend on how much staff and budget an agency allots them. Agencies were asked about the size of the administrative unit dedicated to human subjects protections (see Table 8). Many agencies had difficulty answering this question because duties are shared in part across many individuals. The range for full-time equivalents (FTEs) devoted to human subjects protections was large, from none to 60. FDA responded that it has 287 FTEs dedicated to human subjects protections, because of its mission to monitor and oversee the conduct of clinical trials.
Assurances of Compliance
Many agencies issue their own assurances of compliance (see Table 8). In addition, most rely as well on assurances provided by then OPRR through MPAs with large research institutions that perform a significant amount of research funded by DHHS. If an institution is awarded an MPA by OHRP, the federal agency funding the research must accept that institution’s assurance of compliance with federal requirements and may not impose additional assurance requirements on the institution. This provision is intended to avoid duplicative and potentially contradictory enforcement of the federal protections. A few agencies reported that they do not issue their own assurances of compliance, nor do they rely on those issued by OPRR through DHHS (see Table 8).
Investigating and Acting on Noncompliance
In the event that the Common Rule is violated in the conduct of federally sponsored research involving human subjects, there are various responses that can affect both investigators and grantee institutions, such as withdrawal or restriction of an institution’s or project’s assurance and, with that action, of research funding and suspension or termination of IRB approval of the research. In addition, an IRB is authorized by the Common Rule to suspend or terminate its approval of research that fails to comply with the IRB’s requirements or when a research subject suffers an adverse event. No federal department or agency may continue to fund a project from which IRB approval has been withdrawn or at an institution whose assurance has been withdrawn.
OPRR, in overseeing human subjects protections for DHHS-funded research and for all institutions to which it has issued an assurance, generally investigates the conduct of research only in cases where a complaint has been filed; where an institution, IRB, or researcher has reported a problem or adverse outcome; where a problematic audit finding has been referred to it by the FDA or a DHHS funding agency; or where published research raises concern among OPRR compliance staff.
The FDA, in its role regulating new drugs, biologics, and devices for marketing, enforces the somewhat similar requirements for human subjects protections defined in the Food, Drug, and Cosmetic Act through
J-11
periodic on-site investigations of research institutions (e.g., pharmaceutical firms, university-based research facilities funded by pharmaceutical firms, independent testing laboratories) and their IRBs, as well as clinical investigators, sponsors, monitors, and contract research organizations.
In most agencies, cases of noncompliance would be referred to a high-ranking or fiscally responsible official (the Assistant Attorney General or Legal Counsel, the contracting officer or component director, the Cognizant Human Subjects Officer).
The most common responses to the question about sanctions applied in the case of noncompliance were:
site visits at any facility where there may be an indication that the research is not being conducted in compliance with regulations. The Chief Research and Development Officer and the Chief Officer, Office of Research Compliance and Assurance, make the final determination regarding noncompliance.
At ED, the Director of the Grants Policy and Oversight Staff Education is authorized to investigate allegations of noncompliance with the regulations in extramural research.
Federal agencies may also take disciplinary action against employees involved in human subjects research for failure to follow human subjects protection rules. For example, DOD sanctions for noncompliance by intramural researchers include loss of investigator privileges. For military personnel, potential sanctions are letters of reprimand, nonjudicial punishment, and sanctions under the Military Code of Justice; for civilian DOD personnel, sanctions include reprimands, suspension, or termination of employment. The commander of the military facility is authorized to make final determinations about noncompliance. Depending on the nature of the infraction, the case could result in a general court martial.
At NSF, the Office of the Inspector General investigates allegations of noncompliance.
Human subjects site reviews are conducted at all major DOE laboratories on a “not-for-cause” basis. The DOE Human Subjects Program Manager makes the final determination of noncompliance.
Additional Policies, Statutes, and Regulations
Many agencies must comply with additional requirements as codified in statute or law. In addition, several agencies have imposed additional requirements beyond those specified in the Common Rule. A listing of these requirements appears in Table 9.
Some agencies have to comply with statutes that provide similar, parallel, or somewhat different approaches to subject protection than those that are provided by the Common Rule. For example, NIH has imposed additional guidelines for inclusion of women and children in research. The IHS has requirements about tribal consultation in research activities.
J-12
The Privacy Act allows several agencies to disclose research information about individuals under certain conditions. For example, the Privacy Act permits HCFA to disclose information without individual’s consent if the data is to be used for a purpose that is compatible with the purposes for which it was collected. This is known as routine use, as identified in a System of Record notice. Routine use permits recipients of the information to use data in connection with a matter relating to one of HCFA’s programs. Specifically, HCFA may release data under the routine use for research to an individual or organization for research, evaluation, or epidemiological project related to the prevention of disease or disability, the restoration or maintenance of health, or payment related projects. The Privacy Officer is the point of contact for Privacy Act data requests. Those using data must sign a Data Use Agreement, a legally binding agreement between the requestor of the data and HCFA to ensure the protection of the data. HCFA’s Data Disclosure Review Board is responsible for refining and updating HCFA-wide policies that evaluate access to individually identifiable information, while at the same time ensuring its confidentiality, as well as the privacy of individuals.
Educational Activities
Activities undertaken by agencies to improve staff and grantee/contractor awareness about the system of human subjects protections are listed in Table 10. Activities range from passive dissemination of relevant information about the Common Rule to aggressive requirements that training occur before research is conducted. For example, for its intramural researchers, NIH has a computer-based training program on the protection of human subjects that explains major requirements of its MPA. Registered completion is required of all staff conducting or supporting research involving human subjects and all newly employed NIH researchers. Since 1995, over 4,000 NIH employees have registered completion. DOE has an extensive education program that includes brochures, booklets on special research topics, a handbook for IRBs, large-scale interagency meetings, and a well-used website with information pertaining to human subjects protections.
Emerging Issues and Suggestions for Change
NBAC asked the federal agencies to identify emerging issues that might affect the landscape of human subjects protections in the future. Responses are shown in Table 11. Suggestions for NBAC to consider as it conducts its analysis over the next year are summarized in Table 12.
Conclusions
Several issues are raised by these data. First, many agencies report significantly increased activity in the areas of human subjects protections since they last reported to NBAC in 1996 and 1997. These improvements have ranged from new agency policies and procedures clarifying or enhancing protections, additional staff, establishment of IRBs and other review mechanisms to evaluate research being conducted by agency employees, and increased educational and training activities to educate employees, grantees, and contractors about the federal requirements and the specific policies and procedures of the agency or department.
However, some problems remain, most particularly in inadequacies of review mechanisms, insufficient administrative support, and lack of an assurance process. More generally, the applicability of the Common Rule and its problematic interpretation by some agencies is a central issue that must be addressed. Each of these issues is addressed below.
Appropriateness of the Common Rule
Table 5 shows the wide range of research supported by most federal agencies. Of note, all but one report that they support social science and/or behavioral research. A majority report supporting operational, health services,
J-13
and education research. This is notable because it is the “nonclinical, nonmedical” research communities that most often report the greatest difficulty in interpreting and applying the language of the Common Rule, especially as it applies to defining minimal risk. This leads to a question rather than a conclusion: Because so many agencies support nonclinical research, and because it is these types of research that challenge the paradigm of the Common Rule, is it necessary to consider whether the Common Rule as currently written addresses the unique concerns raised by, for example, behavioral, social science, or educational research.
Subparts B, C, and D of 45 CFR 46
Nearly all agencies reported conducting research that involves vulnerable populations (see Table 6). It is not clear whether these populations are targeted for the research or whether they happen to be subjects in ongoing research focused on a more general population. Of note, all DHHS agencies have adopted Subparts B, C, and D of 45 CFR 46 for funded or intramural research. Nonetheless, most agencies have not adopted Subparts B,C, and D of 45 CFR 46, pertaining to additional protections pertaining to research, development, and related activities involving fetuses, pregnant women, human in vitro fertilization, prisoners, and children. Based on this survey data alone, the implications of this are not clear, but they certainly deserve further consideration.
Lack of an IRB
Much of the success or failure of the federal regulations governing human subjects research depends on the effectiveness of IRBs in carrying out their responsibilities, which include assessing research proposals prior to their funding; stipulating changes in the research protocol or informed consent procedure that strengthen the protections afforded the subjects; disapproving excessively risky research proposals; minimizing risks to subjects; reviewing ongoing research; and taking action quickly to correct or remove threats to subjects’ rights and welfare.
Most agencies have constituted an IRB to review human subjects research conducted by employees or contractors within their purview (i.e., not covered by an IRB at a grantee or contractor institution). However, a few agencies that reported conducting human subjects research in-house have not done so (see Table 4), raising concerns about the processes by which decisions about, for example, exemption or waiver of consent are made, not to mention ensuring adequate protection of the human subjects involved.
Determining Exemptions
Who determines which research is exempt from the federal policy and how the exemptions cited at 46.101(b) are interpreted varies across the agencies. In general, agencies with one or more IRBs or dedicated human subjects protections staff appear to have systems by which such exemptions are determined in a systematic manner. Although the regulations state that “Department or Agency heads retain final judgment as to whether a particular activity is covered by this policy” (46.101(c)), the process by which such determinations are made should be more carefully examined. Some might find it problematic when one individual unilaterally makes a recommendation that research is exempt (even if the recommendation must be accepted by the Department head), given that the individual might be biased, conflicted, or misguided about the meaning of the language.
In addition, it appears that some agencies broadly interpret what is included under the exemptions. Although this does not necessarily indicate a problem, it should suggest that the language and use of the exemptions deserve further consideration.
Assurances
In the past, OPRR was the principal entity responsible for negotiating MPAs with large research institutions that perform a significant amount of research funded by DHHS. If an institution is awarded an MPA by OPRR, the
J-14
federal agency funding the research must accept that institution’s assurance of compliance with federal requirements and may not impose additional assurance requirements on the institution. This provision is intended to avoid duplicative and potentially contradictory enforcement of the federal protections. Many departments indicated that they rely on the DHHS assurance, and in some cases negotiate their own assurance. However, a few agencies appear to have no mechanism in place for issuing assurances (see Table 8). It is not clear from the data whether other mechanisms are in place to offer such assurances.
The implications of this are important in one respect. OPRR (now OHRP), in overseeing human subjects protections for DHHS-funded research and for all institutions to which it has issued an assurance, generally investigates the conduct of research in cases in which a complaint has been filed; in which an institution, IRB, or researcher has reported a problem or adverse outcome; or in which a problematic audit finding has been referred to it by the FDA. In the absence of such an assurance, it is not clear how such an investigation could be conducted by a disinterested party.
Adequacy of Administrative Structures
The adequacy of research oversight and inspection activities at the federal level are likely to depend on how much staff and budget an agency allots them. In overseeing human subjects research conducted by employees or supported extramurally or through contracts, federal agencies have the following responsibilities:
1) Communicating policies and practice guidelines to relevant research institutions and IRBs based on the policies of the Common Rule;
2) Establishing a structure whereby research proposals involving human subjects are peer reviewed for scientific merit as well as for IRB approval and the adequacy of subject protections;
3) Negotiating assurances with research institutions that make certain that adequate protections will be in place for human subjects;
4) Verifying that institutions, their IRBs, and researchers are complying with the federal regulations; and
5) Investigating and following up on complaints of noncompliance.
Agencies that conduct and/or support a large portfolio of human subjects research should have sufficient staffing and resources to assure that these responsibilities are met. Although the data collected in the survey are incomplete and somewhat imperfect, it would appear that some agencies clearly do not devote sufficient resources to these efforts (see Table 8). There is no formula for determining what is adequate for a given agency, but when there are no staff or resources devoted to these activities, one can assume an inadequacy exists.
Notes
1 Until March 31, 1995, the Social Security Administration (SSA) was part of the Department of Health and Human Services (DHHS). Under section 106(b) of P.L. 103-296, SSA is required to continue to follow all DHHS regulations in effect on March 30, 1995, until SSA promulgates its own regulations. Inasmuch as SSA has not issued its own regulations or otherwise amended the Common Rule, those regulations continue to apply to SSA human subject research. NBAC included SSA in this survey. In addition, an Executive Order requires the Central Intelligence Agency (CIA) to follow all the rules and regulations of DHHS pertaining to human subjects protections. Thus, in actuality there are 18 agencies that adhere to the Common Rule.
2 For example, in June 2000, the human research protection activities of OPRR were elevated from the National Institutes of Health to the Office of the Secretary in the Department of Health and Human Services and a new Office for Human Research Protections (OHRP) was created.
J-15
3 Reviewers included former staff members of the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical Research (Barbara Mishkin, then Deputy Director, and Alex Capron, then Executive Director); the former director of OPRR (Charles McCarthy); and a former staff member of the Advisory Committee on Human Radiation Experiments (Anna Mastroianni). The DHHS Office of General Counsel determined that the Phase I survey asked only about the organization, structure, and policies of the departments and thus did not require review by an IRB.
4 The questionnaire language suggested that agencies use the definition of research cited at Section 102(d) of 45 CFR 46, “Research means a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge.” In estimating expenditures for research, NBAC asked the agencies to include all costs for the support of research, such as funds expended through grants, contracts, cooperative agreements and other funding mechanisms; salaries for in-house staff including, program and administrative staff; and other indirect costs. Agencies were encouraged to make best estimates.
5 Agencies were asked to include “exempt research.”
J-16
Appendix A
National Bioethics Advisory Commission
Federal Agency Survey on Policies and Procedures for the Protection of Human Subjects in Research December 21, 1999
| 1. |
Does your agency (i) support, (ii) conduct, or (iii) regulate human subjects research? ____Yes ____No |
| 2. |
What was your agency’s total budget appropriation for FY 1999? __________________________________ In Questions 3–5, NBAC is attempting to get an idea of (1) the proportion of your budget spent on research regardless of the type (e.g., human, animal, weapons) your agency conducts, (2) the proportion of your research budget spent on research involving humans, and (3) the proportion of your human subjects research budget that is conducted within (intramural or in-house) your agency. |
| 3. |
Approximately what percent of your agency’s FY 1999 budget appropriation was dedicated to research activities? (round estimate to nearest ten percent) ___________% If you prefer to provide a dollar amount instead of a percent, please do so here: ___________ |
Please provide a best estimate. NBAC will use the information to describe your research portfolio in terms of a proportion of your total budget. We suggest that the definition of research cited at Section 102(d)1 be used as a starting point, “Research means a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge.” If your agency uses a different definition, please note the definition and include the research activities in your estimate.
In estimating agency expenditures for research, include all costs to your agency for the support of research, such as funds expended through grants, contracts, cooperative agreements and other funding mechanisms; salaries for in-house staff including, program and administrative staff; and other indirect costs.
If appropriate, please describe any limitations or factors that would influence the interpretation of the estimate. (Please note: you will have the opportunity to review NBAC drafts that use these data to ensure that they are not misinterpreted.)
| 4. |
Of the percent provided in your response to Question #3, approximately what percent of the research budget involved human subjects (including exempt research)? (round estimate to nearest ten percent) ___________%. If you prefer to provide a dollar amount instead of a percent, please do so here: _________ |
We refer you to the definition of “human subject” provided at Section 102(f): “A human subject means a living individual about whom an investigator (whether professional or student) conducting research obtains (1) data through intervention or interaction with the individual, or (2) identifiable private information…”
If appropriate, please describe any limitations or factors that would influence the interpretation of the estimate. (Please note: you will have the opportunity to review NBAC drafts that use these data to ensure that they are not misinterpreted.)
1 Please note that throughout this document we refer only to the relevant section of the Federal Policy for the Protection of Human Subjects, since agencies use different numbers in referring to the relevant Title of the Code of Federal Regulations.
J-17
| 5. |
Of the percent provided in your response to Question #4, approximately what percent of your agency’s human subjects research budget was conducted by agency employees or other staff (e.g., students) on-site (i.e., in-house or intramural?) (round estimate to nearest ten percent). ___________% If you prefer to provide a dollar amount instead of a percent, please do so here: ___________ |
With this question NBAC is trying to determine where responsibility for IRB review of research studies lies, i.e., with an agency IRB versus a grantee’s or contractor’s IRB.
| 6. |
What are the policies and procedures of your agency for determining whether a particular activity constitutes human subjects research? Please describe agency procedures for making determinations for (1) research conducted by agency employees or other staff and (2) research conducted by grantees, contractors and other funded entities. Provide a brief description here or attach policies and procedures (please note if they already have been provided to NBAC). |
|
| 7. |
What are the policies and procedures of your agency for determining whether a human subjects research activity is exempt under Section 101 and who makes the determination? Please describe agency procedures for making determinations for research conducted by agency employees or other staff and for research conducted by grantees, contractors and other funded entities. Provide a brief description here, or attach relevant policies and procedures (please note if they already have been provided to NBAC). |
|
| a. |
Approximately what percent of your human subjects research portfolio is determined to be exempt from the Common Rule? |
|
__________% for human subjects research performed by employees or other staff
__________% for human subjects research performed by grantees or contractors or other funded entities
| b. |
In general, of the six categories of research that may be exempt, which categories, if any, does your agency use? Check each category used. (See Section 101(b).) |
_________
“Research conducted in established or commonly accepted educational settings…”
_________
“Research involving the use of educational tests, survey procedures, interview procedures or observations of public behavior, …”
_________
“Research involving the use of educational tests, survey procedures...that is not exempt under the (b)(2)…”
_________
“Research involving the collection or study of existing data…”
_________
“Research and demonstration projects …”
_________
“Taste and food quality evaluation and consumer acceptance studies…”
| 8. |
How many IRBs does your agency have? ___________ |
|
| a. |
Approximately how many protocols did your IRB(s) review in FY 1999? ___________ |
|
| b. |
What criteria are applied, and by whom, to determine that the IRB(s) is/are qualified to review and approve your organization’s intramural/in-house research? Provide brief description here or attach criteria and procedures (please note if they have already been provided to NBAC). |
|
J-18
| 9. |
What is the nature of the human subjects research sponsored by your agency? (Check all that apply) If the categories below do not describe the types of human subjects research conducted by your agency, please provide a listing of categories relevant to your agency, use additional lines below. Add categories if needed. |
a. ________ social science (behavioral) experiments b. ________ social science research, not experimental c. ________ clinical research, experimental (e.g., clinical trials) d. ________ epidemiologic research (excluding clinical trials)
| e. |
________ large population surveys of demographic and other personal data |
| f. |
________ development of new tools or methods to be used in human subjects research |
g. ________ health services research
h. ________ operational, operations, organizational, or management assessments
| i. ________ | demonstration projects |
| j. ________ | educational research |
| k. ________ | community-based intervention research |
| l. ________ | human factors research |
10.Does your agency sponsor or conduct research that targets vulnerable populations (as specified at Section 111(a)(3))? Please check all those that apply.
| a. ________ | children |
| b. ________ | prisoners |
| c. ________ | pregnant women |
| d. ________ | fetuses |
| e. ________ | mentally disabled persons |
| f. ________ | economically disadvantaged persons |
| g. ________ | educationally disadvantaged persons |
| h. ________ | other (please specify) |
11.Does your agency have an administrative unit dedicated to implementing human subjects protections?
_____Yes _____No
| a. |
If so, how many FTEs are working in that unit? ____________ |
|
| b. |
If so, what was the FY 1999 budget for that unit? ____________ |
|
| 12. |
Does your agency comply with additional regulations, policies, or procedures (whether mandated or self-imposed) relevant to the protection of human subjects in research? (apart from implementation of the Federal Policy for the Protection of Human Subjects, 56 Fed. Reg. 28003 (June 18, 1991) (Common Rule)). _____Yes _____No |
|
If yes, please provide copies of regulations, policies or procedures to NBAC if you have not already done so. If already provided, please note here.
J-19
13.Does your agency issue assurances of compliance? _____Yes _____No
If yes, please provide copies of “sample” documents to NBAC. If copies have already been provided to NBAC, please note so here.
| a. |
Who, in your agency, is authorized to negotiate an assurance? (provide title, not name) ____________________________________________________________________________________ |
| b. |
Does your agency rely on other agency assurances, such as the DHHS Multiple Project Assurance? _____Yes _____No |
If yes, please indicate which agency (or agencies) and what type(s) of assurances?
| 14. |
Please describe the policies and procedures, if any, your agency uses to investigate allegations that human subjects research conducted or supported by your agency has not been conducted in compliance with the regulations. Provide a brief description here or attach policies and procedures or note that they have already been provided to NBAC. |
|
| a. |
Who in your organization is authorized to make a final determination about noncompliance? (provide title, not name) _______________________________________________________________________ |
|
| b. |
What sanctions, if any, are available to your agency to impose on individuals or institutions found in violation of the laws, regulations, policies, or procedures for the protection of human subjects in research? Who imposes such sanctions? Provide a brief description here, or attach policies and procedures. (Please note whether they have already been provided to NBAC). |
|
15.Describe any educational or outreach activities undertaken by your agency to inform investigators, institutions, and/or IRBs about the Common Rule. Provide a brief description here or attach descriptions or note that they have already been provided to NBAC
| 16. |
If applicable, please describe emerging research issues that are likely to influence human subjects protection. |
| 17. |
Please provide NBAC with a description of any changes in policies or procedures that have been implemented by your agency since it initially responded to Executive Order 12975. If this information has already been provided to NBAC, please note so here. |
| 18. |
Please provide NBAC with suggestions for changes in the government-wide human subjects protection system, including, but not limited to, changes in or revisions to the Common Rule. |
Please respond by February 15, 2000.
Thank you for your assistance.
J-20
Table 1: Federal Signatories* to the Common Rule**
| Relevant Section of Code of | |
| Federal Regulations (CFR) | Department/Agency |
| 45 CFR Part 46 | Department of Health and Human Services*** |
| 7 CFR Part 1c | Department of Agriculture |
| 10 CFR Part 745 | Department of Energy |
| 14 CFR Part 1230 | National Aeronautics and Space Administration |
| 15 CFR Part 27 | Department of Commerce |
| 16 CFR Part 1028 | Consumer Product Safety Commission |
| 22 CFR Part 225 | International Development Cooperation Agency, Agency for International Development |
| 24 CFR Part 60 | Department of Housing and Urban Development |
| 28 CFR Part 46 | Department of Justice |
| 32 CFR Part 219 | Department of Defense |
| 34 CFR Part 97 | Department of Education |
| 38 CFR Part 16 | Department of Veterans Affairs |
| 40 CFR Part 26 | Environmental Protection Agency |
| 45 CFR Part 690 | National Science Foundation |
| 49 CFR Part 11 | Department of Transportation |
| Not codified in regulation | Office of Science and Technology Policy |
*The Food and Drug Administration adopted a modified version of the Common Rule, codified at 21 CFR, Parts 50 and 56. **The Common Rule only refers to Subpart A of 45 CFR 46.
***Until March 31, 1995, the Social Security Administration (SSA) was part of the Department of Health and Human Services (DHHS). Under section 106(b) of P.L. 103-296, SSA is required to continue to follow all DHHS regulations in effect on March 30, 1995, until SSA promulgates its own regulations. Inasmuch as SSA has not issued its own regulations or otherwise amended the Common Rule, those regulations continue to apply to SSA human subject research. NBAC included SSA in this survey.
J-21
Table 2: Federal Agencies Responding to December 1999 NBAC Survey (Acronyms)
Central Intelligence Agency (CIA)
Department of Commerce (DOC)
National Telecommunications and Information Administration (NTIA)
National Institute of Standards and Technology (NIST) Bureau of the Census (CEN)
Department of Defense (DOD)
Department of Education (ED)
Department of Energy (DOE)
Department of Health and Human Services (DHHS) Administration for Children and Families (ACF) Administration on Aging (AOA) Agency for Health Care Research and Quality (AHRQ)
Centers for Disease Control and Prevention (CDC)/Agency for Toxic Substances and Disease Registry (ATSDR) Food and Drug Administration (FDA) Health Care Financing Administration (HCFA) Health Resources and Services Administration (HRSA) Indian Health Service (IHS) National Institutes of Health (NIH) Office for Protection from Research Risks (OPRR) Substance Abuse and Mental Health Services Administration (SAMHSA)
Department of Housing and Urban Development (HUD)
Department of Justice (DOJ) Office of Justice Programs (OJP)
Community-Oriented Policing Services (COPS) Bureau of Prisons (BOP) Federal Bureau of Investigation (FBI)
Department of Transportation (DOT) Federal Aviation Administration (FAA) U.S. Coast Guard (USCG) Federal Highway Administration (FHA)
National Highway Traffic Safety Administration (NHTSA)
Department of Veterans Affairs (VA)
National Aeronautics and Space Administration (NASA)
National Science Foundation (NSF)
Social Security Administration (SSA)
U.S. Agency for International Development (AID)
U.S. Consumer Product Safety Commission (CPSC)
U.S. Environmental Protection Agency (EPA)
J-22
Table 3: Agency Budget Data, FY 1999 ($ in thousands)
| Amount human | |||
| Agency | Total budget | Amount research | subjects research |
| CIA | classified | classified | classified |
| Commerce | |||
| NTIA | 66,765 | 17,600 | 17,600 |
| NIST | 641,000 | 410,240 | 41,024 |
| CEN | 317,0001 | 317,000 | 158,500 |
| Defense | 252,300,000 | 35,915,600 | 37,100 |
| Education | 39,000,000 | 143,000 | 50,000 |
| Energy | 18,000,000 | 4,000,000 | 27,000 |
| DHHS | |||
| ACF | — | 30,000 | 30,000 |
| AHRQ | 170,955 | 136,764 | 109,411 |
| CDC | 2,638,981 | 433,307 | 167,465 |
| ATSDR | 76,000 | 7,600 | 4,560 |
| FDA | 1,132,974 | 113,297 | 11,329 |
| HCFA | 1,946,0002 | 50,000 | 15,000 |
| HRSA | 4,353,564 | 130,793 | 75,908 |
| IHS | 2,240,000 | 22,400 | 22,400 |
| NIH | 15,602,7003 | 15,600,000 | 8,580,000 |
| SAMHSA | 2,486,787 | 338,344 | 338,344 |
| Housing and Urban | |||
| Development | 24,500,000 | 55,000 | 11,000 |
| Justice | 18,450,850 | 184,508 | 110,705 |
| Transportation | |||
| FAA | 9,750,000 | 150,000 | 25,000 |
| USCG | |||
| FHA | |||
| NHTSA | |||
| Veterans Affairs | 42,625,029 | 316,000 | 175,600 |
| NASA | 13,652,000 | 5,654,000 | 20,000 |
| NSF | 3,737,000 | 2,506,000 | 150,360 |
| Social Security | 421,000,000 | 68,0004 | 40,000 |
| EPA | 7,600,000 | 760,000 | 76,000 |
| AID | 8,342,000 | 200,000 | 60,000 |
| Consumer Product | |||
| Safety Commission | 47,000 | N/A | 100 |
| TOTAL | 10,354,406 | ||
1 This excludes the Census 2000 preparation funding for FY 1999 of $1,071 million. 2 This figure is for program management only.
3 This figure includes $2.7 million for the Office for Protection from Research Risks.
4 Of this amount, $29 million was committed to a single four-year contract to conduct the National Study of Health and Activity.
J-23
Table 4: Review of “In-House” Research at Federal Agencies
| Percent of human | Number of protocols | ||
| subjects research | reviewed in FY 1999 | ||
| conducted by agency | (Total, including new | ||
| Agency | employees or other staff* | Number of IRBs | and continuing) |
| Department of Health and | |||
| Human Services | |||
| ACF | 0 | 0 | 0 |
| AHRQ | 25 | 0 (planned) | 0 |
| CDC | 23 | 6 | 1,031 |
| FDA | <1 | 1 | 14 |
| HCFA | 0 | 0** | 0 |
| HRSA | <1 | 0*** | 0 |
| IHS | 50 | 13 | 200 |
| NIH | 10 | 14 | 1,337 |
| SAMHSA | 0 | 0 | 0 |
| Department of Energy | 0 | 0 | 0 |
| Central Intelligence Agency | 0 | 1 | 2 |
| National Aeronautics and | |||
| Space Administration | 50 | 5 | 200 |
| Department of Commerce | NTIA – 0 | 0 | 0 |
| NIST – 10 | 1 | 37 (including | |
| exempt) | |||
| Census – 100 | 0 | 0 | |
| Consumer Product Safety | |||
| Commission | <10 | 1 | 4 |
| Agency for International | |||
| Development | 0 | 0 | 0 |
| Department of Housing and | |||
| Urban Development | 30 | 0 | 0 |
| Department of Justice | 0–3 | 1 – FBI | 10 |
| 1 – BOJP | 0 | ||
| 1 – BOP | 50 | ||
| Department of Defense | 45–100 | 43 | 3,572 |
| Department of Education | 0 | 1 | 0 |
| Department of Veterans Affairs | 100 | 101 | ? |
| Environmental Protection Agency | 30 | 0 | 0 |
| National Science Foundation | 0 | 0 | 0 |
| Department of Transportation | 0–40 (FAA) | FAA 2 | 40 |
| Social Security Administration | ~66 | 0 | 0 |
*Some agencies reported data on more than one division. Thus, the range of percentages across all components reported is presented. For example, within the Department of Defense, one unit reported that 45 percent of the human subjects research supported was conducted by employees, whereas another unit reported that 100 percent of the human subjects research was conducted by employees.
**HCFA has a Data Disclosure Review Board charged with many of the same functions as an IRB. Similarly, a Beneficiary Confidentiality Board is charged with balancing personal privacy interests with a qualifying public interest.
***HRSA maintains a Human Subjects Committee, which passes on claims of exemptions in accordance with HRSA Policy 96.05, and advises on human subjects protection issues.
J-24
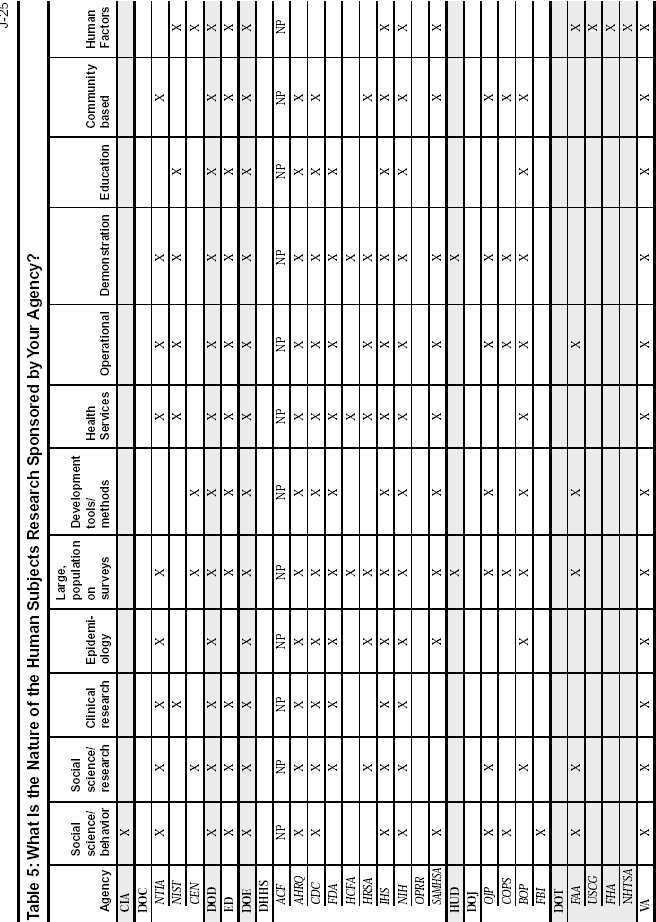
J-26
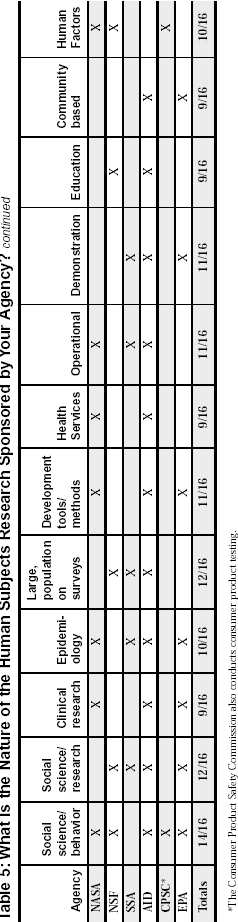
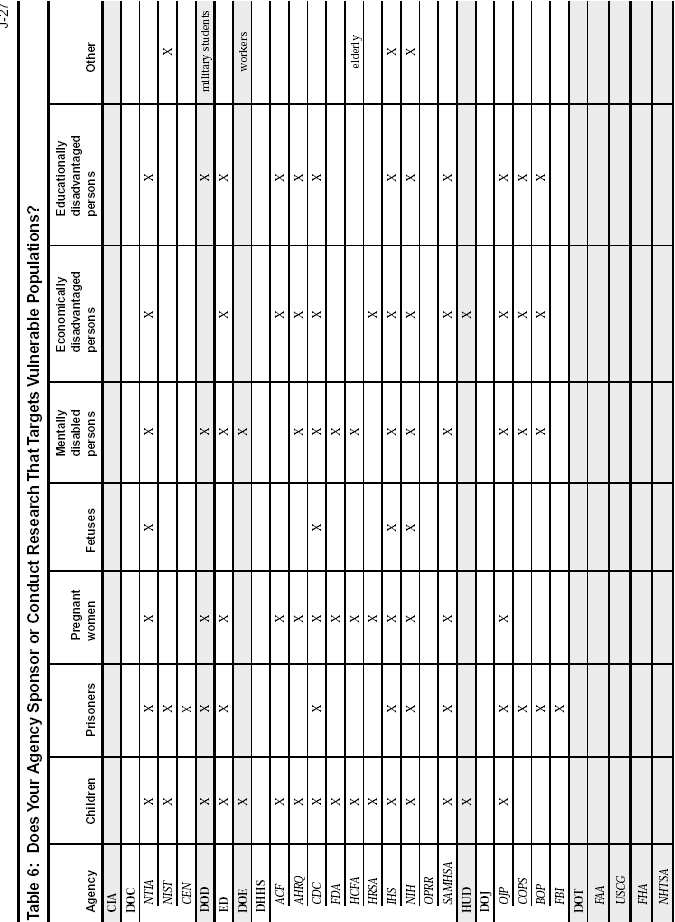
J-28
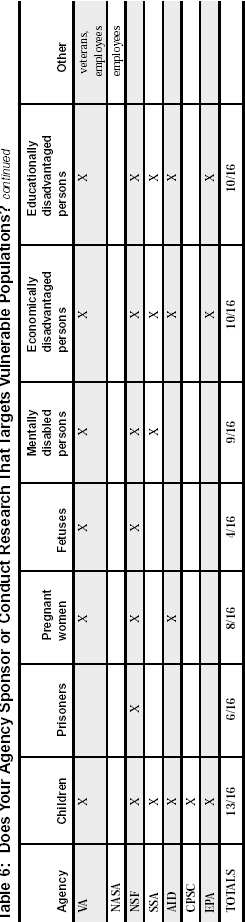
financial standing, employability, or reputation. responses outside the research could reasonably place the subjects at ’ ’ undergo IRB review. 1 Research conducted in established or commonly accepted educational settings, involving normal educational practices, such as (i) research on regular and special education instructional strategies, or (ii) research on the effectiveness of or the comparison among instructional techniques, curricula, or classroom management methods. 2 Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey proce- dures, interview procedures or observation of public behavior, unless: (i) information obtained is recorded in such a manner that human subjects can be identified, directly or through identifiers linked to the subjects; and (ii) any disclosure of the human subjects risk of criminal or civil liability or be damaging to the subjects 3 Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey proce- dures, interview procedures, or observation of public behavior that is not exempt under paragraph (b)(2) of this section, if: (i) the human subjects are elected or appointed public officials or candidates for public office; or (ii) Federal statute(s) require(s) without exception that the confidentiality of the personally identifiable infor- mation will be maintained throughout the research and thereafter.
for payment of levels or methods in changes possible (iv) or procedures; or programs those in such a manner that subjects cannot be identified, directly or through identifiers linked to the subjects. to
5 Research and demonstration projects which are conducted by or subject to the approval of Department or Agency heads, and which are designed to study, evaluate, or otherwise examine: (i) Public benefit or service programs; (ii) procedures for obtaining benefits or services under those programs; (iii) possible changes in or alternatives benefits or services under those programs. 6 Taste and food quality evaluation and consumer acceptance studies, (i) if wholesome foods without addi- tives are consumed or (ii) if a food is consumed that contains a food ingredient at or below the level and for a use found to be safe, or agricultural chemical or environmental contaminant at or below the level found to be safe, by the Food and Drug Administration or approved by the Environmental Protection Agency or the Food Safety and Inspection Service of the U.S. Department of Agriculture.
Table 8: Administrative Aspects of Agency Human Subjects Protection Activities
| Dedicated | Issue assurances | Rely on other | ||||
| Agency | administrative unit | Budget | of compliance | agency assurances | ||
| DOJ | Yes, 2 part time FTEs | N/A | Some components | DHHS | ||
| HUD | No (Office of Lead | |||||
| Hazard Control’s Planning | ||||||
| & Standards Division) | No | |||||
| AID | Yes, Cognizant Human | |||||
| Subjects Officers | Yes | DHHS, DOD | ||||
| CIA | Yes, Chief of Human | |||||
| Subjects Research Panel | ||||||
| and Contracting Officer’s | ||||||
| Technical Representatives | No | No | ||||
| EPA | No | Yes | DHHS | |||
| DOC | NTIA – No | NTIA – No | NTIA – No | |||
| NIST – Personnel in | NIST – Yes | NIST – DHHS | ||||
| Deputy Chief Counsel’s | ||||||
| office and elsewhere | ||||||
| CEN–No | ||||||
| NASA | Yes, 5.9 FTEs1 | $ | 625,000 | Yes | DHHS | |
| VA | Yes, 3 units, 3 FTEs | Yes | DHHS | |||
| CPSC | No | Yes | DHHS | |||
| ED | Yes, 1.5 FTEs | $ | 200,000 | Yes | DHHS | |
| SSA | No | No | DHHS | |||
| DOD | Yes, 60 FTEs | $ | 3,500,000 | Yes | DHHS | |
| NSF | Yes, part time FTE | Yes | Yes | |||
| DOT | No | No | DHHS | |||
| DOE | Yes, 2 FTE | $ | 425,000 | Yes | DHHS | |
| DHHS | Yes* | |||||
| ACF | No | DHHS | ||||
| AHRQ | No | DHHS | ||||
| CDC | Yes, 6 FTEs | $ | 651,505 | DHHS | ||
| FDA | Yes, ~287 FTEs2 | ~$37,000,000 | DHHS | |||
| HCFA | Yes, 6 FTEs | |||||
| HRSA | No | DHHS | ||||
| IHS | Yes, 4.7 FTEs | $ | 1,000,000 | DHHS | ||
| NIH | Yes, 4 FTEs in Office of | $ | 480,000 | DHHS | ||
| Human Subjects Research, | ||||||
| another 10.73 in institute’s | $ | 334,965 | ||||
| administrative units, | $ | 2,700,000 | ||||
| 19 FTEs in OPRR | for OPRR | |||||
| SAMHSA | No | DHHS | ||||
1 During FY 2000 plans are to increase to 8 FTE, and in FY 2001 there will be 12 FTE in Headquarters. There are plans for five field offices to be established in FY 2000.
2 FDA’s Bioresearch Monitoring Program merges human subject protection with good clinical practice. Over 200 FTEs conduct 15,500 inspections in FY 1999, 329 covered IRBs and 604 covered clinical trials. An additional 87 FTEs in the Center for Devices and Radiological Health were devoted to evaluating IDEs and IDE supplements.
*Until June 2000, OPRR was the administrative unit within DHHS that provided assurances for all DHHS agencies as well as other federal departments.
J-30
Table 9: Additional Regulations, Policies, or Procedures Relevant to Human Subjects Protections
Agency/Department Relevant controlling authorities and directives
Department of Justice
Agency for International Development
Central Intelligence Agency
Environmental Protection Agency
Department of Commerce
National Aeronautics and Space Administration
Department of Veterans Affairs
Consumer Product Safety Commission
Department of Education
J-31
Table 9: Additional Regulations, Policies, or Procedures Relevant to Human Subjects Protections continued
Agency/Department Relevant controlling authorities and directives
Social Security Administration
Department of Defense
Department of Transportation
Department of Energy
Department of Health and Human Services
J-32
Table 10: Human Subjects Protections Education and Training Activities
Table 11: Some Emerging Research Issues Likely to Influence Human Subjects Protection (as Reported by Agencies)
Substantive
Procedural
Table 12: Suggested Changes in the Government-Wide Human Subjects Protection System
J-33
LOCAL INSTITUTIONAL REVIEW BOARDS
Commissioned Paper Steven Peckman
University of California-Los Angeles

K-1
When science takes man [sic] as its subject, tensions arise between two values basic to Western society: freedom of scientific inquiry and protection of individual inviolability.1
Jay Katz
Introduction
The Institutional Review Board (IRB), a committee of scientists and nonscientists, is responsible for protecting the rights and welfare of human subjects, people who participate in scientific experiments or research. The IRB conducts a prospective review of proposed research and monitors continuing research in order to safeguard the rights and welfare of human subjects. The term “institutional” refers to the traditional location of the IRB, within the academic research institution or hospital. Local IRBs are geographically close to research sites, to scientists who conduct the research, known as investigators, and to communities of potential human subjects. Human research, however, also takes place beyond the walls of academia and hospitals, and may not be affiliated with such institutions. As a result, there are also IRBs that exist independently of academic research institutions and hospitals and that are not located near the investigators, the research sites, or the human research subjects.
This paper posits that an institutionally based IRB, or local IRB, is ideally situated to create a local culture based on trust and shared responsibility for the ethical conduct of biomedical or social-behavioral research. The first part of the paper will outline the history of local IRB review. The second part will illustrate how local IRB review encourages direct institutional responsibility for and community involvement in the conduct of research. The third section will address whether the current federal regulations provide adequate guidance for local review and whether institutions effectively apply both the letter and spirit of the regulations. For the purposes of this paper, the National Institutes of Health (NIH) definition of clinical research, which includes both biomedical and social-behavioral research, will be used in order to refer without distinction to all research involving human subjects.2 The actions of the local IRB are governed by ethical codes of conduct, federal regulations, local law, and institutional policies. Federal regulations give an IRB the authority to approve, require modification to, or disapprove all research activities that fall within its jurisdiction. “Research that has been reviewed and approved by an IRB may be subject to review and disapproval by officials of the institution. However, those officials may not approve research if it has been disapproved by the IRB.”3 Ultimately, a local IRB functions within a system of self-regulation and oversight on the part of the institution, the investigators, and the Board. A system of self-regulation and oversight requires a highly evolved sense of trust and responsibility from all participants. We trust professionals every day with our health, life, family, money, and property. We also trust scientists to be truthful and ethical in their conduct of research. To trust is to rely on the character, ability, strength, and truthfulness of someone or something. Trust also requires confidence in the truthfulness and accuracy of the information given by an individual or entity. In order to trust individuals or entities we must be assured that they will act responsibly. Therefore, to take responsibility for something makes that person or entity accountable. When we trust an individual, that person becomes responsible for upholding our trust. A discussion of local IRB review, ethical scientific conduct, and the ability to protect the rights and welfare of human subjects requires that we address the ideas of trust and responsibility as essential components of research.
The IRB system has proven “so successful as to set an international standard for monitoring clinical research.”4 Successful IRB review balances the interests of three distinct but inter-related social and political entities: scientists, society, and the individual human subject. The IRB, however, does not balance these interests alone. The IRB functions in a dynamic relationship with federal agencies, research sponsors, institutions hosting
K-3
research, investigators, and the public. The dynamic relationship balances the competing interests of all parties and it facilitates the continued conduct of human research in an ethical and collegial environment. As a result, the local IRB is not the sole party responsible for the protection of the rights and welfare of human research subjects. An effective system of protections is a collective responsibility that requires a collaborative effort from federal agencies, the sponsors, the IRBs, the institution, and the investigators. When all parties acknowledge their shared ethical responsibilities at both the local and national level, and a balance of interests is met, they create a culture of trust that allows for their effective collaboration with the public and the research subjects.
Part I
A Concise History of Local Review and Community Participation
The public has [a] role in monitoring research with human subjects in two distinct areas. The first concerns the selection of particular fields for research programs. These are difficult choices, but with both government money and research charities the public has helped direct research into some fields at the exclusion of others. It is difficult to justify a radical departure from present methods as most important discoveries are made ‘by chance,’ although by researchers with trained and open minds. The second role of the public concerns representatives serving on medical ethical committees. Increased representation of lay members on ethical committees is highly desirable.5 W. E. Waters
The history of IRBs reveals that local review grew out of two major components: 1) ad hoc institutionally based peer review committees that preexisted any systematic notion of human subjects protections, and 2) the federal government’s requirement that grantee institutions take responsibility for the ethical conduct of their research. An in depth history of human subject research review is outlined in many texts.6 Robert Levine, writing for the National Commission for the Protection of Human Subjects in Biomedical and Behavioral Research (National Commission), noted that the first documented suggestion of peer review for research may have originated with Thomas Percival in 1803.7 Though Percival may have seen the future, there is no evidence that his suggestion resulted in widespread adoption of his ideas for the next 150 years.
Prior to 1938, human experimentation was performed without federal restrictions in the United States. Such experiments were self-regulated by professional standards and guidelines such as the Hippocratic Oath of “do no harm,” and a cultural bias that relied upon and trusted the expertise of professionals. The subsequent regulation of human research in the United States consisted of a series of responses to crises rather than a proactive attempt to assure the ethical conduct of research or the protection of the subjects. Essentially, public outcry and political response led to a system of local review and governmental oversight and regulation of human experimentation.
The federal system for the protection of human research subjects developed primarily through major federal agencies, such as the Food and Drug Administration (FDA), the NIH, and the Public Health Service (PHS), all within the Department of Health Education and Welfare (DHEW), now the Department of Health and Human Services (DHHS). The National Cancer Institute (NCI), established in 1937, provided the “first extramural research grants awarded on a competitive basis to medical researchers in the United States.” The PHS was later given the power to fund research at universities and private institutions, and it administered these programs through the NIH.8 The federal Food, Drug and Cosmetic Act of 1938 required the FDA to oversee new drugs and devices for diagnosis, treatment, and prevention of disease unless they were shown to be safe. The Act, the first in the
K-4
United States requiring labeling of new products intended for use with humans, was a response to public outcry over the reported death of more than 100 consumers from “Elixir of Sulfanilamide.” The elixir was tested only for “flavour, appearance, and fragrance” prior to marketing. The legislation, however, exempted regulatory oversight of the experimental use of drugs by qualified scientists9 and only required that they carry a label: “Caution-New Drug-Limited by Federal Law to Investigational Use.”10 Twenty-five years later another drug, thalidomide, was suspected of public harm. As a result of the thalidomide scandal, the FDA’s authority was expanded to encompass oversight of the use of experimental products, including requirements for human testing, and the consent of the human subject.
It appears that the concept of local IRB review grew out of hospital based scientific peer review committees that operated on an ad hoc basis to address difficult ethical patient care issues. The “peers” were other physicians or experts within the institution. By 1953, Jack Masur, Clinical Research Center Director at the NIH, instructed each NIH institute to establish a “disinterested committee of scientists called the Clinical Research Committee” to review human research that involved “unusual hazard.” The committees would review and approve intramural research conducted at the NIH, with “normal” volunteers. The policy also required that “normal” subjects give informed consent.11, 12 Control subjects at the NIH were typically conscientious objectors to military service.13 Recipients of extramural NIH funding were exempted from creating such committees due to the perceived potential interference with the doctor-patient relationship, as well as a “reluctance to interfere with scientific freedom and judgment of researchers and their institutions.” Instead, the NIH relied on professional standards, local laws governing the practice of medicine, and the hope that research institutions would follow the federal lead to assure the ethical conduct of research.14 During this time, there was little differentiation made between research and therapy, between the physician and investigator, and between the patient and subject. Charles McCarthy observes that NIH investigators referred to human research subjects as patients and “research was generically referred to as ‘patient therapy.’ Given that environment, it is not surprising that the NIH had no policy of protections for patient/subjects involved in research.”15 In the introduction to an NIH symposium Thomas Malone observed, “It is unfortunate but true that much of the current progress in protecting the rights of patients and subjects resulted from abuse.”16 The implementation of policies requiring committee review of the ethics of proposed federally funded research was prompted by several crises. Between 1958 and 1968 the country confronted revelations about transplantation of a sheep heart and chimpanzee kidney into humans, radiation experiments on prisoners,17 injection of live cancer cells into unknowing, chronically ill, indigent elderly patients at the Jewish Chronic Disease Hospital, introduction of hepatitis into severely cognitively impaired children at the Willowbrook State School, a placebo controlled crossover study of an oral contraceptive agent where ten women without knowledge of the “dummy pill” became pregnant and were not allowed to seek abortion due to legal restrictions,18 and Henry Beecher’s famous article in the New England Journal of Medicine listing cases of unethical research.19 Some scientists were acutely aware that continued ethical problems in the conduct of human experiments could lead to withdrawal of public support with a concomitant loss of public funding followed by regulation.20 NIH Director James Shannon, concerned about the problems, met with the National Advisory Health Council in September 1965, and proposed an impartial prospective peer review system to address the “risks of the research and of the adequacy of protections of the rights of subjects.”21 The Council accepted his proposal. On the heels of the change in NIH policy, Surgeon General William H. Stewart issued the first comprehensive federal policy for the protection of human subjects in February 1966. The policy required institutions to create local committees to prospectively review new, renewal, supplemental, and continuing grant applications for federally funded biomedical human research. The Surgeon General defined the composition of the committees as “…staff, or consultants to your institution who are at the same time acquainted with the investigator under review, free to assess his judgment without placing in jeopardy their own goals, and sufficiently mature and
K-5
competent to make the necessary assessment. It is important that some of the members be drawn from different disciplines or interests that do not overlap those of the investigator under review.” They would “…provide prior review of the judgment of the principal investigator or program director by a committee of institutional associates.” The local institutions were henceforth responsible for applying “wisdom and sound professional judgment [to] determine what constitutes the rights and welfare of human subjects in research, what constitutes informed consent, and what constitutes the risks and potential medical benefits of a particular investigation.”22
Acknowledgement of Community
Within five months, the Surgeon General’s policy was amended to require from individual grantee institutions an assurance of compliance with NIH human research policies. The assurance meant that, in order to receive federal research funds, the grantee institutions had to accept responsibility for the review and ethical conduct of human subjects research. By December 1966, the policy had undergone further revision. Its jurisdiction was expanded to include social and behavioral research and it now required committee deliberations “…in accordance with the laws of the community in which the investigations are conducted and [with] due consideration to pertinent ethical issues.”23 The amended policy was the first acknowledgment that responsible ethical conduct of research required not only an institutional assurance of human subject protections but also consideration of community standards.
An NIH analysis in 1968 revealed that 73 percent of 142 institutional committees had membership comprised exclusively of scientific peer groups.24 The revised and expanded PHS guidelines of May 1969, formally required institutions to address community acceptance of proposed research. The modification of the guidelines made local institutions responsible for convening committees of sufficiently diverse membership to address scientific issues, local law, institutional policy, and community concerns for the protection of the rights and welfare of human research subjects.25 The DHEW stipulated in 1971 that an IRB should “possess the professional competence to review specific activities, the committee should [also] be able to determine acceptability of the proposal in terms of institutional commitments and regulations, applicable law, standards of professional conduct and practice, and community attitudes. The committee may therefore need to include persons whose primary concerns lie in these areas rather than in the conduct of research, development, and service programs of the types supported by the DHEW.”26
The Tuskegee Study of Untreated Syphilis in the Negro Male marks the dawning of consciousness in the United States regarding ethical obligations in the conduct of human research and a reassessment of the role of peer review in the protection of subjects. The PHS sponsored experiment began in 1932, lasted 40 years, and involved 600 African-American men, 399 with syphilis and 201 who did not have the disease. The subjects were not informed that they had syphilis or that penicillin was available when it was shown to be an effective treatment for the disease in 1947. As a result of the deception, the men lived with the disease and some unknowingly infected their partners. “There was no evidence that researchers had informed the men of the study or its real purpose. In fact, the men had been misled and had not been given all the facts required to provide informed consent.”27 The comments of John Heller, PHS Director of the Venereal Disease Unit, highlighted a lack of ethical obligation on the part of the PHS toward the participants in the syphilis study: “The men’s status did not warrant ethical debate. They were subjects, not patients; clinical material, not sick people.”28 Subsequently, spokespeople for the PHS readily acknowledged that the peer review system failed29 to address fundamental ethical issues. They noted that the syphilis study was not a secret, but rather the subject of numerous reports in medical journals, and it was openly discussed at professional conferences. “An official told reporters that more than a dozen articles had appeared in some of the nation’s best medical journals, describing the basic procedures of the study to a combined readership of well over a 100,000 United States physicians.”30 In 1972, the Associated Press broke the story of the syphilis study. The ethical crimes committed by the federal government against the citizens of Tuskegee, Alabama required deep reflection on the part of bioethicists
K-6
and the research community. The Advisory Committee on Human Radiation Experiments (ACHRE) later noted: “While a slowly increasing number of investigators reflected on the ethical treatment of human subjects during the 1950s, it was not until the 1960s and a series of highly publicized events with names like ‘Thalidomide,’ ‘Willowbrook,’ and ‘Tuskegee’ that it became apparent that a professional code, whether it originated in Nuremberg or Helsinki, did not provide sufficient protection against exploitation and abuse of human subjects in research.”31 Within one year, congressional committees convened hearings to examine human research in the United States.
The National Research Act became law in 1974. It outlined protections for human subjects involved in biomedical and behavioral research, it required the DHEW Secretary to promulgate regulations requiring IRB review for all federally funded biomedical or behavioral research, and it impaneled the National Commission for the Protection of Human Subjects in Biomedical and Behavioral Research.32 The National Commission was charged to assess the state of protections for human subjects around the country and to provide guidance for institutions and investigators when confronting the ethical issues of human subject research. The Commission authored several reports including a document examining IRBs, and in 1979 it issued the definitive American declaration on the ethical conduct of human research, the Belmont Report.33 Following the public revelations of the syphilis study, membership requirements for IRBs were expanded again by the DHEW in regulations issued in 1974. The new regulations emphasized the importance of considering research in the context of community standards. The regulations defined the composition of an IRB as having a minimum of five members and that it should “…include persons whose primary concerns lie in the areas of legal, professional, and community acceptability rather than in the conduct of research, development, and services programs supported by the HEW.”34 In order to ensure a diversity of opinion when considering protocols, membership on an IRB could not come from a single professional or lay group. Furthermore, the regulations now protected against an implicit institutional bias and conflict of interest by mandating that a legally convened meeting must include at least one member not otherwise affiliated with the institution.
The 1978 National Commission Report and Recommendations: Institutional Review Boards, outlined steps necessary to ensure the protection of the dignity and welfare of research subjects. The report defined local IRB review as the cornerstone of the national system for protections and it highlighted the importance of local IRB review:
The Commission believes that the rights of subjects should be protected by local review committees operating pursuant to federal regulations and located in institutions where research involving human subjects is conducted. Compared to the possible alternatives of a regional or national review process, local committees have the advantage of greater familiarity with the actual conditions surrounding the conduct of research. Such committees can work closely with investigators to assure that the rights and welfare of human subjects are protected and, at the same time, that the application of policies is fair to the investigators. They can contribute to the education of the research community and the public regarding the ethical conduct of research. The committees can become resource centers for information concerning ethical standards and federal requirements and can communicate with federal officials and with other local committees about matters of common concerns.35
In 1983, revised DHEW regulations further delineated and refined the broad IRB membership categories described in 1974 by requiring representation of both male and female members and by defining nonscientists as, “…for example: lawyers, ethicists, members of the clergy.”36 The 1991 revision, which is the most recent iteration of the federal regulations, removed the examples for nonscientific members without publication of related or guiding comments.
K-7
By 1993, the concept of local IRB review was firmly entrenched for institutions that receive federal funds.37 The NIH/Office for Protection from Research Risks’ (OPRR’s)38 1993 Protecting Human Research Subjects: Institutional Review Board Guidebook explains the concept of local review, and advises institutions that an IRB:
…must be sufficiently qualified through the experience and expertise of its members and the diversity of their backgrounds, including considerations of their racial and cultural heritage and their sensitivity to issues such as community attitudes, to promote respect for its advice and counsel in safeguarding the rights and welfare of human subjects. In addition, possessing the professional competence necessary to review specific research activities, the IRB must be able to ascertain the acceptability of proposed research in terms of institutional commitments and regulations, applicable law, and standards of professional conduct and practice.39
The federal regulations further require that “if an IRB regularly reviews research that involves a vulnerable category of subjects, such as children, prisoners, pregnant women, or handicapped or mentally disabled persons, consideration shall be given to the inclusion of one or more individuals who are knowledgeable about and experienced in working with these subjects.”40
Assurances: The Foundation of Trust
Since 1966, institutions have been required to provide an assurance of compliance to the federal government. The document assures the federal government that the institution will take responsibility for the ethical and legal conduct of human research done under its auspices. Currently, institutions that receive federal funding for human research must provide the OPRR with an assurance of compliance with ethical codes of conduct and the federal regulations. A Single Project Assurance (SPA) is required for institutions that periodically perform federally funded research. Research proposals and SPAs are reviewed on case by case bases by the OPRR Assurance Branch. Institutions awarded numerous federal research grants and contracts are encouraged to file a Multiple Project Assurance (MPA) with OPRR. The MPAs are negotiated by the institutions and OPRR; they allow institutions to demonstrate responsibility for the ethical conduct of research by creating an IRB responsible for the review and approval of affiliated research. The MPA outlines the types of projects required to undergo IRB review, the responsibilities of the administration at the institution, the reporting lines within the institution, the responsibility of the IRB and investigators, the review process, and the oversight process.
The MPA stipulates that local IRB review of human research will comply with federal regulations and the ethical principles outlined in the Belmont Report: respect for persons, beneficence, and justice. In spite of past and recent problems in the conduct of human subject research, society continues to allow investigators to engage in human research because specific parameters are in place to ensure the protection of the participants. The privilege of conducting human research depends on the ability of the government, sponsors, institutions, IRBs, and investigators to effectively collaborate and ensure the on-going protection of the rights and welfare of the subjects.
The system of assurances for local IRB review is based on trust. The public has entrusted the federal government with its well-being as it relates to human subjects research. The federal government trusts the research institution to impanel an appropriate IRB to review its own research. The trust is based on the acknowledged institutional responsibility for instituting effective mechanisms for the protection of human research subjects. The institution creates an IRB and entrusts it to review research responsibly according to the federal regulations, community standards, and ethical guidelines in order to maximize the protection of the human subjects, and to negotiate the conditions of approval with investigators in a collegial manner. The local IRB review engages the scientist in a dialogue that ensures that the conduct of research is in compliance with the federal regulations and ethical guidelines and is performed according to the agreed upon IRB conditions of approval.
K-8
The subject entrusts the investigator with the protection of his or her rights and welfare beyond any research objectives. The collective trust is built through institutional support of local IRB review and compliance with federal regulations. Without the many levels of trust working together, the system of human subject research and protection falls apart.
Part II
The Concept of Local IRB Review
…To leave the decision entirely to the individual researcher himself, or to a group of his colleagues, would seem to us to violate seriously what some political scientists term the principle of shared or countervailing force. The researcher and his colleagues represent a party at interest—the scientific party: And there is good reason to believe that any party at interest is likely, more often than not, to give himself the ‘benefit of the doubt.’ Whether he does or not, the public generally thinks or suspects that he does. And in our democracy, both theoretically and pragmatically, the views of the public must be recognized as of paramount importance.41 H.S. Conrad
As outlined above, the concept of local IRB review evolved over the last 50 years from a peer review system to one of community participation. Since most research was performed at academic institutions and hospitals, it appeared reasonable to ask such institutions to institute review and monitoring mechanisms. The peer review system, solely consisting of scientific membership and thus insular and isolated, included a potential for the promotion of scientific self-interest. For example, though the syphilis study received multiple reviews at the national level and there were widely published accounts in the scientific literature, scientists did not question the ethics of the study design, which allowed the subjects’ disease to progress without consent and which withheld easily available treatment. Instead, public outcry and a congressional investigation stopped the research. In order to create a more just and representative system, the peer review model was discarded in favor of the local institutionally based IRB system, which includes nonaffiliated or community and nonscientific membership and directly engages the local research institution. It is important to note that, though the local IRB system grew out of earlier peer review programs, it is not a peer review system. As a result, the federal regulations do not recognize the IRB as a peer review committee, nor do they require a majority of scientific experts. Instead, the IRB is an open system that includes members with “varying backgrounds to promote complete and adequate review of research activities commonly conducted by the institution.”42 Ultimately, the federal government achieved sophisticated goals: predicating a research institution’s receipt of research funding on a commitment to ensure both the ethical design of the research and the ethical conduct of its faculty through local IRB review. Such requirements hold an institution’s proverbial “feet to the fire” regarding responsibility for the review and the ethical conduct of research.
The National Commission believed local IRB review offers a distinct advantage over regional or national committee oversight in the review of human research. The local IRB is in a superior position to interact with the institution, the investigator, and the community of potential research subjects, and to assure that the proposed research fulfills the three ethical principles of the Belmont Report: respect for persons, beneficence, and justice. Former OPRR Director Gary Ellis echoed the National Commission’s recommendations and affirmed the importance of local review:
K-9
We embrace the local IRB at the research site as the cornerstone of the American system of protection of human subjects. IRB review is both prospective and a continuing review of proposed research by a group of individuals with no formal interest in the research. It is local review by individuals who are in the best position to know the research at the site, the resources at the institution, the capabilities and the reputations of the investigators and staff, the prevailing attitudes and ethics of the community and most importantly, the likely subject population.43
Implicit in Ellis’ statement is the view that local IRB review provides institutions with an opportunity to demonstrate responsibility and to build a culture of trust and ethical conduct in the performance of human research. The institution’s commitment to local review is manifested through its ethical obligation to provide educational opportunities for investigators, the IRB, and staff, to provide adequate personnel and resources for the IRB, and to ensure oversight of approved research with participation of both the local scientific community and the community of potential research subjects. The institution thereby demonstrates accountability for the conduct of research and the application of regulations and ethical principles that assure the protection of the rights and welfare of the human subjects.
The requirement that each federally funded institution engaged in human experimentation constitute a local IRB encourages the institution to promote an environment that supports the highest ethical standards for the review and conduct of research performed under its auspices. Francis Moore highlighted the importance of “the intellectual and ethical climate of the institution. Such a climate is difficult to regulate or standardize, difficult at times even to recognize or describe. Yet it is more important than any other single consideration in protecting the willing patient from unwise, inexpert, or ill-advised therapeutic innovation.”44 Moore’s 1969 comments on the importance of institutional culture remain largely true today. Most importantly, the imprimatur of the institution makes the local IRB an agent of the highest ethical standards embraced by the institution itself, rather than a foreign agent of the government or an adversary of research. The institution, therefore, is uniquely situated to take responsibility for various aspects of human research, such as: 1) creation of an institutional culture that promotes and upholds the highest ethical standards in the conduct of human research, 2) education and mentoring of the research community and provision of sufficient resources and staff to support the educational mandate of the IRB, 3) involvement of all interested parties in the review process including open communication and interaction with the community (the source of potential research subjects), 4) oversight of the research, and 5) awareness of local resources and standards that may impact proposed research.
Institutional Culture
…The institution should be prepared at all times to question the conduct of research, even though previously approved by both the institution and the [PHS]. The safety and welfare of the subject are paramount.45 U.S. Public Health Service: Protection of the Individual as a Research Subject
The MPA makes the local IRB review more than just a prospective evaluation of proposed research. By compelling institutions to accept responsibility for more than just the economic management of research funds, the MPA extends their responsibility to the ethical conduct of the research. It holds the institutional official, the institutional signatory to the MPA, responsible and accountable for the actions of the investigators and the IRB. The presence of a local IRB is an acknowledgement that the institution plays a vital role in creating and maintaining a culture of ethical behavior among its investigators and staff. An institution that cultivates ethical
K-10
behavior and demonstrates intolerance for unethical behavior ensures a culture of collective responsibility for the protection of the rights and welfare of human subjects and preserves the public trust. The MPA encourages a collective responsibility for the ethical conduct of research by predicating receipt of federal funding for research on the ability of all parties, i.e., the institution, the IRB, and investigators, to comply with the federal regulations. A finding of significant noncompliance can jeopardize the ability of all stakeholders to conduct research. Ultimately, the entire institution is responsible for upholding the MPA and the regulations. The entire research enterprise is balanced on the good conduct of each participant within the institution. The highest level official at the institution, therefore, should sign the assurance and serve as the institutional official, i.e., the person responsible for assuring that all parties acknowledge and carry out their ethical responsibilities in the conduct of research. The assurance thus engages all levels of the institution in a collective commitment to uphold the highest ethical standards.
Local IRB review gives the institution an opportunity to demonstrate a commitment to the highest standards of ethical conduct in human research and to create a community that supports such standards. The research institution is uniquely situated to create a culture where ethics are valued and the importance of IRB review is honored. Levine noted that “in order to function most effectively, the IRB must not only be, but also must be perceived to be, an agent of its own institution.”46 To achieve Levine’s goal, it is incumbent upon the institutional official to use his/her moral and academic authority to require the highest ethical conduct from the faculty and staff. The institution should develop and implement local policies and procedures that reflect the ethical principles of the Belmont Report and the federal regulations to create an internal standard of acceptable behavior. Institutional policies and procedures translate into a demonstration of philosophical and practical support for the autonomy and authority of the IRB, while facilitating a fair, timely, and collegial review of proposed research. An institutional ethos that highlights the importance of ethical principles will also insist upon well-conceived and properly executed research. The requirement should be evident in written institutional policies, in the actions and communications of institutional officials, and the IRB. Research that is designed or conducted so poorly as to be unethical or invalid exposes subjects and the institution to unnecessary risk. The institutional standard for well-conceived and properly conducted research minimizes the potential for conflicts between the IRB and the research community, facilitates local review, and assures the protection of the rights and welfare of the human subjects.
Investigators will perceive such internal standards as an expression of a communal commitment to ethical behavior rather than as an intrusion into academic research by a colonizing federal authority. The IRB is thus perceived by the research community as an expression of its own commitment to human subjects protection, and as the expression of an institutional mandate and policy, rather than as an alien and disembodied review process. As noted by the National Commission, such an environment demystifies the review process and builds the trust of the research community and the public.47 The institution underscores the importance of ethical conduct by convening IRBs with a respected membership that reflects the highest level of scientific expertise and community participation and support. An IRB that has the respect of the research community is better able to fulfill its principal charge as outlined by the National Commission, i.e., education of the research community. The institutional official recognizes that the Board can only carry out its regulatory, educational, and ethical functions when there are sufficient resources and high level support staff to communicate effectively with the research community and to ensure adequate protections of subjects through oversight, including continuing review and monitoring of approved research.
An institution that takes responsibility for the review and conduct of human research positions itself to engage the trust and support of the scientific community, it attracts additional financial support for research because it can assure ethical conduct and safety, and it creates an environment for successful collaboration with the community of potential research subjects. The responsibility of local review obliges all institutional parties
K-11
to acknowledge a collective responsibility for the creation of a culture of intra and extramural community participation, mentoring, and accountability. The local system of review is most effective when the institutional official sets the highest ethical standards for the research community and insists upon an institutional culture that demonstrates support for the charge of the IRB, namely, respect for human dignity.
Education and Mentoring
The most effective protection for all concerned depends upon a recognition and an understanding of the various aspects of the problem.48 Henry Beecher
As previously noted, the local review and conduct of human research is a collective ethical responsibility. The efficacy of the system is predicated on the ability of the institution, the IRB, and the investigators to collaborate in an environment of mutual trust and support to facilitate a common goal: the safe and ethical conduct of research. In order to accomplish this goal, the federal government, the institutional official, the IRB and its support staff, as well as investigators, should view their principal collective charge as educational in nature. An effective system of review and protection requires each party to accept the role of educator and to demonstrate ethical leadership. An institution that accepts responsibility for the review of human research is uniquely situated to demonstrate a commitment to the letter and spirit of the guiding ethical principles and federal regulations by engaging both the scientific and lay community in a shared educational dialogue. The ACHRE highlighted the importance of education: “The historical record makes clear that the rights and interests of research subjects cannot be protected if researchers fail to appreciate sufficiently the moral aspects of human subject research and the value of institutional oversight.”49 An academic research institution such as a university or a research hospital has three responsibilities: to conduct research, educate, and to serve the surrounding community. Education is the foundation for a local culture of trust and shared responsibility. The institution is responsible for ensuring the effective education of its scientists, faculty, staff, and IRB, in the ethical conduct of human research.50 The National Commission highlighted the contribution the local IRB can make to the education of the research community and the public about the ethical responsibilities of human research. An effective education program deepens the awareness of all stakeholders regarding their obligations to protect the rights and welfare of human subjects, it trains them in the regulatory and legal requirements, it promotes ethical conduct, and it builds an institutional culture of shared responsibility that creates trust among all parties. It also builds a participatory process that acknowledges the expertise of all participants, i.e., the IRB, the investigators, the lay community, and the institutional official. An acknowledgement of shared expertise engages the participation of all stakeholders in the creation of policy, in the IRB process, and in the ethical conduct of human research.
The research institution has the resources and the faculty to compose an IRB of respected members from the campus and the community-at-large that will “promote respect for its advice and counsel.”51 By naming senior research faculty members and respected community members to the IRB, the institution reinforces the importance of the IRB process as well as its own commitment to a successful and ethical human research program. Senior scientific experts on the local IRB play two important roles with respect to institutional goals and IRB responsibilities: they serve as scientific and ethical mentors to their colleagues, and they bring their research expertise to the deliberations of the IRB. In addition, the local IRB is a source of information concerning ethical standards and federal requirements, and it facilitates a close working relationship with investigators. The inclusion of faculty and local nonaffiliated lay community members on the local IRB creates a participatory democracy where all stakeholders have a direct voice in the research and review process and control of their own destiny.52
K-12
Many investigators view the human research review process as bureaucratic or mysterious. The local IRB demystifies the review process and creates an environment of collegiality. The local IRB offers investigators the opportunity to attend meetings and to directly discuss with the Board issues regarding specific research projects. Contact with the IRB provides an opportunity to directly address specific concerns of the Board or the investigator, to educate the IRB members and the investigator, and to open the review process to each stakeholder. The direct participation of investigators in the local IRB process empowers both the investigator and the Board. For example, an investigator, in response to requests for ethical justification of a study design, can attend an IRB meeting to clarify the issues. Attendance at an IRB meeting provides both the Board and the investigator with an opportunity to engage in a dialogue about their concerns. The IRB is no longer a disembodied entity making judgements on an investigator and his or her proposed research. Instead, the Board is revealed to be composed of the investigator’s mentors and respected scientific colleagues; ethical questions take on a newly recognized gravity and the IRB has an opportunity to collegially discuss the concerns with the researcher. As a result of direct engagement with the researchers, the IRB is demystified, and both the IRB and the investigators are educated.
A traditional educational paradigm that employs rote memorization may not lend itself to the successful education of an IRB or investigators. A rote memorization of guidelines, principles, and regulations, absent the ability to apply such concepts in practical situations will not ensure the protection of the rights and welfare of human subjects. An effective human research education program, therefore, should also include a dialogue among all stakeholders regarding the application of ethical principles and regulations in the practical research context. The promotion of dialogue among all parties avoids the perception that IRBs claim imperious authority in ethics, regulation, and research standards. Such perception will lead to adversarial relationships between the IRB and the research community, resulting in a breakdown of trust.
Education occurs in the dynamic collaboration between the IRB and the research community. An education program that acknowledges collective expertise among the IRB, the scientific community, and the lay community, and that encourages and supports an engaged dialogue among all parties, prevents adversarial relationships. An institution that adopts a collaborative educational approach draws on collective institutional expertise and knowledge which facilitates an engaged partnership by all parties in the goal of ethical research. Furthermore, the dynamic exchange of experience and knowledge creates an institutional culture that builds awareness of the regulations and sensitivity to collective responsibilities in conducting research with human subjects, and it also enlists all parties in open communication, thereby eliminating any need for conflict resolution or an appeals process outside of the IRB. For example, the institutional official may convene subcommittees of scientific and lay members of the IRB, investigators, and legal counsel to examine issues of concern to the research community for the purpose of creating institutional policy that will guide the actions of the IRB and the investigators. Subcommittees investigate, discuss, and make recommendations to the IRBs, the institutional official, and the research community, on institutional policy regarding such issues, as research with human genetic and biological material, or requirements for ensuring the privacy and confidentiality of human subjects both during recruitment and over the course of the research.
An institutional commitment to IRB review includes a responsibility to supply enough highly educated support staff and resources, such as meeting space, locking files, and computerization, to support the charge of the Board.53 To facilitate the review process, a knowledgeable local IRB staff communicates with the contract and grant administration, the institutional administration, sponsors, investigators, research staff, federal regulators and agencies, and other IRBs. The local IRB staff is an extension of the Board and is a ready and professional source of information for the research community about regulations, current national trends regarding human subject protections, and local laws. It also maintains the “IRB memory” in order to assure consistency in the review of like proposals, and for institutions with multiple IRBs, it assures consistency in the review of like projects among the IRBs.
K-13
Some universities have created human subject research education programs administered by faculty, IRB members, and staff to educate the research community. Professional, educated staff in collaboration with IRB members and faculty provide educational sessions for investigators, on-line tutorials, and manuals that advise investigators on the regulations and ethical standards, and guide them in the mechanics of adequate completion of IRB applications.54 Such educational tools help investigators address significant ethical and regulatory issues prior to IRB review and thus facilitate the review process for all parties. Some IRBs use correspondence, generated as part of the review process, as a way to educate investigators.55 Local educational programs encourage ethical behavior by providing a historical context for the shared regulatory responsibilities of all stakeholders, they sensitize researchers to cultural and community concerns, and they inform investigators of institutional policies to ensure the protection of the human subjects.
Community and Participatory Democracy
Appeals to the principle of respect for persons are often viewed with suspicion not only because they appear to remove people from time but also because they appear to remove people from their communities.56 James Childress
In this section I will discuss the participation of the nonaffiliated community member as a nonscientist.57 A recent NIH IRB survey noted that most nonaffiliated members are nonscientists. The federal regulations require that an IRB include at least one member who is not affiliated with the institution commonly known as the community member or lay member, and one nonscientific member.58 The nonaffiliated membership on the IRB provides a voice for the community of research subjects during the review of research. OPRR suggests that the nonaffiliated member should come from the local “community-at-large….The person selected should be knowledgeable about the local community and be willing to discuss issues and research from that perspective.”59 The OPRR guidance implies that the nonaffiliated member’s charge is to represent community concerns and by extension the concerns of specific subject populations. Recognition of both the implicit scientific bias in the traditional peer review system and the need for community participation in the ethical evaluation of human research, coincides with a societal shift in emphasis from the individual to the social environment in which individuals exist. Through community representation, the IRB is able to acknowledge and address such important issues as the social context and impact of research, the heterogeneity of our society, the impact of scientific paternalism on notions of autonomy, beneficence and justice, the recognition that, in addition to physical risk, scientific inquiry includes potential social, psychological, and economic risk for subjects, and the need to engage the potential subject populations in the decision making process regarding research in their community.
Paul McNeill commented, “The assumption that society (or the community) should have a voice on ethics committees is based on a notion about the role of the lay member.”60 The regulations do not privilege scientific expertise over community participation on the IRB. Instead, the regulations reserve an adequate number of chairs at the IRB table for both scientific expertise and community representation and note that the IRB should be “sufficiently qualified through the experience and expertise of its members, including consideration of race, gender, and cultural backgrounds and sensitivity to such issues as community attitudes, to promote respect for its advice and counsel in safeguarding the rights and welfare of human subjects.”61 The National Commission endorsed a balance of scientific, individual, and community concerns on IRBs in order to guard against scientific self-interest and to demonstrate an “awareness and appreciation for the various qualities, values and needs of the diverse elements of the community served by the institution or in which it is located. A diverse membership will enhance the local IRB’s credibility as well as the likelihood that its determinations will be sensitive to the concerns of those who conduct or participate in the research and other interested parties.”62
K-14
Community, however, consists of several distinct and sometimes intersecting groups, such as the community of potential research subjects, people located in a specific geographical area, people with similar interests, work, culture, or religious, racial, or ethnic background. The letter and the spirit of the National Commission IRB report and the federal regulations require sufficient scientific, cultural, and community expertise, and therefore, appear to support representative or democratic IRB membership, one that includes the participation of representatives of potential subject populations on the IRB. The federal regulations recognize that research is a social act involving particular social relationships. Such awareness underscores an important aspect of the spirit of the regulations and the intent behind local review, that is, the democratic constitution of a local IRB in order to balance the interests of science, society, and the individual. Though a nonscientific community member serves an important purpose on the local IRB, it is important to distinguish between such independent members and community members who are representative of or who directly advocate for subject populations. Representatives of subject populations should have a right to participate in the review process in order to protect and advance their own interests.63,64 The local IRB thus realizes and promotes a form of participatory democracy where “culture [is recognized] as the essence of human endeavor, expressed in respect, recognition of differences, and inclusion.”65 The first principle of the Belmont Report, respect for persons, underscores the importance of autonomy and finds its expression in the process of individual informed consent. Autonomy is derived from the Greek autos or “self” and nomos or “rule,” “governance,” or “law.” Autonomy means that an individual has the right to self-determination or self-governance. The abstract ideal of autonomous existence is a long cherished principle of American freedom. Our idea of autonomy, however, is not necessarily actualized in the real world. It is difficult to view individuals as isolated beings living outside any social context because our complex social settings do not permit us to act in an isolated way or in a purely autonomous fashion. Our actions intrinsically link us to other people in a complex web of social interactions and dependencies. The nomos or governance, therefore, is expressed through social, political, and professional interactions. James Childress reminds us, “People are not as distinct and as separate as some interpretations of the principle of respect for persons appear to suggest. This point not only suggests that ‘no man is an island’ because actions have so many effects on others; it also implies that an individual’s wishes often reflect the social context in ways that are sometimes overlooked.”66 Celia Fisher critiqued the common understanding of autonomy through the lens of relational ethics which “conceives personhood and autonomy as social constructions which best can be respected through mutual understanding and dialogue between scientist and subject.”67 The principle of respect for persons, therefore, requires us to balance the abstract concept of autonomy with the functional reality of lived relationships and community.
The local IRB balances respect for individual autonomy, through requirements for individual informed consent, with respect for the individual’s social context, through the participation of community representatives of possible subject populations in the review process. The 1996 revised joint FDA/NIH regulations for waiver of informed consent in emergency research reveal a paradigm shift in our concept of respect for persons, traditionally expressed through individual informed consent. The revised regulations reflect a more nonindividualistic interpretation of autonomy that involves the community of the potential subject population. The regulations effectively allow a community to consent for individuals who participate in emergency room research, such as research on traumatic brain injury.68 The radical shift in emphasis from the primacy of individual autonomy to group consent embodied in the regulations is predicated on the presumption that such research would not be reviewed by regional or central IRBs.69 Instead, the regulation requires the local IRB and investigator to discuss implementation of the waiver of informed consent for specific projects within the community of the potential subjects. Annette Dula suggests the important role community plays in local IRB review when noting that the majority of emergency research performed under the waiver of informed consent regulations will include
K-15
disproportionate numbers of African-American, Latina/o, and poor subjects, “…because of the location of trauma centers and because of the disproportionately high rate of certain kinds of trauma… and a large proportion of trauma centers are located in public hospitals in or near inner cities.”70 The importance of community participation in the local IRB process was also illustrated during a national FDA human research meeting on implementation of the waiver rule. High level FDA representatives underscored the importance of community participation in the IRB process: “if the community response reveals substantial concerns, the [IRB] should ask for a redesign of the study, and if that is impossible, the research may not be appropriate for that community.”71 The role of community members on local IRBs evolved out of concern that a committee comprised exclusively of institutional representatives would be biased toward research and the interests of the institution. The membership requirements were expanded in order to address such issues as relevant local law and community acceptance of the research. The local IRB is uniquely situated within the community of the proposed research to be able to provide potential subject populations with sufficient seats at the table to represent effectively their interests and inform the Board of prevailing attitudes and issues in the community. Community agencies, local advisory counsels, and other groups can serve as resources of information about their members’ concerns, they can advocate for subjects, and they can open channels of communication with the institution and the IRB by serving as Board members. A local IRB can maximize interaction with and access to community representatives and organizations in order to adequately reflect the concerns of potential research populations and cultures, and ensure that they are treated with respect and justice during the review and ultimately during the conduct of the research. The inclusion of community members who are representative of the potential subject populations can help assure the safety of the subjects by providing a window onto the local culture. They can help educate the IRB, the investigators, and the institution to the unique needs of the community, to the social and cultural implications of the research, and to local cultural nuances that will permit investigators to recruit subjects more successfully.72, 73 Community members, therefore, bring an expertise to IRB deliberations. Their collaboration with the scientific community helps the IRB identify areas of concern in the community and helps to “construct a scientific enterprise based upon mutual respect, accommodation, and trust” with the potential subject population.74 Adequate community representation ensures that the IRB is able to address local cultural, religious, and language issues, and to make provision for respectful and understandable informed consent, privacy, and confidentiality. Federal regulations require that an investigator provide subjects with legal, written informed consent except in specifically delineated exceptions. The consent process, including the informed consent document, must be in a language understandable to the subject.75 OPRR advised investigators and IRBs “to safeguard the consent process and to promote open and free communication between the researcher and the participants. Investigators and IRBs must seek to understand cultural nuances and types of foreign languages inherent in the populations to be enrolled.”76 Consideration of local language and cultural differences play a significant role in the IRB review of the scientific design of proposed research, in the assessment of the risks and benefits of the proposed research, and in the application of principles such as respect for persons and justice. Consider the population of Los Angeles. The Los Angeles Unified School District identified 80 different language groups in its student population. The largest groups with non-English language backgrounds are those that speak Spanish, Armenian, Korean, Cantonese, Vietnamese, and Russian.77 Los Angeles represents a community of diverse and highly distinct cultural groups with different community practices and needs beyond translated consent documents. The local IRB, through community awareness and by virtue of its local setting, is better able to ensure that an investigator is sensitive to cultural differences that will affirm the dignity, as well as the rights and welfare of the subject population.
K-16
Leslie J. Blackhall and colleagues noted that examining a “…family-centered decision-making style does not mean abandoning our commitment to individual autonomy or its legal expression in the doctrine of informed consent. Rather, it means broadening our view of autonomy so that respect for persons includes respect for the cultural values they bring with them to the decision-making process.”78 Community membership on the IRB can help the investigator address different cultural paradigms such as family centered decision making and consent. In order to assure safety and autonomy, a local IRB with knowledge of the local community can also address specific safety concerns, such as how socio-economic factors including hunger or food insecurity may affect health related behavior and priorities,79, 80 the risks of combining traditional remedies with experimental drugs for populations who may access herbal medicine when participating in clinical trials,81 the inability of certain subject populations to obtain basic palliative pain relief from their local pharmacies due to societal racial inequities or alienation from the health care system,82, 83 suspicions and fears resulting from cultural and historical abuses and racial factors in mental health care,84 genetics,85 and language barriers and requirements for translated consent documents and informational materials. Additionally, the broad membership of a local IRB can address the “cultural differences in beliefs and values, language and communication difficulties, issues of transportation and immigration status, and lack of familiarity with the United States health care system.”86 An IRB without adequate representation from the potential subject population may fall prey to the misconception that it can generalize decision making processes, social and psychological risks, and the rights and welfare of research subjects based on a paradigm of a homogenous society. In order to create a system of human subject protection that honors and respects differences among cultures and groups, the local IRB can include representatives from research populations and provide them with an opportunity to participate in the decision making process. For example, investigators may note that the geographical location of a woman with breast cancer is irrelevant to IRB deliberations about their participation in research. This type of comment reveals a world view that sees subjects as members of a homogenous population. It also reveals an ignorance of the scope of concerns besides diagnosis and treatment that a subject and an IRB may have, including consideration of the social, psychological, and economic risks that may be involved with identification, contact, and recruitment of the potential subject, as well as from the subject’s ultimate participation in research. The Jewish newspaper, Forward, illustrated the point in an article about how orthodox Jewish women, fearful of stigmatization resulting from community knowledge of having the BRCA1 genetic link for breast cancer, were “travelling far from home for treatment and disguising their hospital stays as out-of-town visits, lest the news of their affliction poison the marriage prospects of their daughters.” Some people in the orthodox Jewish community also expressed concern about the potential social ramifications of the discovery of the BRCA1 gene and its impact on Ashkenazi Jews.87 The article articulates concerns held by orthodox Ashkenazi Jewish women that may be different from those of other women and which require, therefore, different consideration by the local IRB and the investigator in order to ensure the respect and dignity of the subject and to minimize risk.
It is important to remember that the definition of risk includes physical, social, psychological, and economic risks.88 Human research may involve minimal physical risk, but significant potential social, psychological, and economic risks may result from a breach of confidentiality or an invasion of privacy. Research on the conditions of industrial workers, for example, may pose nonphysical risks to subjects that IRB members who do not live or work in the area may never contemplate. A national or regional IRB may not have ready access to sufficient expertise in local management/labor issues, workplace relations, etc., in order to adequately assure the protection of the subjects. A local IRB with membership from the labor community has the expertise to take the necessary steps to secure the protection of the rights and welfare of worker subjects that may not be readily apparent to the investigator, scientific members, or nonworker members. Such expertise could determine whether the scientific design of the study increases the risk to workers by linking identified individuals with specific job conditions, hazards, and adverse health effects, thus potentially disqualifying the subject from his or her job,
K-17
limiting access to medical or life insurance, or excluding the worker/subject from other careers or trades. The inclusion of worker representatives on IRBs that review workers studies helps IRBs balance societal benefits against personal risks to the participants.89 Additionally, minority, immigrant, and poor populations may be vulnerable to risks that are not readily apparent to faculty at academic institutions or to members of an IRB located thousands of miles from the community of subjects. Therefore, the ivory tower may pose very real dangers for research subjects and must be a concern for an IRB reviewing the adequacy of protections for the rights and welfare of research subjects.
Jason Karlawish posited that community participation represents a basic point of justice:
It is a maxim of research ethics that a poorly or improperly designed study is unethical. The claim that a trial is potentially beneficial ought to rest upon a consensus that the trial measures benefits that the community values and acceptably balances potential risks and benefits…. Clinical research needs to be responsive to the needs and values of the community that will participate in the clinical trials and use of drugs. This community comprises not just physicians and industry, but also patients. This is a basic point of justice that should govern right reason in the republic of science, and it is an agenda for bioethics research.90
Robert Fullilove suggested that the principle of beneficence may also require an IRB to examine and balance community interests. Fullilove compared a proposed epidemiological study on the “frightening excess mortality rates reported for Harlem residents” with a 1990 study that reported “the life expectancy of black males in Harlem [as] approximately 49 years.” He noted that community members expressed “antipathy” towards the new study and indicated that the community needed doctors and not research. Fullilove wondered whether IRBs should ask investigators proposing to work with minority communities “…to demonstrate how their work will have benefits (or create risks) for the community at the same time that they are describing the risks and benefits that such research will pose for participating individuals.”91 Many communities approach research with skepticism due to past inequities. Some communities express concern about colonization and appropriation of their culture for research purposes without concomitant benefit to the community.92, 93 The Native American community, for example, highlights complaints about participation in research that may resonate with many other communities, such as participation in research without “fully understand[ing] the risk to their health and safety,” miscommunication that results in potential subjects feeling obligated to participate in research, research conducted without “respect [for] the basic human dignity of the individuals or their religious and cultural beliefs,” demeaning the dignity of individuals and communities due to the supposed “purity” of their gene pool, “researchers [pursuing] issues of importance to the larger society but of marginal interest to Indian people,” and manifesting a disinterest “in problems of more urgent concern to the Indian community.”94 The local IRB, with sufficient representation from potential subject groups, can sensitize both its own members and the investigator to issues regarding local fears, attitudes, problems, concerns, and practice. The local IRB that functions as a participatory democracy will apply the principles of the Belmont Report within a community context to ensure respect, beneficence and justice for all research subjects.
David Hayes-Bautista noted that previous conceptions of majority and minority populations are quickly changing. He outlined that California in the 1940s, was 89.9 percent European-American, and California of the 1990s, “…as a result of rapid and unprecedented demographic changes, reverse[d] the traditional demographic structure and [has] a minority Anglo population.” Similar demographic trends exist in Texas, New York, Arizona, New Mexico, Florida, and the Chicago area. The changing demographic trends challenge us to “question assumptions” of minority versus majority as well as homogenous European-American value systems that may not be applicable to communities of color.95 The recognition that American society consists of multiple value
K-18
systems requires that the IRB understand and honor subjects, their cultures, and preferences. By including community representatives on the Board, the research institution can ensure that the local IRB consistently and adequately applies the principles of respect for persons. It is important to acknowledge that we live in a country with a history of profound social inequity and that an individual’s conception of the society is informed by variables, such as race, culture, ethnicity, and class. The local IRB, working with the institution, the sponsor, and the investigator is positioned to provide representational participation in the review process and to allow communities to have an impact on decisions regarding their needs and concerns, and their individual and collective welfare as research subjects.
Oversight
…The ultimate goal of any institutional commitment to monitoring of research must be the education of its research staff. An effective institutional monitoring program should be coupled with an institution wide program to educate researchers and other staff about the proper and ethical conduct of research. A monitoring program can help an institution develop an educational program that is responsive to its own needs, to ‘fill in the gaps.’96 Charles Weijer, et al.
The authority of the local IRB to review, approve, require modification to, or disapprove research is founded on the institutional mandate described in its MPA, on the acceptance by the scientific community of the tradition of peer review, and on the applicable regulations. The ACHRE noted, “…the IRB is the enforcing agent of federal protections that is situated closest to the conduct of the research.”97 The local IRB, in collaboration with investigators and the institutional official, is responsible for the adequate review of research protocols. In addition, it must ensure appropriate conduct of the informed consent process, that the research design includes adequate monitoring of the safety and data, any additional safeguards necessary to protect the welfare of particularly vulnerable subjects, and the continuing review of research at intervals appropriate to the degree of risk.98 The institution plays a vital role in ensuring adequate oversight or monitoring and review of research activities. Weijer and his colleagues proposed four categories of research monitoring: 1) continuing review; 2) monitoring of the consent process; 3) monitoring for adherence to [the] protocol; and 4) monitoring of data integrity.99 Such monitoring coupled with a human research education program ensures the responsible conduct of research.
The research institution has a vested interest in maintaining a culture of compliance. An institution that cannot demonstrate adequate protection of human subjects is at risk of losing its MPA, of incurring the suspension of all federally funded research, and of losing the trust of the community. In order to assure compliance, the institution may wish to make use of various existing committees, including the local IRB, to proactively monitor approved research. Whatever committee or mechanism the institution or the IRB employs should report directly to the IRB to ensure adequate, on-going protection of the subjects.
Biomedical institutions typically have more than one committee engaged in the review and oversight of research with human subjects. An institution may have a radiation safety committee, responsible for the review of proposals that use radiation producing machines and radioactive isotopes; institutional biosafety committees, who oversee the use of biohazardous material and recombinant DNA products, hospital or discipline quality assurance committees; and various scientific peer review committees, such as those required by the NCI for Comprehensive Cancer Centers. The institutional official can assure active collaboration and collegial relations amongst the various committees and with the IRB in order to effectively ensure the protection of the subjects. Collaborative review and monitoring by different institutional oversight entities brings diverse expertise to bear on the goal of protecting subjects.
K-19
The IRB responsibility for continuing review of approved research, if it is both thorough and substantive, is the easiest form of monitoring.100 IRBs may approve research for less than one year or require periodic reports regarding the research. An IRB that requests periodic reports from an investigator within an approval period may serve as its own Data and Safety Monitoring Board (DSMB). Some local IRBs go further and actively monitor approved research. The UCLA IRB has created Independent Safety Monitoring Boards to monitor data and the informed consent process for specific projects. At least two institutions were mandated by OPRR to create a DSMB to monitor the rights and welfare of cognitively impaired subjects enrolled in research. The DSMBs report directly to the local IRB, which may request clarifications or modifications of the research as a result of the reports.101 The UCLA IRB has trained consent monitors to serve as subject advocates and to observe the consent process. They ensure adequate communication between the researcher and the subject upon presentation of the consent form. The Department of Health and Human Services/Office of Inspector General (DHHS/OIG) report, Institutional Review Boards: Promising Approaches, outlined several approaches, other than the regulatory requirement for continuing review, to monitor the rights and welfare of subjects. One institution assigns a post-informed consent research intermediary for all subjects in psychiatric research. The research intermediary “discusses the consent form with a subject after the form has been signed to ensure that the subject understood its terms and that upon reflection the [subject] continues to want to participate in the research.” The research intermediaries report to the IRB every few months about subjects’ concerns.102 The federal regulations require that the legally effective written informed consent document provide a subject with contact information should a subject have questions about their rights.103 Local oversight includes the responsibility to follow-up on inquiries or complaints from subjects or other concerned parties about the conduct of the research. For local IRBs, the contact person is commonly the IRB administrator or the IRB chair. The IRB administrator or chair can conduct an initial inquiry, provide information to the IRB, request clarifications from the investigators, and respond to the concerns. As a result of an inquiry or complaint, the local IRB may initiate investigations into the conduct of research and engage in educational and corrective actions to ensure an ethical research environment. The availability of a local contact person, such as an IRB administrator, is a demonstration to subjects of institutional accountability and responsibility for the research and sensitivity to the surrounding community.
Some investigators, claiming a right to academic freedom, have challenged the authority of an institution or their IRB to monitor or suspend human research. A federal court, however, determined that the conduct of human subject research is a privilege and not a right, such as the right of intellectual inquiry embodied in academic freedom. The court ruled that human subject research is a privilege granted by the institution to individuals who are willing to work within the federal guidelines and state law.104 An institution that suspends an investigator’s privilege to conduct human research sends a message to the entire research community that anything less than the highest ethical standards in the conduct of human research will not be tolerated.
Local Resources and Standards
The local IRB is ideally situated to know the available resources for the conduct of human research. The local IRB is well acquainted with institutional standards, such as hospital policies and procedures, it can adequately assess the scientific design of a proposal in light of those standards, and it can ascertain how the availability of resources or lack thereof, may impact potential subjects. The local IRB is familiar with the qualifications of the members of the research community and is able to assess their ability to perform research procedures. The local IRB is also able to assess the adequacy of proposed protections for the privacy and confidentiality of the subjects.
Many academic institutions and hospitals are close-knit communities where very little happens that is not common knowledge. The communal nature of such institutions allows the local IRB to have an intimate
K-20
knowledge of institutional resources and standards. Members of the local IRB will know whether the institution has the facilities to conduct the research, such as sufficient beds in the clinical research center, or whether investigators have access to certain procedures, such as PET imaging. Members of a hospital IRB are intimately familiar with the procedures for engaging emergency response teams in case of serious adverse events. Some institutions have buildings spaced across a large campus. Such geographical problems play a role in the local IRB review. For example, at one institution affiliated medical buildings are across the street from the university hospital. If a subject experiences an adverse event in the affiliated medical building, it is well known to the IRB that a city ambulance service is required to move the subject from the research site across the street to the hospital. A delay on the part of the city ambulance may place the subject at undue risk. The local IRB, therefore, commonly requests that investigators conduct certain types of research in the hospital in order to secure the safety of their subjects.
The IRB is required to ensure that the investigator has the qualifications to perform the proposed research. To the local IRB the qualifications of an investigator are more than a paper curriculum vitae. Members of the local IRB have personal knowledge of the investigator’s capabilities, training, and reputation. The local IRB is also in a good position to make decisions about appropriate personnel, administration of experimental agents, and responsibility for assessing side effects. Because of lack of familiarity with the research setting a nonlocal IRB may allow unqualified staff to monitor symptoms and side effects which could place subjects at increased risk.105 The IRB must ensure the protection of privacy and confidentiality of research subjects. Medical record review is commonly used to identify potential research subjects, but it may also compromise a subject’s confidentiality or privacy and violate local law. The local IRB with its intimate knowledge of institutional policy and local law can assess the adequacy of proposed protections for the privacy of subjects and the confidentiality of student or medical records. Additionally, the local IRB is familiar with local standards and policies regarding systems of patient referral for research, and can avoid invasions of privacy. Because of its knowledge of local standards and practice the local IRB can help the institution create systems to facilitate the ethical use of research tools, such as medical records, while ensuring the protection of patient confidentiality. IRB members typically participate in institutional discussions regarding the creation of data systems that can effectively and prospectively de-identify medical records for the purposes of health services and epidemiological research.
PART III
The Status of Local IRBs
Regardless of whether one believes that the ultimate justification for government policies is the goal of promoting welfare and minimizing harms or respect for self-determination, one can agree that policies represent commitments to action and hence generate obligations.106 Advisory Committee on Human Radiation Experiments
Local IRB review is a self-regulating system that depends on the honesty and integrity of the participants, namely, the institution, the researchers, and the IRB. The efficacy of the system is predicated on the ability of the institution, IRB, and investigators to work together in a supportive environment of mutual trust towards a common goal: the safe and ethical conduct of research. The system works when each stakeholder trusts the others to act responsibly in their respective roles. The design of the local IRB system holds the research institution responsible for the conduct of human research and the creation of a culture of trust and responsibility in order to achieve the goals of science. The public and the federal government rely on institutional responsibility and accountability, and its support of the local IRB review, to ensure the protection of human subjects. The
K-21
system is designed to ensure a fair review by a disinterested committee that includes the participation of the scientific and local lay community. Its single purpose is to protect the rights and welfare of the research subjects by creating a healthy and ethical research environment. The system also ensures that all parties receive adequate education to perform their ethical duties, that mechanisms are in place to assess available resources to conduct the research, to assess the qualifications of investigators, and to embrace community involvement. Yet, recent events and reports highlight questions about the efficacy of the system. Problems in the implementation of the recommendations of the National Commission, the federal guidelines, as well as the letter and spirit of the federal regulations have hampered local IRBs and prevented them from achieving their full potential.
The ability of the local IRB to assure human subject protection is predicated on four factors: 1) institutional support, 2) appropriate composition of the Board, 3) education, and 4) engagement of investigators. The IRB alone cannot create a culture that recognizes the importance of human subject protection. The responsibility for creating a culture of respect for the regulations and the research review process rests in the collaborative efforts of the institutional officials, the community of investigators, and the IRB. Recent OPRR review of several academic institutions during 1998–99 noted systemic institutional deficiencies in human research protection programs that resulted in suspension or restriction of institutional assurances. Furthermore, three recent unrelated reports by independent groups highlighted that an IRB working without both moral and economic support from the institution cannot ensure the protection of research subjects. The General Accounting Office (GAO) reported in 1996, that IRBs are overworked and vulnerable to pressure. The problems include pressure to “mute concerns” about multicenter trials, pressure to recruit subjects that can lead IRBs to overlook informed consent deficiencies, and the pressure of volume: the “sheer number of studies necessitates that IRBs spend only 1 to 2 minutes of review per study.”107 The DHHS/OIG made similar findings in June 1998, reporting that “expanded workloads, resource constraints, and extensive federal mandates contribute to a rushed atmosphere where sufficient deliberation often is not possible.”108 In December 1998, the Journal of the American Medical Association published a report by the Human Research Ethics Group which noted, “Many IRBs are overburdened by the quantity and the complexity of proposals they review and by the oversight demands of federal agencies….If current trends continue, there is a distinct danger that IRBs will be considered as little more than protection against legal liability.” Ultimately, the authors warned that “research institutions and especially sponsors of research must accept the expenses of human subjects review as part of the cost of doing business. The burden on IRBs could be reduced if sufficient staff could be recruited. A system of recognition and rewards for IRB service should be implemented by local institutions…Work on the IRB should be recognized as the invaluable professional activity it is.”109 The Human Research Ethics Group reflected the concerns of the National Commission and the DHHS/OIG, noting that IRB service may be a thankless job with a tremendous workload and no recognition or compensation.
All three reports came to the same conclusion: there is insufficient institutional support for local IRBs and their function. The reports implicitly state that systemic institutional problems exist throughout the country and that these problems threaten the IRB system and, by extension, threaten the ethical conduct of research. An objective observer could view the systemic problems as a lack of institutional commitment to the importance of IRB review and oversight of research. This lack of commitment can be interpreted as a sign of institutional disrespect for the IRB process, the investigators, and the rights and welfare of subjects; disrespect that ultimately places the system of protection and the ability to conduct human research at undue risk.
K-22
IRB Membership, Participatory Democracy, and the Balance of Influence
…We commit to increase our community involvement so that we may begin restoring lost trust. The study at Tuskegee served to sow distrust of our medical institutions, especially where research is involved. Since the study was halted, abuses have been checked by making informed consent and local review mandatory in federally funded and mandated research.110 William Jefferson Clinton
The IRB system was developed from the peer review model. The National Commission warned that it is important when constituting an IRB to “guard against self-interest influencing or appearing to influence IRB determinations.” To this end, according to the National Commission, no more than two-thirds of the IRB members should be scientists, “including members of the disciplines in which research is customarily reviewed by the IRB.”111 The National Commission’s recommendations were widely ignored and not incorporated into the federal regulations, the OPRR IRB Guidebook, or the FDA Information Sheets112 for IRBs. The result of this rejection is that many IRBs still reflect the peer review model with only a nod to community membership that could balance the influence of scientific expertise.113 The federal regulations define a legally constituted IRB as a minimum of five members, with at least one nonscientist and at least one nonaffiliated member. Interestingly, the regulations require the attendance of a nonscientist in order to vote on human research proposals, but not the attendance of the nonaffiliated member. That regulatory loophole effectively allows for exclusion of a nonscientist community representative from IRB decisions.114 In order to satisfy the regulatory requirement for a legally convened IRB meeting, some institutions rely on an IRB administrator, institutional lawyer, or contract and grants officer to serve the role of nonscientist. The OPRR submits that the nonaffiliated member should come from the local “community-at-large….The person selected should be knowledgeable about the local community and be willing to discuss issues and research from that perspective.”115 The OPRR language implies that the nonaffiliated member should be a nonscientific community representative. Many institutions comply with the OPRR recommendation and the regulations through a common practice of simultaneously fulfilling the requirements for the nonscientist and nonaffiliated member by engaging one or more individuals from the community, known as the community or lay member.
The lasting influence of the peer review paradigm continues today to affect the membership of IRBs, where little recognition is given to the importance of community participation. The NIH-sponsored James Bell and Associates 1998 IRB survey clearly indicates that IRBs still reflect the peer review model without adequate voting representation from the local nonscientific community. A 1995 national IRB survey indicated IRB membership was 66 percent male and 90 percent white, nonHispanic.116 The Bell survey also showed little change in the composition of IRB membership with 92 percent white and 58 percent male. Institutionally affiliated members comprised 83 percent of IRB membership.117 IRBs based in large academic institutions in some cases have three to four times the minimum required membership, leaving one nonscientist community member to fend for him/herself.118 The statistics reveal that institutions have largely ignored the IRB membership expectations of the National Commission, i.e., community representation of subject populations, and the spirit of the regulations.
The disparity of nonaffiliated lay members to affiliated employees and scientists presents many community members with an imbalance of influence or power during IRB meetings. Mildred K. Cho and Paul Billings outlined issues of power within the IRB structure and indicated that “many of the shortcomings in the function of IRBs have been attributed to the imbalance of power among its medical/scientific and lay members. The composition of IRBs heavily favors biomedical scientists over social scientists and research oriented professionals over lay people. Such a composition often results in a deference to medical expertise and social-behavioral scientific knowledge, which may lead the IRB process away from ethical or social questions and create an
K-23
inherent bias in favor of scientific activities for their own sake.”119 An IRB composed of a large majority of scientists, academic scholars, and ethicists may, therefore, apply “federal and professional guidelines, abstract moral principles, and values situated within the cultures of academia, institutionalized medicine, or science.”120 It is important to question whether such an imbalance of influence maximizes the local IRB’s potential to serve as an advocate for the community and to demonstrate a commitment to justice and respect for the public as well as for scientists.121 The IRB membership regulations imply at least a 4:1 ratio of affiliated scientists to non-affiliated community members. The authority of scientific expertise may be difficult to overcome even with a more broad interpretation of the regulation, e.g., a 3:2 ratio of scientific to nonscientific/nonaffiliated members. As a parallel to democratically constituted IRBs, McNeill notes that management representatives are not allowed to exceed the number of employee representatives on occupational health and safety committees.122 George Annas recently proposed a more complete democratization of the IRB process by requiring a majority of community members and by opening all meetings to the public.123 The concern of Annas and Cho and Billings resonates with the experience of many lay community members who can feel quite overwhelmed or intimidated discussing research from a lay perspective with an IRB that includes a majority of scientific experts.124 Some commentators question whether the inclusion of nonaffiliated members on an IRB is “prompted by a genuine desire [on the part of the institution] to incorporate a community perspective,” or only serves to address the letter of the regulations. The comment is not intended to dismiss the contribution of such members on IRBs but rather to highlight that a nonaffiliated member does not necessarily represent the interests of specific or general research subject populations.125 The Bell survey also noted that respondents suggested a top priority should be strengthening the IRB membership through additional ethnically and racially diverse community representatives, thus suggesting a need for fundamental community consultation by the Boards.126 The survey revealed an imbalance of power favoring scientific expertise over community participation. This is consistent with the OPRR Common Findings and Guidance, that IRB membership lacks both sufficient knowledge of the local research context and sufficient diversity of its membership with respect to gender, race, and cultural backgrounds to sensitively and effectively address community attitudes.127 The DHHS/OIG saw the need for more extensive representation of community members on IRBs as a vital matter, noting that “individuals not associated with the institution or with the research enterprise can provide a valuable counterbalance to pressures that threaten IRB independence.”128 The concept of participatory democracy suggests human research review should include representatives of subject populations in sufficient numbers to balance the spheres of influence on an IRB. Subjects might thereby protect and advance their own interests.129 There is precedent for the inclusion of subject advocates on IRBs. For example, the federal regulations require an IRB to include a prisoner representative among its members when it reviews research with prisoners.130 At least two large biomedical institutions were required by OPRR to include subject representatives on their IRBs when reviewing research that includes cognitively impaired subjects.131 Many institutions created AIDS/HIV community advisory groups that participate in discussions of the scientific design of proposed research. Some federal human research peer review panels, such as those at the Department of Defense and the NIH, successfully include patient advocates.132 This is a form of participatory democracy. That is, the subject population is given an opportunity to voice its interests in any discussion of research that will impact it. Rebecca Dresser notes, “Representation of affected groups can reduce improper bias in planning and conducting research [with] human subjects.”133 Institutions could approach community advisory groups when seeking appropriate community representatives for the IRB. They should be careful not to assume, however, that scholars or community leaders selected to “represent” a particular group can speak for the local subject populations. Fisher provides the example of the NIH Violence Initiative where a panel of African-American leaders was appointed to review a scientific protocol
K-24
on the effects of pharmacologic interventions on urban violence. The appointed group, however, did not include community representatives who would have been affected by the research, i.e., “African-American men and women living in impoverished ghetto communities, whose sons, based on current statistics, have a devas-tatingly high probability of entering the juvenile justice system before they reach adulthood.” She wonders how advocates from the community of subjects may have addressed such issues as group stigmatization and how their concerns might have better ensured the protection of vulnerable subjects as well as community support for the research.134 Dula warns that the exclusion of community representatives “…from decision making results in paternalistic decisions made for the ‘good’ of the powerless. At worst, it victimizes the powerless.”135 The federal regulations and guidelines clearly require an IRB to be sensitive to community attitudes and to promote respect for its advice and counsel through membership with diverse racial, gender, and cultural background. The regulations, however, give the institution broad flexibility in their choice of IRB members and they give no interpretation of who, scientists or lay people, may best fulfill the requirements. Such flexibility and lack of definitions may have resulted in diminishing community participation and may have ultimately created an imbalance of power on IRBs. The lack of diversity among IRB members may diminish the ability of the Board to achieve its educational charge. Disparity of power and inadequate community membership limits the IRB’s ability to engage in a free exchange of ideas where different points of view and expertise are honored and respected.
Education
Henry Beecher, President Clinton, ACHRE, and the DHHS/OIG emphasize that an education program is important for assuring the protection of the rights and welfare of human subjects. An institution can only assure the protection of human subjects through an effective education program for IRBs, investigators, and staff that includes sufficient training regarding their ethical responsibilities. Nevertheless, the DHHS/OIG notes that training of IRB members and investigators is minimal and “they face significant obstacles which include not only sufficient resources, but the reluctance of many investigators, especially experienced ones, to participate.” The report also identifies the specific need to train community members in order to increase their participation and effectiveness in IRB deliberations.136 A common compliance concern noted by OPRR is insufficient education of the IRB, staff, and investigators.137 As discussed above, the local IRB is uniquely situated to contribute to the education of all stakeholders regarding the ethical conduct of research. A well-educated faculty, administration, staff, and IRB is key to a successful human subject research program. A 1995 survey of IRBs indicates that one quarter of Category 1 universities (those graduating at least 30 doctorally prepared individuals in three unrelated disciplines each year) offered no training to their IRB members. Of those institutions that offered training, 84 percent offered four hours or less, while 26 percent of the institutions provided less than one hour of training for their members. Training covered the scope of Board functions, IRB responsibilities, the process of group decision making, or other issues.138 The survey did not specify who was responsible for the training. Interestingly, the Bell and Associates survey did not assess whether respondents felt that the institutional administration or individual officials required more education about human subject protections. Most studies have focused only on the education of IRBs and have ignored the responsibility to educate the institutional administration and investigators.
Who is responsible for training the local IRB, investigators, and institutional officials, and for offering ongoing guidance on the nature of their charges? Neither the regulations nor the OPRR guidelines give specific direction on how to educate the institutional parties regarding their mandate or the appropriate execution of their charge. The ACHRE report leans towards making the education of all members of the scientific community a shared responsibility between federal agencies and institutions.139 An institutional commitment to education is the responsible approach for ensuring a safe research environment where all parties acknowledge their responsibilities and are aware of their shared and individual obligations.
K-25
IRB members are commonly chosen for their research expertise and not because they are knowledgeable about the federal regulations, the ethics of human research, or IRBs. Some institutions have proactively addressed the educational needs of their community by creating staff positions for human research education as well as programs that apply a spectrum of educational approaches, such as orientation and training for all IRB members, and web based training, didactic sessions, manuals, and guidebooks for investigators and research staff. Some IRBs allow time during meetings for educational sessions on current topics of concern or distribute relevant articles to the membership. Some institutions budget annual funds to allow IRB staff, IRB members, both affiliated and nonaffiliated, and investigators to attend national and regional meetings sponsored by OPRR, FDA, and national organizations such as Public Responsibility in Medicine and Research (PRIM&R) and Applied Research Ethics National Association (ARENA). IRB members and staff who attend such meetings then return to their institution and serve as educators of their own community. An institutional commitment to education helps create an open forum where all parties can share their collective expertise and can educate each other, thereby reinforcing the importance of protecting human dignity.
Staff and Resources
The most common compliance citations issued to research institutions by OPRR include: 1) overburdened IRBs; 2) inadequate resources, including lack of space and privacy for administrative staff “sufficient to conduct sensitive IRB duties;” and 3) lack of professional educated IRB support staff.140 The OPRR findings mirror the DHHS/OIG report calling for increased human resources, computerization, “and other elements essential to an efficient and effective IRB.”141 Federal regulations require that the institution provide sufficient infrastructure, space, and staff to carry out the charge of the IRB.142 The Bell report notes, “Additional staff—both professional and clerical—was the resource most commonly mentioned….”143 OPRR cited at least five institutions in the last two years for systemic deficiencies in their human research programs, including inadequate staff and resources to successfully implement a protection and review program.144 In order for the IRB to successfully fulfill its charge, it must have a sufficient staff of educated professionals. The DHHS/OIG reports that increasing IRB workloads have not resulted in increasing staffing levels and budgets in support of the Board. A lack of institutional support can negatively impact an IRB’s ability to adequately perform its charge: “With limited personnel and few resources, IRBs are hard pressed to give each review sufficient attention.”145 One institution’s post-OPRR site visit evaluation of its human subject protection programs resulted in a 300 percent increase in staffing and budget in order to address deficiencies effectively. The institution acknowledged that too few people understood the regulations, that the administrator responsible for the effective management of the IRB operations was overburdened, that there was too much administrative presence on the IRB, and that insufficient participation in national/regional IRB workshops resulted in problems.146 Without sufficient infrastructure and professional level staff, an IRB for a large academic institution or hospital is isolated, paralyzed, and cannot carry out basic functions, such as creating appropriate letters of condition to investigators, tracking and reviewing adverse event reports, addressing complaints from subjects, conducting appropriate annual review, and receiving guidance regarding changing interpretation of regulations.
The institution should also consider the burdens of IRB membership on affiliated members. The National Commission recognized that IRB membership requires a significant commitment of time and energy. The Commission called on institutions and the government, with little response, to allow direct costing of an IRB, “…to provide at least a portion of the salary of the IRB chair[person] or of the cost of administrative support for the IRB. Recognition of IRBs by providing earmarked funds for their operation would complement compliance and education activities of the DHEW in promoting quality performance by IRBs.”147 There is a need to compensate IRB members for their commitment and work. IRB meetings may last three to seven hours per meeting, one to four times per month. The time spent in meetings does not include additional effort devoted to reading protocols, analyzing adverse event reports, conducting expedited reviews, creating letters of condition
K-26
for investigators, among other responsibilities.148 Affiliated members are also responsible for their own clinical activities, research, teaching, and generating departmental income.149 In order to maintain adequate scientific representation on the local IRB, it is imperative that an institution implement a just system for the recruitment of members to ensure the equal distribution of the burden of service. Additionally, membership on an IRB should be honored and respected in a way that maximizes benefits for the member and demonstrates an institutional commitment to the best possible human protection program.
Institutional Influence and the IRB
The institution must create mechanisms that assure that mandates other than maintaining the protection of the rights and welfare of human subjects do not interfere with the enduring independence of the local IRB. The DHHS/OIG reports that research is an “important revenue source for most academic health centers…For decades, under the fee-for-service system research expenditures were subsidized by patient-care revenues; under managed care, however, traditional financial support for research activities has been diminishing. In the process, commercial sponsorship has become increasingly important.”150 Annas notes that medicine “...is currently faced with a new dominate ideology—the ideology of the marketplace, which puts profit-making (sometimes denoted by its method, cost-containment) as its highest priority....Both scientific truth and the best interests of patient-subjects can often find themselves sacrificed in the name of the bottom line....To do science you need money, but to raise money competitively you need to project illusions that are the antithesis of science.”151 Cho and Billings explain that “interference by IRBs with large or well-funded projects may be perceived by others in the institution as adverse to the institution and therefore inappropriate....IRBs have a large and direct impact on an institution’s ability to obtain funding for its research activities.”152 The above comments clearly suggest that the institutionally based IRB plays an integral role in the search for research funding. The regulations require IRB review and approval before an institution can initiate a clinical trial or accept federal funding for human subject research. As a result, IRB decisions potentially affect millions of dollars in research funds at an institution. The institution’s commitment to scientific inquiry and to the quest for extramural funding for research may create a conflict between the institution’s obligation to promote the ethical conduct of research and its need to attract research funds. That conflict may ultimately impact the charge of the IRB. It may pose serious questions regarding the protection of research subjects, and it may prove a fundamental challenge to the autonomy of the IRB process.153 The local IRB may struggle under overt and covert institutional pressure to approve research. The OPRR warns that “the IRB must be and must be perceived to be fair and impartial, immune from pressure either by the institution’s administration, the investigators whose protocols are brought before it, or other professional and nonprofessional sources.”154 The selection of the institutional official is crucial to the success of a local IRB program and to its ability to address internal and external pressures, as well as assuring the protection of the rights and welfare of the research subjects. The regulations, however, do not specify the necessary qualifications of the institutional official, the person responsible for ensuring the independence of the IRB, its support, and its standing within the institution. Clearly, the institutional official should have enough authority within the institution to ensure the proper support and respect for the IRB. The OPRR guidelines describe the institutional official as a person “…who has the legal authority to act and speak for the institution, and should be someone who can ensure that the institution will effectively fulfill it research oversight function.” The official, however, may delegate the authority to “…the director of research and development, a dean or assistant dean, or hospital administrator.”155 Bell and Associates noted that 35 percent of IRBs reported directly to a provost or vice president for research, with only 7 percent reporting to the highest level official such as a president or the next highest level such as an executive vice chancellor.156 Yet, reasoned consideration of the concerns expressed by federal agencies, professional groups, Annas, Cho, and Billings requires one to question whether an individual who is directly involved and responsible for research funding, such as a director of research and development,
K-27
is immunized against financial pressures; and whether an assistant dean or a hospital administrator has sufficient authority to avoid institutional conflicts and to ensure that an IRB is given the necessary respect and authority. An institution that successfully addresses such conflicts and supports the independence and authority of the IRB can avoid the common systemic problems found by OPRR in 1999. For example, OPRR expressed concern that the “placement of the IRB at a relatively low [institutional] level…contributes to the diminished status and support of the system for the protection of human subjects.” The Office strongly recommended elevation of the IRB to a higher level within the institutional hierarchy in order to demonstrate a “greater institutional commitment to human subject protections.”157 The Bell report indicates that IRBs continue to try to do their jobs without institutional support, staffing, resources, and education. In spite of the perceived conflicts and pressures on local IRBs, the NIH-funded Bell Report indicates that local IRBs are not approving research without due consideration of scientific and human protection issues. Bell and Associates found that “…in 73 percent of IRBs, one-quarter or fewer protocols were approved as submitted. In fact, 34 percent of IRBs did not approve any (zero) protocols as submitted in 1995.” They noted four categories of protocol deficiencies: 1) consent form, 2) consent process, 3) risk/benefit, and 4) scientific design. IRBs could not approve protocols as submitted due to informed consent document deficiencies such as technical language, understatement or omission of the risks and benefits, and omission of cost information and alternatives. Additionally, IRBs reported problems with the proposed consent process where comprehension was not promoted and did not ensure voluntariness, with scientific and ethical justification for the placebo control design, and with the adequacy of provisions for the protection of privacy and maintenance of confidentiality.158 The Bell report findings are consistent with OPRR site visit letters indicating that, by and large, local IRB chairpersons, members, and staff are sincerely committed to their charge: the protection of the rights and welfare of human research subjects. The Bell statistics also indicate the inadequacy of research proposals submitted to the local IRB and, probably more profoundly, a lack of communication and education within the institutions about the requirements for the protection of the subjects. These findings, as well as reports from the DHHS/OIG and the GAO, lead to the conclusion that there is too little institutional support for the protection of the rights and welfare of human subjects.
Part IV
Conclusion
When so much can be accomplished medically for those who are ill, it would be tragic if the patient could not be completely confident that his [sic] welfare was the physician’s only concern.159 Herman L. Blumgart
The principles that inform and guide local IRB review engage the trust and require the responsible behavior of all parties involved in human subject research. Guidelines, ethical principles, and federal regulations create a system of human subject protection that, when responsibly implemented, addresses the concerns of all stakeholders: the government, sponsors, research institution, the investigator, the IRB, the community, and the human subjects.
The application of democratic principles to the composition of local IRBs and the review of human research acknowledges, as noted by Lawrence Gostin, that “genuine respect for human dignity requires deeper understanding of the patient’s values, culture, family, and community.”160 The system of local IRB review represents a fundamental societal and regulatory shift from reliance on scientific expertise and self-interest as represented by
K-28
peer review, to acknowledgment of the expertise in ethical matters that is held within the community of research subjects. The local IRB provides the community of potential human subjects with a venue where it can actively contribute to the research review process.
The efficacy of the system of local IRB review is predicated on improved federal guidance on the role of the institution and the institutional official, and on the inclusion of community. Institutional responsibility requires more than compliance with the letter of the regulations; it also requires a willingness to apply the ethical principles that are the spirit of the regulations: to educate the research community and to create an institutional ethos that governs the actions of all stakeholders in the protection of human subjects. The research institution with support from the federal government has the authority and the responsibility to create a culture that is sensitive to the ethical imperative of protecting the rights and welfare of human research subjects. As noted by the National Commission, the local IRB, with support from its institution, is perfectly situated to ensure collegial interactions, the effective review and oversight of research, the participation of the scientific community and the community of potential research subjects in the education of all stakeholders. A system that encourages education, participation, and dialogue and calls on all parties to uphold the highest ethical standards will earn trust and support for its enterprise.
Acknowledgments
I owe many thanks to Marina Moevs for her support, time, and commitment to this project as well as to Judith L. Brookshire, John Dreyfuss, and Christian Moevs for their insight, comments, and assistance.
Notes
1 Jay Katz, introduction, Experimentation with Human Beings (New York: Russell Sage Foundation, 1972) 1.
2 Department of Health and Human Services Part VIII, NIH Guidelines on the Inclusion of Women and Minorities as Subjects in Clinical Research (Federal Register 59.59: March 28, 1994) 14509.
3 Office for Protection from Research Risks (OPRR), Protecting Human Research Subjects: Institutional Review Board Guidebook
(Washington D.C: U.S. Government Printing Office, 1993) 1-1.
- 45 CFR 46.109 & 112.
4 Harold Edgar and David J. Rothman, “The Institutional Review Board and Beyond: Future Challenges to the Ethics of Human Experimentation,” The Milbank Quarterly 73.4 (1995):489.
5 W. E. Waters, “Role of the Public in Monitoring Research with Human Subjects,” Issues in Research with Human Subjects,
Symposium at the National Institutes of Health, March 20–21, 1978 (Bethesda: NIH Publication No. 80-1858, March 1980) 129.
6 William J. Curran, “Governmental Regulation of the Use of Human Subjects in Medical Research: The Approach of Two Federal Agencies,” Experimentation with Human Subjects, ed. Paul A. Freund (New York: George Braziller, 1969). Robert J. Levine, “The Institutional Review Board,” (1976) Appendix to Report and Recommendations: Institutional Review Boards, National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (USDHEW No. (OS) 78-0009, 1977). Dennis M. Maloney,
Protection of Human Research Subjects (New York: Plenum Press, 1984). Paul M. McNeill, The Ethics and Politics of Human Experimentation (New York: Press Syndicate, 1993).
7 Levine, 4-4. 8 Curran, 431–432. 9 McNeill, 54. 10 Curran, 410.
11 Thomas Malone, Issues in Research with Human Subjects (Washington, DC: Department of Health, Education, and Welfare: NIH Publication No. 80-1858, March 1980) 2.
K-29
12 Charles R. McCarthy, “Reflections on the Organizational Locus of the Office for Protection from Research Risks,” National Bioethics Advisory Commission, 1997:2.
13 Curran, 418. 14 McNeill, 56. 15 McCarthy, 2. 16 Malone, 3.
17 McNeill, 34 and 57.
18 “The Jewish Chronic Disease Hospital,” 9–65; Stephen Goldby, “Experiments at the Willowbrook State School,” 1007; “Placebo Stirs Pill “Side Effects,” Medical World News 18–19 (April 16, 1971) 792, Experimentation with Human Beings, ed. Jay Katz (New York: Russell Sage Foundation, 1972)
19 Henry Beecher, “Experimentation in Man,” New England Journal of Medicine 274.2 (1966).
20 McCarthy, 4. “…NIH officials [were] aware that if research was to continue to enjoy public confidence, and if it was to continue to be funded with public dollars, then a policy for the protection of research subjects must be implemented.”
21 McNeill, 58.
22 William H. Stewart, “Clinical Research and Investigation Involving Human Beings,” Experimentation with Human Beings, ed. Jay Katz (New York: Russell Sage Foundation, 1972) 855.
23 Curran, 437.
24 Curran, 443.
25 Levine, 4–7, 8. Levine cites the May 1, 1969 revised PHS guidelines “…the committee must be composed of sufficient members of varying backgrounds to assure complete and adequate review… [and possessing] not only broad scientific competence to comprehend the nature of the research, but also other competencies necessary in the judgment as to acceptability of the research in terms of institutional regulations, relevant law, standards of professional practice, and community acceptance.”
26 Bernard Barber, John J. Lally, Julia Loughlin Makarushka, Daniel Sullivan, Research on Human Subjects (New York: Russell Sage Foundation, 1973) 194.
27 Centers for Disease Control, The Tuskegee Syphilis Study: A Hard Lesson Learned, http://www.cdc.gov/nchstp/od/tuskegee/time.htm.
28 James H. Jones, Bad Blood (New York: The Free Press, 1993) 179.
29 Advisory Committee on Human Radiation Experiments (ACHRE), Final Report, 1995: http://tis.eh.doe.gov/ohre/roadmap/achre/chap3_2.html.
30 Jones, 7.
31 ACHRE, http://tis.eh.doe.gov/ohre/roadmap/achre/chap2_7.html.
32 The National Research Act; Public Law 93-348, July 12, 1974, Protection of Human Research Subjects, ed. Dennis M. Maloney (New York: Plenum Press, 1984) 23.
33 National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research (Washington, DC: DHEW No. (OS) 78-013: April 18, 1979).
34 Levine, 4–8.
35 The National Commission for the Protection of Human Subjects in Biomedical and Behavioral Research, Report and Recommendations: Institutional Review Boards (Washington, DC: DHEW No. (OS) 78-0008) 2.
36 45 CFR 46.107: March 8, 1983.
37 The FDA did not propose regulations specific to IRB review of experiments under their authority, including drugs, medical devices, and biological products until 1978. The regulations would also apply to independent investigators who did not receive federal funding but wished to conduct research on products under FDA authority. In 1981, final IRB regulations were codified and published by DHEW and FDA as 45 CFR 46 and 21 CFR 56 respectively.
38 The office was recently renamed as the DHHS/Office of Human Research Protections (DHHS/OHRP).
K-30
39 OPRR, Protecting Human Research Subjects: Institutional Review Board Guidebook, 1–3.
40 45 CFR 46.107(a).
41 H.S. Conrad, “Clearance of Questionnaires with Respect to ‘Invasion of Privacy’ Public Sensitivities, Ethical Standards, Etc.” Experimentation with Human Beings, ed. Jay Katz (New York: Russell Sage Foundation, 1972) 824.
42 45 CFR 46.107(a).
43 Gary Ellis, “Perspectives from OPRR,” Changes and Choices for Institutional Review Boards: The Inclusion of Women and Minorities, and Other Emerging Issues in Research (Boston: Public Responsibility in Medicine and Research, October 31 and November 1, 1994)
31 –32.
44 Francis D. Moore, “Therapeutic Innovation: Ethical Boundaries in the Initial Clinical Trials of New Drugs and Surgical Procedures,” Experimentation with Human Subjects, ed. Paul A. Freund (New York: George Braziller, 1969) 375.
45 Public Health Service, “Protection of the Individual as a Research Subject,” Experimentation with Human Beings, ed. Jay Katz (New York: Russell Sage Foundation, 1972) 887.
46 Robert J. Levine, Ethics and Regulation of Clinical Research (Baltimore: Urban and Schwarzenburg, 1986) 342. 47 National Commission, Report and Recommendations, 2. 48 Beecher.
49 ACHRE, http://tis.eh.doe.gov/ohre/roadmap/achre/chap18_2.html. 50 National Commission, Report and Recommendations, 2. 51 45 CFR 46.107(a).
52 McNeill, 199.
53 45 CFR 46.103(b)(2). The institution will assure the federal government of the “Designation of one or more IRBs established in accordance with the requirements of this policy, and for which provisions are made for meetings space and sufficient staff to support the IRB’s review and recordkeeping duties.”
54 June Gibbs Brown, Institutional Review Boards: Promising Approaches (Office of the Inspector General, Department of Health and Human Services: OEI-01-91-00191, June 1998) 8–11. University of Minnesota, http://www.research.umn.edu/consent/. University of California, Irvine, http://www.rgs.uci.edu/rig/hstrain.htm. University of California, Los Angeles, www.oprs.ucla.edu. University of Rochester, http://www.urmc.rochester.edu/urmc/rsrb. University of Washington in Seattle, http://depts.washington.edu/hsd/.
55 Brown, Promising Approaches, 10.
The UCLA IRB also uses correspondence in this manner.
56 James F. Childress, Practical Reasoning in Bioethics (Bloomington: University of Indiana Press, 1997) 132.
57 “Most of the non-affiliated members were also non-scientists (10 percent of total members).” James Bell, John Whiton, and Sharon Connelly (James Bell Associates), Final Report: Evaluation of NIH Implementation of Section 491 of the Public Health Service Act, Mandating a Program of Protection of Research Subjects (Washington, D.C: The Office of Extramural Research, National Institutes of Health, June 15, 1998):26, http://ohrp.osophs.dhhs.gov/hsp_report/hsp_final_rpt.pdf.
58 45 CFR 46.107(c) & (d).
59 OPRR, Protecting Human Research Subjects: Institutional Review Board Guidebook, 1–4.
60 McNeil, 197.
61 45 CFR 46.107(a).
62 National Commission, Report and Recommendations, 14.
63 McNeill, 199.
64 Celia B. Fisher, Relational Ethics and Research with Vulnerable Populations, In Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity, National Bioethics Advisory Commission, March 1999; www.bioethics.gov.
65 Harold DeRienzo, “Beyond the Melting Pot: Preserving Culture, Building Community,” National Civic Review 84.1 (Winter 1995):5.
K-31
66 Childress, 132.
67 Fisher, 31.
68 Food and Drug Administration and National Institutes of Health, 45 CFR 46 & 21 CFR 56, Protection of Human Subjects; Informed Consent and Waiver of Informed Consent Requirements in Certain Emergency Research; Final Rules (Washington, D.C:
Federal Register: October 2, 1996).
69 “FDA anticipates that this type of research will usually be performed in an institution with an IRB.” Food and Drug Administration and National Institutes of Health, Protection of Human Subjects (Federal Register: October 2, 1996).
70 Annette Dula, “Bearing the Brunt of the New Regulations: Minority Populations,” Hastings Center Report, January–February 1997:11.
71 Paulette Walker Campbell, “Debate Swirls Around New Rules for Informed Consent in Trauma Research,” Chronicle of Higher Education, October 24, 1997:A30.
72 Fran Ansley and John Gaventa, “Researching for Democracy and Democratization Research,” Change 29.1, January–February, 1997:46.
73 Colleen Cordes, “Community-Based Projects Help Scholars Build Public Support,” Chronicle of Higher Education, September 18, 1998: A38.
74 Fisher, 33.
75 45 CFR 46.116 and 21 CFR 56.116.
76 Gary Ellis, OPRR Reports: Inclusion of Women and Minorities in Research, Number 94-01, April 25, 1994.
77 Los Angles Unified School District, www.lausd.k12.ca.us./lausd/offices/bilingual/senate_pres/note011.html. The Los Angeles Unified School District has identified 80 languages, within the district, with 8 languages comprising the largest percentage of students with these language backgrounds. The cultural diversity of the southern California region is reflected through the predominant language groups: Spanish, Armenian, Korean, Cantonese, Vietnamese, and Russian and may impact different considerations for the protection of the subjects.
78 Leslie J. Blackhall, Sheila T. Murphy, Gelya Frank, Vicki Michel, Stanly Azen, “Ethnicity and Attitudes Toward Patient Autonomy,” Journal of the American Medical Association 274.10, September 13, 1995:825. The authors indicated, “…It is vital to uncover the usually unspoken beliefs and assumptions that are common among patients of particular ethnicities to raise the sensitivity of physicians and others who work with [Korean American and Mexican American] groups. Understanding that such attitudes exist will allow physicians to recognize and avoid potential difficulties in communication and to elicit and negotiate differences when they occur.”
79 Paula M. Lantz, James S. House, James M. Lepkowski, David R. Williams, Richard P. Mero, Jieming Chen, “Socioeconomic Factors, Health Behaviors, and Mortality,” Journal of the American Medical Association 279.21, June 3, 1998.
80 Karin Nelson, Margaret E. Brown, Nicole Lurie, “Hunger in an Adult Patient Population,” Journal of the American Medical Association 279.15, April 15, 1998:1213.
81 Grace Xueqin Ma, The Culture of Health: Asian Communities in the United States (Westport: Bergen and Garvey, 1999) 112.
82 R. Sean Morrison, Sylvan Wallenstein, Dana K. Natale, Richard S. Senzel, Lo-Li Huang, “‘We Don’t Carry That’ —Failure of Pharmacies in Predominantly Nonwhite Neighborhoods to Stock Opioid Analgesics,” New England Journal of Medicine 342.14, April 6, 2000:1025. The authors reported that African-American, and Latino communities receiving “palliative care at a major urban teaching hospital are unable to obtain prescribed opioids from their neighborhood pharmacy….Two thirds of the pharmacies that did not carry any opioids were in neighborhoods where the majority of the residents were nonwhites. This finding, together with reports that nonwhite patients are significantly less likely than white patients to receive prescriptions for analgesic agents recommended by the AHCPR, suggests that members of racial and ethnic minority groups are at a substantial risk for the under-treatment of pain.”
83 Alexis Jetter, “Breast Cancer in Blacks Spurs Hunt for Answers,” New York Times, February 22, 2000.
84 William R. Macklin, “Breaking a Silence about Mental Health: A Summit will Focus on the Needs of African Americans,”
Philadelphia Inquirer, September 8, 1998: D01. “African Americans mental health needs historically have been ignored or mishandled, and that African Americans are often suspicious of the therapeutic community.” Additionally, “African Americans will remain skeptical until psychologists, social workers, and counselors demonstrate a broader understanding of the impact of racial, historical, and cultural factors on mental health.”
K-32
85 Morris W. Foster, Ann J. Eisenbraun, Thomas H. Carter, “Communal Discourse as a Supplement to Informed Consent for Genetic Research,” Nature Genetics, November 1997.
86 Ma, 112.
87 E.J. Kessler, “The Secret Shake-Up in the Shiduch,” Forward, July 26, 1996.
88 OPRR, Protecting Human Research Subjects: Institutional Review Board Guidebook, 3-1.
89 Robert Pleasure, “Union Perspectives on Workplace Studies, Protecting Human Subjects, Office of Biological and Environmental Research,” Protecting Human Subjects (Department of Energy, Winter 1999/2000):5–6.
90 Jason Karlawish, “Building Right Reason in the Republic Science: Why We Must Include Community Values in the Design and Conduct of Clinical Research,” PennBioethics 5.2, Center for Bioethics-University of Pennsylvania, Summer 1998.
91 Robert Fullilove, “Justice, Access, and Scientific Discrimination: Recruiting and Retaining Women and Minorities in Research,”
Changes and Choices for Institutional Review Boards: The Inclusion of Women and Minorities, and Other Emerging Issues in Research,
(Public Responsibility in Medicine and Research, Boston, October 31 and November 1, 1994) 84.
92 Fisher, 39.
93 Ansley and Gaventa.
94 American Indian Law Center, Model Tribal Research Code (Albuquerque: American Indian Law Center, September 1999) 1.
95 David Hayes-Bautista, “Formulating Health Policy in a Multicultural Society,” Health Policy and the Hispanic, ed. Antonio Furino, (Boulder: Westview Press, 1992) 147.
96 Charles Weijer, Stanley Shapiro, Abraham Fuks, Kathleen Cranley Glass, Myriam Skrutkowska, “Monitoring Clinical Research: An Obligation Unfulfilled,” Canadian Medical Association Journal 152.12, June 15, 1995:1975.
97 ACHRE, http://tis.eh.doe.gov/ohre/roadmap/achre/chap14_5.html.
98 J. Thomas Puglisi, memorandum to Division of Human Subjects Protections, OPRR, Knowledge of Local Research Context, Rockville, August 27, 1998. http://grants.nih.gov/grants/oprr/humansubjects/guidance/local.htm.
99 Weijer, 1975.
100 Gary B. Ellis, “OPRR Reports: Continuing Review—Institutional and Institutional Review Board Responsibilities,” (Rockville, MD: Number 95-01, January 10, 1995).
101 Office for Protection from Research Risks, Evaluation of Human Subject Protections in Schizophrenia Research Conducted by the University of California Los Angeles, May 11, 1994.
- Michael A. Carome, “To Smith Jenkins, Veterans Administration Greater Los Angeles Healthcare System,” March 22, 1999 (Obtained through Freedom of Information Act Request).
102 June Gibbs Brown, Institutional Review Board: A Time for Reform (Washington D.C: Office of the Inspector General, Department of Health and Human Services: OEI-01-97-00193, June 1998) 12.
103 45 CFR 46.117(a)(7).
104 Dale Hammerschmidt, “There is No Substantive Due Process Right to Conduct Human Subject Research: The Saga of the Minnesota Gamma Hydroxybutyrate Study,” IRB 19.3:4 May–August 1997).
105 Vicki Brower, “Vulnerable Groups at Risk from ‘Commercial’ Ethical Review Boards,” Nature Medicine 3.7, July 1997:705.
106 ACHRE, http://tis.eh.doe.gov/ohre/roadmap/achre/chap4_2.html.
107 Sarah F. Jagger, “Scientific Research: Continued Vigilance Critical to Protecting Human Subjects,” General Accounting Office, Testimony before the Committee on Governmental Affairs, U.S. Senate, March 12, 1996.
108 June Gibbs Brown, introduction, A Time for Reform, ii.
109 Jonathan Moreno, Arthur L. Caplan, Paul Root Wolpe, and the Members of the Project on Informed Consent, Human Research Ethics Group, “Updating Protections for Human Subjects Involved in Research,” Journal of the American Medical Association 280.22, December 9, 1998:1956.
110 William Jefferson Clinton, “Remarks in Apology to African-Americans on the Tuskegee Experiment” (transcript), (Weekly Compilation of Presidential Documents: 33.20, May 19, 1997).
K-33
111 National Commission, Report and Recommendations, 14.
112 Food and Drug Administration, FDA Information Sheets (Rockville, August 1998).
113 Bell, 26. “Most of the non-affiliated members were also non-scientists (10 percent of total members).”
114 45 CFR 46.108(b).
115 OPRR, 1993 Protecting Human Research Subjects: Institutional Review Board Guidebook, 1-4.
116 Gregory Hayes, Steven Hayes, Thane Dykstra, “A Survey of University Institutional Review Boards: Characteristics, Policies, and Procedures,” IRB 17.3, May–June 1995:2.
117 Bell, 25–26. According to OPRR records from the summer of 1995, the 491 surveyed “IRBs had 6,923 members, with membership ranging in size from 5 to 44 members. The mean number of members for IRBs in the lowest volume decile was 10.5, compared to a mean of 19.7 members in the highest volume decile…Seventy percent of members were affiliated scientists; the remainder were about equally divided between affiliated non-scientists (13 percent), such as persons educated in law and business, and non-affiliated members (14 percent). Most of the non-affiliated members were also non-scientists (10 percent of total members).”
118 Brown, A Time for Reform, 8. “It is not unusual for an IRB of 15 to 20 or more members to include only one or two noninstitutional members.”
119 Mildred K. Cho, Paul Billings, “Conflict of Interest and Institutional Review Boards,” Journal of Investigative Medicine 45.4 April 1997:155.
120 Fisher, 29.
121 Rebecca Dresser, letter, “Is Informed Consent Always Necessary for Randomized Controlled Trials,” New England Journal of Medicine 341.6, August 5, 1999. Dresser notes, “most [IRBs] are composed primarily of employees of the research institution, together with one or two members of the community. This group is not sufficiently representative to judge when a reasonable person would want to be consulted about participation in a clinical trial.”
122 McNeill, 207.
123 George J. Annas, “Questing for Grails: Duplicity, Betrayal and Self-Deception in Postmodern Research,” Journal of Contemporary Health Law and Policy 12.100:125.
124 McNeill, 195. “Even if we accept for a moment that lay members represent the community, they are typically the minority, participate less in the committee discussion, are seen (and see themselves) as relatively unimportant and have consequently less influence in the decisions reached by the committees.”
125 McNeill, 196.
126 Bell, 68 and 70.
127 OPRR, “OPRR Compliance Activities: Common Findings and Guidance,” http://grants.nih.gov/grants/oprr/findings.pdf: 11.
128 Brown, A Time for Reform, 17.
129 McNeill, 199.
130 45 CFR 46.304(b).
131 Office for Protection from Research Risks, Evaluation of Human Subject Protections in Schizophrenia Research Conducted by the University of California Los Angeles, May 11, 1994.
- Michael A. Carome, “To Smith Jenkins Veterans Administration Greater Los Angeles Healthcare System,” March 22, 1999 (Obtained through Freedom of Information Act Request).
132 Collen Cordes, “Breast Cancer Survivors Play an Unusual Role in Reviewing Grant Applications,” Chronicle of Higher Education, December 19, 1997, A29.
133 Rebecca Dresser, “Mentally Disabled Research Subjects: The Enduring Policy Issues,” Journal of the American Medical Association 276.1, July 3, 1996:71.
134 Fisher, 40.
135 Annette Dula, “Bioethics: The Need for a Dialogue with African Americans,” It Just Ain’t Fair: The Ethics of Health Care for African Americans, ed. Annette Dula and Sara Goering (Westport: Praeger, 1994) 11.
K-34
141 Brown, A Time for Reform, 19.
142 45 CFR 46.103(2).
143 Bell, 68.
144 The following letters from OPRR to research institutions were obtained through a Freedom of Information Act request: Michael Carome, OPRR, “To Arthur Rubenstein,” Mount Sinai School of Medicine, March 4, 1999. Michael Carome, OPRR, “To Joan F. Lorden,” University of Alabama at Birmingham, January 19, 2000. Carol J. Weill, OPRR, “To David C. Borski,” University of Illinois, Chicago, August 27, 1999. Michael Carome, OPRR, “To Ralph Snyderman,” Duke University Medical Center, June 4, 1999. Michael Carome, OPRR, “To Leo M. Henikoff,” Rush-Presbyterian-St. Luke’s Medical Center, October 23, 1998.
145 Brown, A Time for Reform, 6.
146 David Clark, “The Top Ten Compliance Problems and How to Avoid Them,” IRBs Through the Looking Glass, Public Responsibility in Medicine and Research Conference, Sheraton Boston Hotel and Towers, Boston, December 6, 1999.
147 National Commission, Report and Recommendations, 9.
148 Cho, 155. “...IRBs have a tremendous workload and are continually being proposed as a mechanism by which to implement more oversight and enforcement activities.”
See also Brown, A Time for Reform.
149 Brown, A Time for Reform, 6. “At the same time, managed care cost pressures have constrained the time that IRB members have to devote to reviewing protocols.”
150 Brown, A Time for Reform, 7.
151 Annas, 118.
152 Cho, 156.
153 Brown, A Time for Reform, 5. The report noted that the expansion of managed care with an emphasis on cost control may result in institutional “pressures to accommodate research sponsors who can provide research related revenues for the parent institution.”
154 OPRR, 1993 Protecting Human Research Subjects: Institutional Review Board Guidebook, 1-4–1-5. 155 OPRR, 1993 Protecting Human Research Subjects: Institutional Review Board Guidebook, 1-7. 156 Bell, 27.
157 Michael A. Carome, OPRR, “To Arthur Rubenstein,” Mount Sinai School of Medicine, March 4, 1999 (Obtained through Freedom of Information Act Request) 2.
158 Bell, 62.
159 Herman L. Blumgart, “The Medical Framework for Viewing the Problem of Human Experimentation,” Experimentation with Human Subjects, ed. Paul A. Freund (New York: George Braziller, 1969) 39.
160 Lawrence O. Gostin, “Informed Consent, Cultural Sensitivity, and Respect for Persons,” Journal of the American Medical Association 274.10, September 13, 1995:844.
K-35

INSTITUTIONAL REVIEW BOARD ASSESSMENT OF RISKS AND BENEFITS ASSOCIATED WITH RESEARCH
Commissioned Paper
Ernest D. Prentice and Bruce G. Gordon University of Nebraska Medical Center
L-1
Introduction
The assessment of risks and benefits is arguably the most important responsibility of an Institutional Review Board (IRB). In accordance with DHHS regulations 45 CFR 46.111(a),* IRBs are required to determine that risks to subjects are minimized and are reasonable in relation to anticipated benefits. Toward this end, the description, quantification, and analysis of risks and benefits are critical to the performance of both initial review and continuing review of research by IRBs. In this paper, we review the concept of risk in the research context, types of risk, identification and quantification of risks, the minimal risk standard, minimization of risk, assessment of benefit, risk-benefit analysis, and the ongoing assessment of research. All are an integral part of the IRB’s review of medical, behavioral and social science research. In addition, problems that IRBs encounter in the interpretation and application of pertinent sections of the federal regulations will be discussed and recommendations made that will, hopefully, assist IRBs in performing risk-benefit analysis of research.
Definition of Risk
Risk is the likelihood that harm may occur. Typically, harm is thought of as physical damage, such as a broken bone or sprained ankle,1 but harms extend beyond physical injury. Feinberg suggested that someone is harmed when his or her interests have been thwarted, defeated, invaded, or set back.2 This definition, then, would include a broad spectrum of harms that persons may experience. In the context of research, however, the concept of risk is usually focused and limited by setting the scope of harms to be considered. A risk associated with research is a potential harm, discomfort, or inconvenience that a reasonable person in the subject’s position would likely consider significant in deciding whether or not to participate in the study.
The above definition of research risk arose to a large extent from the medical malpractice case of Canterbury v. Spence.3 This case established the “reasonable person” or “material risk” standard for disclosure of risks associated with medical therapy. Using this standard, the court would decide whether a reasonable person in the patient’s position would have undergone the proposed treatment if he or she had been adequately informed. In the context of research, at the very least, the same standard would apply, i.e., the risks of harms occurring must be considered by the IRB if a “reasonable person” would want to know them before deciding whether or not to participate in the research. However, it has been argued that this standard, although adequate for patients, may not be sufficient for disclosure of risks to research participants. The National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, hereinafter, the National Commission, suggests in the Belmont Report that the material risk standard “seems insufficient since the research subject, being in essence a volunteer, may wish to know considerably more about risks gratuitously undertaken than do patients who deliver themselves into the hand of a clinician for needed care.” The National Commission further suggests that “a standard of the reasonable volunteer should be proposed: the extent and nature of information should be such that persons, knowing that the procedure is neither necessary for their care nor perhaps fully understood, can decide whether they wish to participate in the furthering of knowledge.”4 Thus, it is clear that IRBs should go beyond the material risk standard when assessing the significance of risks associated with research. The reasonable volunteer standard applies equally to research involving sick patients who are seeking a health benefit as well as to nonclinical research involving normal volunteers.
L-3
Types of Risk
In general, risks may be categorized as physical, psychological, social, or economic.5 Another type of risk, which is less commonly associated with research in general, is legal risk. Risks of any of these types may occur in the setting of various biomedical or behavioral research projects, but physical risks, and to a lesser extent psychological risks, are most common in biomedical studies, while social, economic, and legal risks are often limited to behavioral or social science research. IRBs should recognize that these categories of risk are somewhat fluid in that a given risk may fall into two or more of the categories or multiple types of risk may be present in a single study.
Physical Risks
Physical risks are usually thought of as the possibility of pain, suffering, or physical injury. Such harms may be easy to identify in certain biomedical studies, such as phase III clinical trials of antihypertensive drugs, or may be yet unknown, as in the case of phase I dose and toxicity finding studies. Nonetheless, the pharmacology of a drug, and its similarity to other drugs, often provides enough information to predict some potential harms with reasonable certainty. Similarly, physical harms associated with other research interventions are often clear, e.g., the risk of ecchymosis with venipuncture, pain associated with lumbar puncture, myocardial infarction related to a maximal exercise treadmill test, sore throat as a consequence of bronchoscopy.
Physical risks may arise from withholding or withdrawal of effective therapy. For example, subjects in a trial of a new oral hypoglycemic drug may suffer harm if the new drug is not as effective as their standard therapy. Similarly, evaluation of new drugs often requires discontinuation of standard therapy followed by a so-called “wash out period” during which time the subject receives no treatment. Although these periods are usually short, they are not without risk.6 Physical risk also includes the possibility that a subject may experience discomfort or mere inconvenience which may not rise to the level of an actual harm, such as pain or injury. These risks can easily be overlooked during the process of risk assessment. For example, the requirement to lie still in an MRI machine for an extended period of time during an imaging study may have associated discomfort or boredom for some subjects. A cardiac device study may require the subject to wear a Holter monitor for 48 hours, which would more than likely represent inconvenience. These less obvious risks should be considered by IRBs during their review.
Psychological Risks
Psychological risks may be readily apparent, although they are often less quantifiable. For example, withdrawal of antidepressant therapy in a wash-out period, or administration of placebo to subjects in a trial of a new therapy for depression may precipitate an episode of depression. Research involving genetic testing may have psychological risks associated with disclosure of a subject’s likelihood of developing a disease for which there is no treatment or cure such as Huntington’s chorea. Administration of a sensitive survey to adult subjects regarding domestic violence may provoke feelings of guilt, distress, and anger. There may also be a concomitant risk of precipitating further incidents of spousal abuse.
In some studies, the generation of psychological distress is expected and may be an end point of the study itself. A classical case that illustrates this point would be the “Obedience to Authority” experiments conducted by Milgram.7 In these studies, subjects were asked to deliver “electric shocks” of increasing intensity to another individual. The research relied upon deception of the subject, i.e., the shocks were, in fact, not real, and the other individual, an actor. The intent of the research was to determine the degree to which subjects would follow instructions “increasingly in conflict with conscience.” As a result of participating in this study, many subjects experienced severe and prolonged anxiety due to what has been termed “inflicted insight,” that is, insight into the fact that they were capable of cruel actions in their obedience to authority.8 The risks of such
L-4
a study would not be acceptable today under contemporary ethical standards but deception is still a fairly common component of behavioral research and, accordingly, there may be risks which must be considered by the IRB.
Some psychological risks may be more nebulous or not even related to the research procedure per se. For example, a prospective subject may be asked to donate allogeneic bone marrow to be used in an experimental manner to treat a patient with AIDS. He or she, however, may feel guilty for not wanting to participate in the research, especially when such a refusal is associated with a risk of harm to another party. Although the individual being asked to donate bone marrow is not yet a research subject, the psychological harm, i.e., feelings of guilt is certainly associated with the process of consent for the study. IRBs, therefore, should be cognizant of this kind of risk.
Social Risks
Participants in research may experience social risks, that is, risk of harm to a person in the context of his or her social interactions with others. Examples include the risk of stigmatization as a result of testing positive for HIV, or the risk that genetic studies will disclose nonpaternity. Social risks are particularly associated with studies of private aspects of human behavior. For example, the description of homosexual practices revealed through the controversial “Tearoom Trade” studies in the 1960s could have placed unknowing subjects at considerable social risk should their unacknowledged homosexuality be disclosed.9 Indeed, the possibility of a breach of confidentiality is often the most significant risk of social science research. The degree of risk, however, is related to the sensitivity of the research data from the subject’s perspective and the likelihood that unauthorized individuals could gain access to the data. In other words, discovery or disclosure of meaningless albeit personal information about a subject is certainly a “wrong” but does not rise to the level of a “harm.” While the IRB should be more concerned about the possibility that a subject could suffer a harm, nonetheless, the Board should not dismiss a potential wrong.
Economic Risks
Research may pose economic risks to subjects. Participants in “high tech” clinical research may incur financial obligations for treatment which are significantly higher than those associated with standard therapy and, ultimately, no health benefit is realized. In some studies, subjects may need to take time off from work, or pay costs of transportation to the study center, which could impose a significant economic hardship. Subjects may also be exposed to the possibility of loss of insurability, associated with diagnosis of a chronic or life-threatening disease. Economic risk may even extend to a research participant’s livelihood. For example, sociologic studies of employer-employee relationships may carry the risk of loss of employment if confidentiality is breached.
Legal Risks
Participation in research may present legal risks to the subject. For example, one IRB was asked to approve a study of paroled felons which assessed the effect of time since release from prison on the incidence of repeat offenses. Subjects were asked to complete a survey of crimes committed three months and one year after release. The surveys contained linked codes so the responses of each subject could be compared at the two time points. Had the investigator been compelled by judicial order to provide data with subject identifiers to the court, the study participants would have incurred significant legal risk. Similar risks can easily be incurred in studies of possession and use of illicit drugs, sexual or physical abuse, or workplace theft. As Wolf points out, assuring confidentiality of research records may require the investigator and the IRB to be aware of various legal protections, such as Certificates of Confidentiality, for sensitive research data.10
L-5
Risk To Others
Although the previous discussion focused upon risks of harm to research subjects, risks may also accrue to other persons not directly involved in the research. For example, in a study of a new live virus vaccine, there may be risks to family members or contacts of the subject who may be immunosuppressed and who could potentially contract the attenuated disease. Or, in some states, there may be legal risk to parents in a study of illegal activity by their minor children. Studies involving genetic testing are particularly problematic with regard to the involvement of family members who may be exposed to risks without their knowledge or consent.11 Even society, itself, may incur risk as a consequence of research involving procedures such as xenotransplantation or studies of viruses where there is a danger of unleashing pathogenic organisms.
Identification of Risks
As a starting point, most IRBs require the investigator to present a comprehensive review of the potential harms that may arise as a consequence of research participation. He or she should assist the IRB by identifying those procedures performed solely for research purposes versus those that would be carried out regardless of the research, i.e., the therapies the subject would otherwise receive if not participating in the research. It is incumbent, however, on the IRB to review this information for accuracy and completeness, and ultimately determine which risks are germane to the IRB’s charge to protect the rights and welfare of research subjects. IRBs traditionally look to their own members for expertise first, and many institutions choose IRB members based on the organization’s research profile. For example, an IRB at a university with an active cancer center is well advised to include medical, pediatric, and perhaps radiation oncologists. Similarly, an IRB that reviews behavioral and social science research should include members that have the knowledge necessary to adequately assess the risks inherent in this type of research.
In medical research, particularly that which is conducted with the intent of providing a clinical benefit to the subject (so-called therapeutic research), research interventions may take place concurrently with clinical procedures, or may in fact be identical to those procedures. Therefore, potential harms may not entirely be associated with the research. In other words, research data may also be obtained during the performance of clinically warranted procedures. An argument could be made, consistent with 45 CFR 46.111(a)(2), that such risks are not part of the research and, accordingly, need not be included in the IRB’s analysis of the risk-benefit relationship of the research or disclosed to prospective subjects, at least not in the context of research participation.
The differentiation of research versus therapy in terms of the IRB’s consideration of risk can, however, be problematic particularly in the field of oncology where therapy considered to be standard is often evaluated in a research context. For example, allogeneic unrelated donor bone marrow transplantation is the standard of care at most centers for treatment of chronic myeloid leukemia (CML). A study, therefore, might consist of an evaluation of this type of bone marrow transplantation in the treatment of CML with an assessment of long-term survival as the research objective. Should the risk analysis of this study include the known risks of graft-versus-host disease, therapy related toxicity and relapse? Should these risks be disclosed in the informed consent document? The answer is not clear, but most IRBs would likely consider these risks in their review and opt for complete disclosure in the consent form. On the other hand, such disclosure increases the length of the consent document and may negatively impact its readability. In addition, in some studies, the difference between research and therapy can become obscured, which could be misleading, and can affect the validity of the consent.
L-6
Quantification of Risk
Once identified, insofar as possible, risks must be quantified. Risk quantification considers both the likelihood of occurrence and the potential severity of the harm. Severity, in turn, depends upon the amount of damage, the duration, the permanency of the consequences as well as subjective considerations, such as the extent to which it may alter or affect the subject’s lifestyle.12 Quantification of risk is important not only to help the IRB accurately assess risk but also to ensure the adequacy of disclosure in the informed consent document. It is often the only way that a subject can assess the significance of potential risks. Indeed risk disclosure without any concomitant quantification may not fully inform the subject and, accordingly, lead to either an underestimation or overestimation of the importance of a given risk. FDA reinforces the need for risk quantification in the preamble to the agency’s informed consent regulations which state “where such descriptions or disclosures may contain quantified comparative estimates of risk or benefits they should do so.”13 Quantification of risk, however, is often difficult or even impossible. In many cases, such as in phase I dose and toxicity finding studies, the likelihood and severity of harm, or even the possibility of harm, may be unknown. In other cases, the potential harms may be known, but quantification is difficult, such as the risk of falling off a treadmill during an exercise physiology study. Alternatively, the risks may be known and quantified, but not for the population undergoing the research intervention. For example, a colonoscopy carries a definable risk of bowel perforation, based on aggregate data from “normal” patients undergoing this procedure. The risk may, however, be higher and less definable in a population of patients who have recently undergone high dose chemotherapy. All of these factors make quantification difficult. Nevertheless, the IRB should quantify research risks whenever possible and apply the “reasonable volunteer” standard in ensuring that risks are appropriately characterized in the consent document.
Classification of Research According to the Minimal Risk Standard
After the IRB completes the quantification of risk, the next task is to classify the research as minimal or greater than minimal. This classification, though artificial, and of limited value to the prospective subject, is important because the minimal risk standard serves as a threshold level of risk for the purpose of IRB review. As the risks of research increase above this threshold, the criteria for IRB review and approval become more stringent. As will be discussed below, the minimal risk threshold serves as one determinant used by the IRB in deciding the type of review required (expedited or full Board), the acceptability of a waiver of informed consent, and whether additional protections are needed for certain subject populations who are considered vulnerable.
Minimal risk means that “the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests” [45 CFR 46.102(I)]. Some IRBs have interpreted “daily life” as referring to the daily life of a normal healthy person, a so-called “absolute standard.” Clearly, there is nothing absolute about “daily life,” and the risks inherent in the daily life of a person from rural Iowa are not the same, quantitatively or qualitatively, as those inherent in a person from inner city New York. This definition, nonetheless, sets a standard of the daily life of a “healthy person.” An equally common, as well as defensible interpretation, however, sets minimal risk as reflecting the daily life and experiences of the “research subject.” This is a much lower risk threshold and is referred to as a “relative standard of minimal risk.” For example, a bone marrow aspiration would not be considered minimal risk in relation to the daily life of a normal person, but the risks associated with this procedure might well be viewed as not greater in and of themselves than those risks routinely encountered in the daily life of a patient with acute leukemia. Thus, the same intervention may be classified as minimal risk or greater than minimal risk depending on the health status of the research subject and his or her experiences. Indeed, some IRBs use “previous experiences” of
L-7
normal subjects to justify a sliding scale of minimal risk. For example, it may be reasonable to classify a bone marrow aspiration performed on a “normal child” who is cured of his/her cancer as a minimal risk procedure, based on the child’s previous experience as a cancer patient.
The difficulty in interpreting “minimal risk” is compounded by conflicting messages from federal agencies. The preamble to the DHHS regulations 45 CFR 46 states, “HHS in the proposed regulations used the terminology, ‘healthy individuals.’ In light of public comment on this, however, HHS reworded the final regulation to reflect its intention that the risks of harm, ordinarily encountered in daily life means, ‘those risks encountered in the daily lives of the subjects of research.’”14 The preamble to the FDA regulations (21 CFR 56) is similar.13 However, the Office for the Protection of Research Risks (OPRR)** is on record as choosing to apply the term “minimal risk” using an absolute standard which the Office feels is “…defined in the policy itself, rather than to rely upon the introductory language of the regulations, the reading of which by some would adversely affect the core of federal human subjects protections dangerously and unnecessarily.”15 The end result of these conflicting statements is confusion.
A further difficulty with the interpretation of the definition of minimal risk and its application during IRB review arises with the nature of the comparison of risks. To qualify as minimal risk, must the research procedures only be those encountered in routine tests or can procedures with equivalent risks in terms of the probability and magnitude of harm be considered minimal risk? For example, routine physical examination may involve venipuncture. The risks associated with venipuncture include pain, bruising, and rarely, infection and syncope. A template bleeding time is a test whereby a small, shallow (1 mm deep) cut is made on the volar surface of the forearm, for purposes of assessing the function of the early stages of hemostasis. A template bleeding time is not a feature of a routine physical examination, but the probability and magnitude of harm and discomfort are equivalent to those associated with venipuncture. It would, therefore, appear reasonable to classify this procedure as “minimal risk,” using a risk equivalency rationale. In addition, the risks of a template bleeding time test clearly do not exceed those encountered in daily life.
As mentioned previously, the importance of the threshold of “minimal risk” is that it sets standards for IRB review and approval of the study. Certain types of research, which involve no more than minimal risk, may be reviewed by an expedited review procedure (45 CFR 46.110). IRBs are also empowered to “approve a consent procedure which does not include, or which alters, some or all of the elements of informed consent…or waive the requirement to obtain informed consent providing…the research involves no more than minimal risk and meets the other conditions specified under 45 CFR 46.116(d).” An IRB may also waive the requirement for the investigator to obtain a signed consent form “if it finds that the research presents no more than minimal risk of harm to subjects and involves no procedures for which written consent is normally required outside the research context” [45 CFR 46.117(c)(2)]. FDA allows a waiver of the documentation of informed consent under the same circumstances described above [21 CFR 56.109(c)(1)]. However, FDA has no provision for a waiver of informed consent which is tied to the minimal risk threshold as does DHHS. Once again, the issue of using an absolute versus a relative standard of minimal risk becomes an important factor in the IRB’s review of research.
The “minimal risk” threshold is particularly important for IRB review of research involving vulnerable subjects. For example, 45 CFR 46 Subpart C provides additional protections for prisoners which limits most studies to those that present no more than minimal risk to the subjects [45 CFR 46.306(2)]. Interestingly, Subpart C specifically defines “minimal risk” as the “probability and magnitude of physical or psychological harm that is normally encountered in the daily lives, or in the routine medical, dental, or psychological examination of healthy persons” [45 CFR 46.303(d)]. Thus, the standard of minimal risk applicable to research with prisoners is “absolute” as opposed to “relative,” which affords this subject population a greater degree of protection.
L-8
45 CFR 46 Subpart D, which provides additional protections for children involved in research, utilizes the threshold of “minimal risk” to determine the necessity for additional conditions to be met to assure protection of subjects. Research posing no greater than minimal risk to subjects is allowable without additional protections (45 CFR 46.404). Research posing greater than minimal risk may be permitted if there is the “prospect of direct benefit for the individual subject,” i.e., if the risk is counterbalanced by a direct benefit (45 CFR 46.405). Research posing greater than minimal risk and not presenting the prospect of direct benefit may involve children only if “the risk represents a minor increase over minimal risk;” the interventions are “reasonably commensurate with those inherent in their actual or expected medical, dental, psychological, social, or educational situations;” and the research is “likely to yield generalizable knowledge about the subjects’ disorder or condition which is of vital importance…” (45 CFR 46.406). In other words, when the research poses more than minimal risk and no direct benefit, it must offer the possibility of indirect benefit, i.e., it must yield generalizable knowledge about the subject’s disorder or condition which is of significant value. The research procedures must also be “reasonably commensurate” with the subject’s daily life and experiences and present only a “minor increase over minimal risk.” The regulations, however, do not define these terms, further confusing the application of the minimal risk standard in this context.
Problems in interpretation and application of the minimal risk standard during IRB review of research involving children also arise because of conflicting guidelines and commentary from both the National Commission and DHHS. The Report of the National Commission on Research Involving Children defines “minimal risk” as “the probability and magnitude of physical or psychological harm that is normally encountered in the daily lives, or in the routine medical or psychological examination, of healthy children,”16 thereby espousing an absolute rather than a relative standard. The DHHS, however, in codifying these recommendations as 45 CFR 46 Subpart D, chose not to adopt this definition, instead defaulting to the loosely interpretable definition of minimal risk in Subpart A. Furthermore, as mentioned previously, the wording of 46.406 adds to the confusion. To be approvable under this section, among other things, the IRB must find that the intervention or procedure presents experiences to subjects that are reasonably commensurate with those inherent in their daily lives. This standard of commensurability is justified in the National Commission’s Report by the statement that it should “assist children who can assent to make a knowledgeable decision about their participation in research, based on some familiarity with the intervention or procedure and it’s effects.” Commensurability, according to the National Commission, also “assures that participation in research will be closer to the ordinary experiences of the subjects.”17 This would, however, seem to suggest a relative standard of minimal risk, which is not consistent with OPRR’s stated position.
In consideration of the importance of using a well defined and consistently interpreted risk threshold in determining the level of protections for human subjects, further clarification and guidance concerning the minimal risk standard is needed. IRBs should not be making decisions about the protection of human subjects in an inconsistent manner.
Minimization of Risk
Once the risks are identified and quantified, and the risk magnitude (minimal versus greater than minimal) assigned, IRBs are required to ensure that risks are minimized to the greatest extent possible within justifiable limitations imposed by the nature of the research. To approve a research protocol, the IRB must determine that the probability of occurrence and severity of the risks are minimized by using “procedures which are consistent with sound research design and which do not unnecessarily expose subjects to risk” [45 CFR 46.111(a)(1)]. Investigators and IRBs may reduce risks to the subject through various means. Risk may be reduced by assuring that the investigator and study personnel are qualified to perform the procedures involved in the research. This
L-9
may require the inclusion of study personnel or consultants with relevant expertise. For example, a study involving colonoscopy in children and adults with cytomegalovirus (CMV) enteritis after liver transplantation should include both pediatric and adult gastroenterologists. Risk may also be reduced by assuring that procedures with less associated risks are substituted whenever possible. For example, in a study assessing the relationship between a particular phenotype and expression of a gene, a protocol using cells obtained by buccal swab is preferable to one using cells obtained by skin biopsy. Certainly, attention should be paid to the possibility of using a procedure already scheduled for routine clinical purposes in order to obtain data for research, thus reducing the number of interventions and the risk to which the subject is exposed.
Risk may be reduced by making sure that subjects are properly monitored, that subject withdrawal criteria are appropriate, and a timely treatment plan is in place. For example, subjects participating in a study of a new oral hypoglycemic drug may require a wash out period without standard treatment. The study should, therefore, be designed to allow frequent monitoring for episodes of hyperglycemia, there should be specific criteria in the protocol for taking a subject off-study if persistent or severe hyperglycemia occurs, and a plan should be available to treat these patients in an expeditious manner. Risks are also minimized by assuring that adverse events are promptly reported to the IRB and the sponsor of the research. Observation of an unanticipated side effect, or increased frequency of a known adverse effect, may require modification of a protocol, more frequent monitoring, or other action to reduce the risk.
In the course of clinical research, certain situations arise where the obligation to reduce risk may be particularly difficult to implement. Use of placebo in clinical trials of new pharmacologic agents can be quite problematic. For example, a placebo-controlled trial of a new antihypertensive drug may be the most scientifically valid study, but the administration of a placebo to a subject with known hypertension carries a risk associated with discontinuation of effective treatment for a serious disease. The matter is complicated further by investigators who state that use of a placebo-controlled trial design is an FDA requirement. The FDA, in fact, has no such general requirement, specifying only that a drug sponsor must show, “through adequate and well-controlled clinical studies, that it is effective,” and that “the study design chosen must be adequate to the task.”18 In some cases, use of an active control trial design may be scientifically valid, and reduce risks to the subjects.
While IRBs should scrutinize the study design to minimize risk to subjects, it should be recognized that pharmaceutical companies who sponsor multicenter trials are resistant to drastically altering study design. In addition, investigators may certainly apply pressure on the IRB to approve the protocol if other IRBs have already approved it, and may insist that the sponsor will just contract with the next “more reasonable” site if the study is not expeditiously approved. Thus, as a practical matter, IRBs should carefully consider the seriousness of the risks to the subjects, and whether these risks can be minimized in a manner other than by a change in the research design. If not, then the IRB may find the research unapprovable.
The obligation to minimize risks to subjects also includes the duty to exclude prospective subjects who are at undue risk of harm. Exclusion criteria are part and parcel of the research plan. It is the ethical and legal responsibility of the investigator to adhere to these criteria,19 and the IRB is obligated to ensure that the exclusion criteria are appropriate. In order to exemplify this point, Levine states “in planning research designed to test the effects of strenuous exercise in normal humans, one would ordinarily plan to perform various screening tests to identify individuals with coronary artery disease in order to exclude them.”20 The IRB must, therefore, be assured that the screening tests in place are sufficient to identify potential subjects who should be excluded from participating in the research.
L-10
Assessment of Benefits
Research is commonly divided into “therapeutic” and “nontherapeutic.” Although the distinction between the two is not as clear as it would seem, nonetheless the division has some value, particularly in the IRB’s assessment of study related benefits. Typically, therapeutic research or “research on therapy,” as it has been more correctly described, is not performed solely to produce generalizable knowledge, but also has the intent of providing a direct medical benefit to the subject. Benefits in this setting are usually direct health benefits, such as treatment of the subject’s disease or condition, however, other potential benefits may also exist. Participants in therapeutic research may benefit by receiving reduced cost or free care. For example, many patients with HIV infection participate in drug studies sponsored by the manufacturers and the AIDS Clinical Trial Group (ACTG) since the costs of these expensive drugs are borne by the sponsor. Subjects may even have access to care that they otherwise would not have had available, e.g., investigational drugs or devices. IRBs must be cognizant of the fact that these benefits may be so significant that they have the potential of unduly influencing a patient to participate in high risk research that he/she would not otherwise consider.
Nontherapeutic research, in contrast, has no intent of producing a diagnostic, preventive, or therapeutic benefit to the subject. Although subjects of nontherapeutic research are usually healthy volunteers, this is not always the case. For example, a study of the salivary cortisol levels in patients hospitalized after severe trauma would be considered “nontherapeutic” research without any direct health benefit to the subject. In other cases, participation in nontherapeutic research may yield information that is of benefit to the subject. For example, a study that measures maximal oxygen consumption during sustained exercise may be of value to a long-distance runner in establishing training goals or a study that includes comprehensive educational and psychological testing may be of value to a subject planning his or her career path.
Financial compensation for participation in research may also be considered a potential benefit to subjects. Payment of research subjects is, however, the source of much controversy, raising concerns of “undue inducement,”21 and of the burden of research being borne by economically disadvantaged populations.22 Indeed, it has been suggested that any payment of subjects violates the ethical norms of the investigator-subject relationship by turning it into a commercial relationship.23 For these reasons, many IRBs do not consider monetary compensation as a benefit to be weighed in the risk-benefit relationship, although it should be recognized that most individuals view compensation as a benefit. In addition, it can be argued that the amount of compensation should not be determined by the level of risk to which a subject is exposed.
Finally, subjects may benefit from a feeling of satisfaction at having assisted in scientific research that may be of value to others. This may be motivation for many subjects and ought not to be dismissed by the IRB. Indeed, the principles of “justice” and “respect for persons” requires that people not be deprived of an opportunity to participate in research unless there is a compelling reason to do so. It should also be recognized that research participation is now commonly viewed as beneficial instead of harmful, which was the prevailing attitude in 1974 when the National Commission was established. Indeed, patient advocacy groups are demanding more research and greater access to clinical trials and have established a sense of entitlement to their perceived right to participate in research.
In addition to benefits that accrue to the subject of the research, IRBs must also consider potential benefits to society at large or to special groups of subjects in society. In the setting of nontherapeutic research, benefits to society in terms of knowledge to be gained may be the only clearly identifiable benefit. Societal gain without direct benefit to the subject, however, may not be sufficient justification, especially when vulnerable populations are involved. For example, because of their vulnerable status, prisoners cannot be participate in a study which involves possible assignment to a control group that offers no benefit from the research unless approval from the DHHS Secretary has been obtained [45 CFR 46.306(a)(2)]. Similarly, as mentioned previously, children may participate in research involving greater than minimal risk with no direct benefit only when the research is
L-11
likely to yield generalizable knowledge about the children’s disorder or condition which is of vital importance to the understanding or amelioration of the disorder or condition (45 CFR 46.406). That is, the research “must hold out the promise of a significant benefit in the future to children suffering from or at risk for the disorder or condition (including, possibly the subjects themselves).”17
Risk-Benefit Analysis
The IRB is obligated to ensure that the risks to subjects are reasonable in relation to anticipated benefits. This requirement, termed the risk-benefit relationship, stems from the moral principle of “beneficence,” which is emphasized by the National Commission in its many reports and recommendations. Application of the principle of “beneficence” to research demands that the risks be minimized and the benefits maximized to the greatest extent possible. In other words, the risk-benefit relationship of the research should be as favorable as possible and the IRB’s review should be designed to achieve this goal. A favorable risk-benefit relationship in research is required by a number of national and international codes governing human subject research, including the World Medical Assembly’s Declaration of Helsinki, the guidelines of the Council for International Organizations for Medical Sciences (CIOMS), the Medical Research Councils of Canada and the United Kingdom, as well as by the DHHS and FDA Regulations for the Protection of Human Subjects.
In many studies, a comparison of risks and potential benefits is fairly straightforward. For example, the risks of taking a new antibiotic for pneumonia in a phase III clinical trial may be known, e.g., nausea, rash, small risk of mild renal toxicity, and the benefits relatively clear, i.e., the potential for more rapid improvement in symptoms and reduced likelihood of progression to respiratory failure. In some circumstances, however, the balancing of risks and benefits is a difficult task. At least part of this problem arises from what has been termed “incommensurability,” that is, the risks and benefits lack a basis for comparison. As pointed out by Martin and colleagues, this incommensurability may arise because risks and benefits for subjects affect different domains of health status, or because risks and benefits may affect different people.24 They present the case of living donor lung transplant, where risks accrue mainly to the donor, and benefits mainly to the recipient. Further, among donors, the risk is physical, but the potential benefit is psychological, in that they may derive satisfaction by helping another person.
The analysis becomes even more difficult when either benefits or risks accrue to society rather than to the subjects of the research. Phase III vaccine trials are particularly problematic in this regard.25 In most cases, the risk of contracting the disease for any one individual is low, so most of the benefit is to society, i.e., for the small number of other persons who might contract the disease. The risks, however, accrue almost entirely to the subject of the research. This problem becomes particularly difficult when the subjects of the research are children, and the stringent requirements of Subpart D must be satisfied.
Although less common, situations may arise, as alluded to earlier, where benefits accrue to the subject but society bears some of the risk. Recent studies of porcine hepatocyte transplantation for patients with end-stage liver disease present the possibility of maintenance of hepatic function, at least until a donor human liver is available. However, concern has been appropriately raised regarding the possibility of transfer of porcine viruses such as the porcine endogenous retrovirus (PERV). Once introduced into humans, zoonotic viruses, such as the Ebola virus, that are not particularly pathogenic in their host species have resulted in outbreaks of disease. Compelling arguments suggest that the epidemics of HIV types 1 and 2 resulted from the adaptation of simian retroviruses introduced across species lines into humans.26 Therefore, the risks of porcine hepatocyte transplantation may include the theoretical risk of developing new pathogenic viruses, with consequential harm to society. IRBs reviewing such a trial would need to attempt to balance the benefits to the individual subject against the risks to the subject and to society in general.
L-12
IRBs must also examine the risk-benefit relationship of research in the context of best available therapy. No patient should be allowed to participate in research if a standard treatment exists which offers a better prospect of overall benefit. Indeed, a comparison of the risks and benefits of the research versus the available therapeutic alternatives is a very important as well as difficult component of the risk-benefit analysis. An acceptable risk-benefit relationship in research demands there be at least an equal prospect of benefit in consideration of the relative risks associated with the research and the alternative standard therapy. Standard care, however, is often a matter of debate and the IRB may not have sufficient expertise to perform a valid risk-benefit comparison. Thus, IRBs should require investigators to provide a comparative risk-benefit analysis and seek the advice of consultants as necessary.
Ongoing Assessment of Research
The IRB’s responsibility regarding assessment and minimization of risk does not end with the initial approval of the research protocol. IRBs perform continuous, ongoing assessment of adverse events (AEs). Both DHHS (45 CFR 46.103) and FDA (21 CFR 56.103) regulations require institutions to establish written procedures for the “prompt reporting to the IRB of any unanticipated problems involving risks to subjects...” Sponsors of FDA regulated research are required by 21 CFR 312.32(c) to “notify FDA and all participating investigators in a written IND safety report of any AE associated with use of the drug that is both serious and unexpected.” Although this regulation does not require the sponsor to notify IRBs at participating study sites, it is routine for sponsors either to instruct investigators to provide a copy of the safety report to the IRB or to send a copy of the report directly to the IRB.
The occurrence of AEs, either as new side effects, or an increase in the severity or frequency of known toxicities, requires the IRB to reassess the risk-benefit relationship of the research. Such reassessment may necessitate modification of the consent form for prospective subjects, reconsent of current subjects, or modification of the research plan to reduce risk. Under certain circumstances, serious AEs may require termination of the study if they impact unfavorably on the risk-benefit relationship. IRBs, however, seldom have sufficient data to make this determination independently. Individual AE reports are usually reviewed without adequate knowledge of multicenter data and, therefore, incidence and often causal relationship cannot be ascertained.
IRBs are also required by both DHHS and FDA regulations to conduct periodic continuing review of approved protocols “at intervals appropriate to the degree of risk, but not less than once per year” [45 CFR 46.109(e)]. The criteria for IRB reapproval are the same as for initial review, including the requirement that the risks to subjects are minimized and reasonable in relation to anticipated benefits. Therefore, continuing review of ongoing research requires the IRB to identify any changes in the risk profile of the research, as well as to reassess the potential benefits of the research. Accordingly, the IRB needs to examine interim results of the study. The justification for performing the research is the intent to produce generalizable knowledge which will be of value to the subject and/or society. When the research has completed this goal, i.e., when statistically significant data have been obtained, there is no a priori reason to continue the research.
For example, in the late 1980s, several studies were ongoing which evaluated the efficacy of warfarin in decreasing the risk of embolic events in patients with atrial fibrillation. In the early 1990s, two of these studies were published, both of which showed a statistically significant decrease in embolic events in patients treated with warfarin and a low rate of major bleeding events. Investigators associated with the Canadian Atrial Fibrillation Anticoagulation (CAFA) study, which was not yet completed, decided that the evidence of benefit with warfarin, from the two published studies, was sufficiently compelling to stop recruitment into CAFA. The risks to the subjects had not changed, but the potential benefits of continuing the study were reduced or eliminated, making the risk-benefit relationship no longer favorable.27
L-13
Finally, the presence of internal study monitoring, such as Data and Safety Monitoring Boards (DSMBs) can greatly assist IRBs in their ongoing assessment of the risk-benefit relationship. The National Institutes of Health (NIH) requires monitoring of clinical trials to assure safety of human subjects of the research. The type of monitoring is dependent upon the size, complexity, and the risks associated with the study. NIH funded phase III trials are required to have a DSMB. These DSMBs are responsible, among other things, for “periodic assessment of participant risk and benefit.” This assessment includes consideration of “scientific and therapeutic developments which may have an impact on the safety of the participants or the ethics of the study.” DSMBs are also expected to make recommendations to the investigators and to the IRBs concerning continuation or termination of the trial.28 In addition, DSMBs are required to provide summary reports of adverse events to each IRB involved in the study.29 NIH funded phase I and II studies must also have, at a minimum, a mechanism in place for reporting adverse events to IRBs.30 Although DSMBs are not routinely required by FDA for studies involving investigational drugs and devices, many phase III studies include such formal monitoring. Summary reports from these Boards can be of assistance to the IRB in performing a continuing assessment of the risk-benefit relationship of research.
Conclusion
This paper has addressed the cardinal responsibility of the IRB which is the assessment of the risk-benefit relationship of research in accordance with the principle of “beneficence.” While any available data regarding potential risks and possible benefits of a proposed study are certainly of value to the IRB, it must be recognized that at the time of initial review, the data are limited and, therefore, the judgement of the Board, at times, may necessarily be more subjective than objective. There are no computer programs or formulas that the IRB can use to establish the acceptability of research. Instead, such judgements are initially made by the investigator and then by the men and women who serve on the IRB. IRB members, individually and collectively, apply their knowledge, experience, wisdom, and moral values in rendering decisions concerning the approvability of research. Society and research subjects can ask no more and should expect no less.
Recommendations
| 1. |
IRBs should perform a thorough evaluation of the research risks to which a subject may be exposed, including those risks not likely to rise to a level of “harm.” IRBs should also consider risks to others that may occur as a consequence of the research. |
| 2. |
IRBs should be given guidance concerning the extent of disclosure of risks to research subjects. These guidelines should include use of the “reasonable volunteer” standard for both “therapeutic” and “nontherapeutic” research. In the setting of therapeutic research, these guidelines should also address criteria which can be used by IRBs in determining the necessity for disclosure of the risks associated with clinically indicated procedures which would not be performed solely for research purposes. Such criteria should be based upon the degree and type of risks associated with the therapy, and the relationship of the research question to the routine therapeutic intervention. In many cases, it may be appropriate to have the risks associated with clinically indicated procedures detailed in the informed consent document in order to facilitate the subject’s full understanding of the study. |
| 3. |
Investigators should be required to quantify the risks related to the research, insofar as possible, when negotiating consent with prospective subjects. This quantification should be reflected accurately in the consent form, utilizing language that is understandable and relevant to the subject. |
L-14
| 4. |
IRBs should be provided with clear and consistent guidance regarding the definition of “minimal risk,” as a threshold level of risk. A relative standard of minimal risk seems appropriate for research involving competent adults. However, an absolute standard of minimal risk, based on the daily life of a “healthy” person, may be more appropriate for research involving vulnerable subjects such as children and the cognitively impaired. Although such guidance is essential to consistent application of the minimal risk standard, IRBs will still need to exercise judgement based on the characteristics of the targeted subject population. That is, even use of the absolute standard demands an element of relativity with regard to what constitutes a “healthy” person. |
| 5. |
The requirement for IRBs to evaluate research protocol design and inclusion/exclusion criteria should be emphasized in order to assure that risks to subjects are minimized. FDA, NIH, cooperative groups and commercial sponsors should be receptive to reasonable recommendations from IRBs for modification of clinical trials in order to satisfy the requirements for minimization of risks. A mechanism should exist for IRBs reviewing the same study to share significant findings which negatively impact the risk-benefit relationship of the research. |
| 6. |
The Regulations for the Protection of Human Subjects should mandate the use of DSMBs where appropriate. DSMBs should be required to share data with IRBs on a scheduled basis in order to facilitate an ongoing assessment of risk and benefit. |
Acknowledgment
The authors gratefully acknowledge the assistance of Gwenn S.F. Oki, M.P.H., Director of Research Subjects Protection at the City of Hope National Medical Center and the Beckman Research Institute, who reviewed this paper and provided invaluable critique and comment.
Notes
*For the purpose of clarity, when DHHS and FDA Regulations for the Protection of Human Subjects are considered equivalent, only the former will be referenced. It should also be noted that FDA regulations do not provide any specific additional protections for vulnerable subjects, except in the context of emergency research.
**As of June 13, 2000, OPRR has been reorganized as the Office of Human Research Protection (OHRP) under DHHS [Fed Reg 65(114):37136, June 13, 2000], but for purposes of this paper the former acronym (OPRR) will be used.
References
1. Meslin EM: Protecting human subjects from harm through improved risk judgements. IRB: Rev Human Subjects Res 12(1):7–10, 1990.
| 2. |
Feinberg J: Harm to Others. Oxford University Press, New York, 1984, pp. 31–64. |
| 3. |
Canterbury v. Spence 464 F.2d 772, 785 (D.C. Cir. 1972). |
| 4. |
The National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research: The Belmont Report: |
Ethical Principles and Guidelines for the Protection of Human Subjects of Research. DHEW Publication (OS) 78-0012, Washington D.C., 1978, pp. 5–6.
| 5. |
Levine RJ: Ethics and Regulation of Clinical Research (2nd ed). Yale University Press, New Haven, 1986, p. 42. |
| 6. |
Houston MC: Evaluating risk when antihypertensive medications are abruptly discontinued in research subjects. IRB: Rev Human |
Subjects Res 6(1):9–11, 1984.
L-15
| 7. |
Milgram S: Obedience to Authority: An Experimental View. Harper and Row, New York, 1974. |
| 8. |
Baumrind D: Nature and Definition of Informed Consent in Research Involving Deception. In: The National Commission for the |
Protection of Human Subjects of Biomedical and Behavioral Research: The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research. DHEW Publication (OS) 78-0014, Washington D.C., 1978 Appendix II, pp. 23.1–23.71.
| 9. |
Warwick DP: Tearoom trade: Means and ends in social research. Hastings Center Rep 1(1):24–38, 1973. |
| 10. |
Wolf LE, Lo B: Practicing safer research. Using the law to protect the confidentiality of sensitive research data. IRB: Rev Human |
Subjects Res 12(5):4–7, 1999.
11. Clayton EW, Steinberg KK, Khoury MJ, Thomson E, Andrews L, Kahn MJ, Kopelman LM, Weiss JO: Informed consent for genetic research on stored tissue samples. JAMA 274(22):1786–1792, 1995.
| 12. |
Levine RJ: Ethics and Regulation of Clinical Research, p. 57. |
| 13. |
Fed Reg, 46(17):8946, 1981. |
| 14. |
Fed Reg, 46(16):8373, 1981. |
| 15. |
Ellis GB: Federal policy for the protection of human subjects. In: Report on the Public Forum on Informed Consent in |
Clinical Research Conducted in Emergency Circumstances. U.S. Food and Drug Administration/National Institutes of Health, Washington D.C., 1995.
16. The National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research: Research Involving Children. Report and Recommendations. Fed Reg, 43(9):2085, 1978.
| 17. |
Ibid, p. 2086. |
| 18. |
Food and Drug Administration: Information Sheets: Guidelines for Institutional Review Boards and Clinical Investigators. |
US Government Printing Office, Washington D.C., 1998, p. 58.
| 19. |
Weijer C, Fuks A: The duty to exclude: excluding people at undue risk from research. Clin Invest Med 17(2):115–122, 1994. |
| 20. |
Levine RJ: Ethics and Regulation of Clinical Research, p. 58. |
| 21. |
McGee G: Subject to payment? JAMA 278:199, 1997 |
| 22. |
Macklin R: “Due” and “undue” inducements: on paying money to research subjects. IRB: Rev Human Subjects Res 3:1–6, 1981. |
| 23. |
Murray TH: Gifts of the body and the needs of strangers. Hastings Center Rep 17(2):30–8, 1987. |
| 24. |
Martin DK, Meslin EM, Kohut N, Singer PA: The incommensurability of research risks and benefits: practical help for Research |
Ethics Committees. IRB: Rev Human Subjects Res 17(2):8–10, 1995.
25. Bjune G, Gedde-Dahl T: Some problems related to the risk benefit assessment in clinical testing of new vaccines. IRB: Rev Human Subjects Res 15(1):1–5, 1993.
26. Chapman LE, Folks TM, Salomon DR, Patterson AP, Eggerman TE, Noguchi PD: Xenotransplantation and xenogeneic infections. N Engl J Med 333(22):1498–1501, 1995.
27. Laupacis A, Connolly SJ, Gent M, Roberts RS, Cairns J, Joyner C: How should results from completed studies influence ongoing clinical trials? The CAFA Study experience. Ann Intern Med 115(10):818–22, 1991.
| 28. |
NIH Guide: NIH Policy for Data and Safety Monitoring (http://www.grants.nih.gov/ grants/notice-files/NOT98-084.html). |
| 29. |
NIH Guide: Guidance on Reporting Adverse Events to Institutional Review Boards for NIH-Supported Multicenter Clinical |
Trials (http://www.grants.nih.gov/grants/ notice-files/NOT99-107.html).
30. NIH Guide: Further Guidance on Data and Safety Monitoring for Phase I and Phase II Trials (http://www.grants.nih.gov/ grants/ guide/notice-files/NOT-OD-00-038.html).
L-16
OVERSIGHT OF HUMAN SUBJECT RESEARCH: THE ROLE OF THE STATES
Commissioned Paper Jack Schwartz
Office of the Maryland Attorney General

M-1
Introduction
Federal regulations establish basic protections for human subjects that cannot be diminished by state law.
That is, a researcher or institution subject to a federal requirement must comply with it. This obligation exists whether or not a comparable requirement is imposed by state law. Moreover, a researcher may not engage in an activity prohibited by federal law even if state law were to allow it. As the Supreme Court recently reiterated, “even if Congress has not occupied the field, state law is naturally preempted to the extent of any conflict with a federal statute.”1 The same principle, grounded in the Supremacy Clause of the United States Constitution, leads to preemption of state laws that conflict with federal regulations.
Yet, state law is hardly irrelevant to the research enterprise. First, to the extent that research is not subject to federal law, pertinent state law (if any) becomes the only legally applicable regulatory regime. Second, federal law, when it does apply to research, expressly preserves any additional state protections. The Common Rule contains the following non-preemption language: “This policy does not affect any State or local laws or regulations which may otherwise be applicable and which provide additional protections for human subjects.”2 In addition to this general provision, the Common Rule more specifically recognizes additional state requirements for informed consent: “The informed consent requirements in this policy are not intended to preempt any applicable Federal, State, or local laws which require additional information to be disclosed in order for informed consent to be legally effective.”3 Identical language appears in the regulations of the Food and Drug Administration.4 This policy decision, to preserve a role for states in the regulation of the research enterprise, is unsurprising, for the protection of human subjects may be seen as an application of a state’s core function, protecting its citizens against harm. As the Supreme Court observed many years ago, “The police power of a state...springs from the obligation of the state to protect its citizens and provide for the safety and good order of society....It is the governmental power of self-protection and permits reasonable regulation of rights and property in particulars essential to the preservation of the community from injury.”5 Yet, state regulation of research has not escaped criticism. Some contend that, especially as research increasingly involves multi-site collaborations, a regulatory system inefficiently increases compliance costs if it imposes requirements or restrictions that differ from one site to the next. These critics insist that federal regulation is sufficient and state regulation unnecessary or even harmful. For example, lobbyists for the Pharmaceutical Research and Manufacturers of America, commenting on a legislative proposal in Maryland to regulate research involving decisionally incapacitated subjects, argued that “creation of additional state laws that add to the federal common rule may inhibit the conduct of research on a multi-state level” and, they asserted, would “delay approval of lifesaving and life-altering drugs.”6 Others, while accepting that states can play an appropriate role in the overall regulatory system, have condemned specific state activities as short-sighted and inimical to research. For example, critics of the intermediate appellate court decision in T.D. v. New York State Office of Mental Health, discussed later in this paper, suggested that its “repercussions may prevent numerous studies from going forward and ultimately put a halt to research that may have provided participants with access to effective treatment and significantly improved the well-being of other individuals suffering from mental illness and other cognitive impairments” (Oldham, Haimowitz, and Delano 1998, 154).
The Commission’s consideration of the appropriate role of the states in an overall regulatory system should be informed by a survey of current state regulatory efforts. This paper presents such a survey. It discusses state statutes and regulations concerning research activities or the information used in research, the limited case law involving research, and a few particular issues for Commission attention.
M-3
Statutes and Regulations: Imposing Limits or Prerequisites Regulating All Research
One of the oft-noted voids in the current federal regulatory system is that some privately funded research is unregulated. That is, research is subject to federal regulation only if it is conducted or funded by agencies that have subscribed to the Common Rule; is the basis on which the Food and Drug Administration (FDA) will be asked to approve a drug or device; or is conducted at an institution that, as an aspect of its assurance to the Office for Protection from Research Risks (now the Office for Human Research Protections), has agreed to conduct all research at the institution in accordance with the Common Rule. Otherwise, research is free of federal regulation. Consequently, as the Commission has pointed out, “the absence of Federal jurisdiction over much privately funded research means that the U.S. government cannot know how many Americans currently are the subjects in experiments, cannot influence how they are recruited, cannot ensure that research subjects know and understand the risks they are undertaking, and cannot ascertain whether they have been harmed.”7 If Congress sought to assert federal jurisdiction over all privately funded research, its ability to do so would depend on the scope of its Commerce Clause power in this context.8 “Every law enacted by Congress must be based on one or more of its powers enumerated in the Constitution.”9 By contrast, a state need invoke no particular grant of constitutional authority to regulate privately funded research or any other activity within its borders; state legislatures have plenary power, subject only to constitutional limitations. As the Supreme Court wrote more than century ago, “The legislative power of a State extends to everything within the sphere of such power, except as it is restricted by the Federal Constitution or that of the State.”10 A state legislature that deemed it appropriate to regulate research beyond the scope of existing federal regulations is unquestionably free to do so as an aspect of its sovereign power.
Hence, the concerns expressed by the Commission about the consequences of limited federal jurisdiction might be ameliorated if privately funded research were subject to state laws that provided for the twin protections of informed consent and independent review. Only two states, however, New York and Virginia, have applied these protections to biomedical research generally.11 A third state, California, has applied a vigorous informed consent requirement comprehensively.
The New York law, enacted in 1975, applies to “human research,” defined as “any medical experiments, research, or scientific or psychological investigation, which utilizes human subjects and which involves physical or psychological intervention by the researcher upon the body of the subject and which is not required for the purposes of obtaining information for the diagnosis, prevention, or treatment of disease or the assessment of medical condition for the direct benefit of the subject.”12 The law excludes studies limited to tissue or fluid specimens taken in the course of standard medical practice, epidemiological studies, and “human research which is subject to, and which is in compliance with, policies and regulations promulgated by any agency of the federal government for the protection of human subjects.”13 By means of this last exclusion, the New York law avoids any additional regulatory burden on research that is already subject to the Common Rule or FDA regulations.
The New York law extends to privately funded research the core requirements of the federal scheme: informed consent and institutional oversight. “No human research may be conducted in this state,” the law directs, “in the absence of the voluntary informed consent subscribed to in writing by the human subject” or, in the case of a minor or other individual “legally unable to render consent,” by someone who is “legally empowered to act on behalf of the human subject.”14 “Voluntary informed consent” is consent that is “knowing,” given by someone “so situated as to be able to exercise free power of choice without undue inducement or any element of force, fraud, deceit, duress or other form of constraint or coercion.”15 The law’s itemization of the “basic elements of information necessary to such consent” parallels those in the Common Rule’s informed consent
M-4
provision:16 an explanation of the research procedures; a description of risks and benefits, if any; a disclosure of alternatives to research participation; an offer to provide additional information; and notice of the subject’s right to discontinue participation at any time.17 The law requires each research institution, public or private, to establish a “human research review committee,” the membership requirements of which are similar to those in the Common Rule’s specification for IRBs.18 The committee is to secure from its parent institution a “statement of principle and policy in regard to the rights and welfare of human subjects,” which is subject to approval by the New York commissioner of health.19 The committee is then to review each proposed instance of human subject research within the institution for “its necessity,” adequate protection of the subjects’ rights and welfare, a favorable ratio of benefits to risks, “adequate and appropriate” informed consent methods, and sufficiently qualified researchers.20 Although no annual or other set requirement for continuing review is specified, the committee is to “periodically examine each existing...research project with regard to the proper application of the [previously] approved [institutional] principles....” and is to report any violation to the commissioner of health.21 In addition to this oversight role, indistinguishable in its main elements from that of an IRB operating under the Common Rule, a committee is also called on to play a more direct role when vulnerable subjects are to be involved. Beyond the informed consent of the subject or legally authorized representative, “the consent of the committee and the commissioner [of health] shall be required with relation to the conduct of human research involving minors, incompetent persons, mentally disabled persons, and prisoners.”22 The Virginia law, enacted in 1979, applies to “human research,” defined as “any systematic investigation utilizing human subjects which may expose such human subjects to physical or psychological injury as a consequence of participation as subjects and which departs from the application of established and accepted therapeutic methods appropriate to meet the subjects’ needs.”23 The law exempts some of the same activities as are exempt from the Common Rule: educational testing, survey and interview procedures, and observation of public behavior, if the resulting data are not linked to personal identifiers or if, in any event, disclosure would not be damaging to the subjects;24 the surveying or interviewing of public officials or candidates for public office;25 and research involving existing, unlinked data or specimens.26 In addition, the law exempts the disease prevention and control activities of the Virginia Department of Health.27 Finally, research is exempt if it is “subject to policies and regulations for the protection of human subjects promulgated by any agency of the federal government....”28 For human research that is not exempt, the law requires both informed consent and approval by a “human research review committee.”29 If the subject is not competent, informed consent may be given by a legally authorized representative.30 Proxy consent may not be given, however, for participation in “nontherapeutic research unless it is determined by the human research committee that such nontherapeutic research will present no more than a minor increase over minimal risk”31—a description of risk drawn from the federal regulation on pediatric research32 but criticized by the Commission (National Bioethics Advisory Commission 1998, 40–43). The elements of informed consent are an abbreviated version of the Common Rule requirements.33 Likewise, the law’s standard for protocol approval by a human research review committee is an abbreviated version of the Common Rule requirements.34 The law omits the Common Rule’s reference to an IRB’s authority to impose additional safeguards when the research subjects are drawn from vulnerable populations.35 The law’s continuing review provision simply states the committee’s obligation to “require periodic reports from each existing human research project to ensure that the project is being carried out in conformity with the proposal as approved”;36 unlike the Common Rule, there is no mention of a maximum one-year interval between reviews or of third-party observation of the consent process or the research itself.37 Virginia seeks to promote compliance with its research law through licensing laws for physicians and other health care professionals; failure to comply “shall constitute unprofessional conduct,” a ground for discipline against licensees.38
M-5
California enacted its Protection of Human Subjects in Medical Experimentation Act in 1978. The law’s scope is narrower than New York’s or Virginia’s, for it contains no provisions for IRB review. Instead, it gives strong legal force to the informed consent requirement.
The law applies to “medical experiments”: invasive procedures or the use of a drug, device, or radiation “in a manner not reasonably related to maintaining or improving the health of the subject or otherwise directly benefitting the subject.”39 The term also includes the withholding of medical treatment “for any purpose other than maintenance or improvement of the health of the subject.”40 Research that would otherwise be subject to the law is exempted, however, if it is conducted within an institution that holds an assurance of compliance with the Common Rule.41 Also exempt, by virtue of a 1997 amendment, is emergency research that meets the main requirements of the FDA’s exception from informed consent requirements.42 The California law requires informed consent from the subject or one of several specified legally authorized representatives.43 If a representative provides consent, the research must be “related to maintaining or improving the health of the human subject or related to obtaining information about a pathological condition of the human subject.”44 Conducting a medical experiment without first obtaining informed consent subjects the principal investigator to civil damages, if the failure is negligent, or to criminal prosecution, if the failure is willful.45 The elements of informed consent largely parallel those of the Common Rule. The California law also requires explicit identification of the research sponsor or funding source, or the name of the manufacturer if a drug or device is involved,46 and contact information for “an impartial third party, not associated with the experiment, to whom the subject may address complaints about the experiment.”47
Regulating Research Involving Particular Procedures or Subjects
Human Cloning
California,48 Louisiana,49 Michigan,50 and Rhode Island51 have enacted prohibitions on efforts to clone a human being.52 Contrary to the Commission’s recommendation (National Bioethics Advisory Commission 1997), none of these enactments contains a sunset clause. Laws of this kind are analyzed in detail in a previous commissioned paper (Andrews 1997).
Genetic Research
Since 1978, New York law has required those who engage in “recombinant DNA activity” to be certified by the commissioner of health. The commissioner in turn, is required to adopt regulations that are the “substantial equivalent of [the ‘containment’ and ‘experimental guidelines’ sections, including any revisions] of the recombinant DNA research guidelines of the National Institutes of Health....”53 Rather more directly, Oregon requires anyone “carrying out recombinant DNA research [to] comply with the recombinant research guidelines adopted by the National Institutes of Health and any subsequent modifications thereof.”54
Fetal Research
The Department of Health and Human Services (DHHS) regulations that apply to research involving fetuses, pregnant women, and in vitro fertilization, 45 CFR Part 46, Subpart B, contain language that, although close to a parody of legalistic style, does unmistakably affirm the primacy of state law: “Nothing in this subpart shall be construed as indicating that compliance with the procedures set forth herein will in any way render inapplicable pertinent State or local laws bearing upon activities covered by this subpart.”55 Consequently, although Subpart B allows research of this kind to go forward with certain additional protections, states are free, as many have, to enact highly restrictive or prohibitory legislation. For example, as reported by Professor Andrews in an earlier commissioned paper, nine states have banned research involving in vitro embryos (Andrews 2000, A-4).
M-6
Amid the wide variety of state laws in this area are those that seemingly attempt to codify aspects of the federal regulations. For example, Subpart B limits DHHS-supported research involving pregnant women to studies that are intended “to meet the health needs of the mother and the fetus will be placed at risk only to the minimum extent necessary to meet such needs” or, for other research, “the risk to the fetus is minimal.”56 New Mexico has incorporated substantially the same language in a statute applicable to all clinical research, whatever its funding source.57
Prisoners
The DHHS regulations that apply to research involving prisoners, 45 CFR Part 46, Subpart C, also contain language explicitly preserving more restrictive state law: “Nothing in this subpart shall be construed as indicating that compliance with the procedures set forth herein will authorize research involving prisoners as subjects, to the extent such research is limited or barred by applicable State or local law.”58 Some states do indeed bar research involving prisoners if the research has no prospect of direct medical benefit. For example, regulations in at least four states prohibit “the use of an inmate for medical, pharmaceutical, or cosmetic experiments,” although the regulations allow, to quote the Massachusetts provision, for participation in research that is “medically appropriate for a specific inmate.”59 Similarly, Oregon law states flatly that, “There shall be no medical, psychiatric, or psychological experimentation or research with inmates in Department of Corrections institutions of the State of Oregon.”60 The pertinent definitions limit the ban, however, to what the law terms “nontherapeutic” procedures.61 Laws in some states, like Arizona, generally allow the conduct of whatever research is approved by the prison director and chief of inmate health services.62 Georgia regulations vest broad approval discretion in the commissioner of corrections but add the requirement of “periodic reports
| [ |
to the commissioner] during the course of the project.”63 Other states distinguish in more detail permissible from impermissible research. California law, for example, |
prohibits all “biomedical research,” except for participation in investigational new drug research deemed to be in the prisoner’s best medical interest.64 “Behavioral research,” on the other hand, is permissible if it concerns certain penological matters and presents “minimal or no risk and no more than mere inconvenience to the subjects of the research.”65 This language is drawn from Subpart C;66 other reasons for prisoner research that are at least theoretically permissible under the federal regulation are omitted from the California law and, accordingly, are prohibited.67 Virginia law draws lines based on benefit and risk: “Nontherapeutic research using institutionalized participants shall be prohibited unless it is determined by [a] research review committee that such nontherapeutic research will not present greater than minimal risk.”68
Children
Unlike the other subparts of 45 CFR Part 46, the DHHS regulations concerning children as research subjects, Subpart D, do not expressly preserve state law on the subject, apart from deferring to state law for determining “the legal age for consent to treatments or procedures involved in the research....”69 Nevertheless, Subpart D does not purport to preempt any state laws and, since the regulations are framed in terms of research that DHHS “will conduct or fund,”70 they have no direct effect on state regulatory efforts.
No state has enacted a local analogue to Subpart D. Rather, states have focused on the need to enact protections when children are receiving social services from the state or are in state facilities. The Oregon law on the care and treatment of indigent children, for example, states flatly that, “No child shall be used for the purpose of experimentation.”71 Massachusetts law, by contrast, allows unrestricted research participation with parental consent or, for children in state custody after a termination of parental rights, with court approval.72 Somewhere in the middle are laws like the Illinois regulations governing research involving children served by the Department of Children and Family Services; under these regulations, research risk may not exceed minimal;73
M-7
“the purely experimental use of drugs in research” is prohibited;74 and the selection of research subjects “will not be based solely on administrative convenience, availability of a population living in conditions of social or economic deprivation, or convenient access to the population.”75 Most state laws do not directly address the question whether, under some circumstances, minors may themselves consent to research participation. An exception is Virginia’s general regulatory statute, which provides that informed consent must be obtained from both “a minor otherwise capable of rendering informed consent” and the minor’s legally authorized representative.76 Most states have statutes or common law decisions identifying the criteria for a minor to become “emancipated” and so fully able to make his or her own health care decisions (under Maryland law, for example, if a minor is married or the parent of a child).77 Other state laws identify particular conditions for which minors are granted the right to consent to treatment–typically, sexually transmitted diseases and substance abuse.78 Laws of this kind might be construed as authority for a minor to consent to expected-benefit research within the scope of the statutory grant of decision-making authority, although the application of these laws to research remains largely unsettled (Hershey and Miller 1976, 118). Oklahoma law is unusually explicit in this regard: Under the circumstances specified in the law, a minor may consent to “health services,” which do not include “research or experimentation with minors except where used in an attempt to preserve the life of that minor, or research as approved by an appropriate review board involved in the management of reportable diseases.”79
Patients in Psychiatric Facilities
Many states have acted on the view that residents in psychiatric facilities are in need of special protection against the risk of research abuse. As the Secretary of the then-Department of Health, Education and Welfare observed in an early draft of regulations to protect “the institutionalized mentally infirm in research,” these individuals “might lack the...capacity to comprehend relevant information, and to make informed judgments concerning their participation” in research.80 In addition, “they experience a diminished sense of personal integrity as a result of confinement in an institution.”81 As the federal government struggled, unsuccessfully, to find an appropriate policy response to these considerations (Hoffmann, Schwartz, and DeRenzo 2000), states adopted a variety of provisions. One approach is prohibitory: Missouri law declares that, “No biomedical or pharmacological research shall be conducted in any mental health facility...or in any public or private residential facilities...unless such research is intended to alleviate or prevent the disabling conditions or is reasonably expected to be of direct therapeutic benefit to the participants.”82 A more permissive approach, typified by a Montana law, codifies the patient’s “right not to be subjected to experimental research without the express and informed consent of the patient, if the patient is able to give consent,” and of the patient’s guardian or other appointed proxy.83 In addition, the law calls for notice of the proposed research involvement to the patient’s next of kin and the attorney who represented the patient.84 “The proposed research must have been reviewed and approved by the mental disabilities board of visitors before consent may be sought. Prior to approval, the board shall determine that the research complies with the principles of the American Association on Mental Deficiency and with the principles for research involving human subjects required by the United States Department of Health and Human Services for projects supported by that agency.”85 Michigan’s law similarly requires the responsible agency to establish a review process that complies with federal law.86 Some state laws, like South Dakota’s, merely require a review process without standards: “No person may be the subject of any experimental research or hazardous procedure unless the research or procedure is approved and conducted in the manner prescribed by the secretary of human services.”87 Delaware’s law on pharmaceutical research involving patients in its facilities is particularly interesting as a model of a detailed protective regime. Among its requirements are that research be limited to patients who can
M-8
give informed consent;88 that capacity to consent be determined by, and the consent process itself be observed by, a “health care professional who will receive no financial benefit from the research”;89 and that a patient “participating in double blind research shall be advised both verbally and in writing that the patient may receive a placebo for the duration of the research instead of medication. The term placebo shall be fully defined both verbally and in writing.”90 The informed consent requirement may be waived only if “no accepted pharmaceutical or other therapy exists for the type of illness affecting the patient or the patient has not responded to accepted pharmaceutical or other therapies,”91 and only then with court approval.92 Involuntarily admitted patients are ineligible.93 Patients who are eligible are to be diagnosed prior to study entry and are to be monitored by “psychiatrists who will receive no financial benefit form the research.”94 Finally, pharmaceutical research may be approved only by a two-third affirmative vote of the IRB.95
Residents in Facilities for the Developmentally Disabled
State laws range from a prohibition in Texas96 to general requirements for informed consent by the resident or legally authorized representatives (District of Columbia97 and Montana98) to a detailed regulatory regime that replicates key elements of the Common Rule (California99).
Nursing Home Residents
Many states have codified a nursing home residents’ “bill of rights.” These statutes or regulations invariably include a “right to refuse to participate in experimental research”100 or a requirement that informed consent be obtained prior to research participation.101 Rhode Island and Wisconsin have extended similar protection to home health care patients;102 Maryland, to patients in ambulatory care facilities;103 and New York, to hospice patients.104
Statute and Regulations: Facilitating Research
States provide tangible economic support to the research enterprise in a variety of direct and indirect ways: for example, grant programs to encourage private research on particular topics of concern,105 tax abatements and other economic development incentives for research enterprises or facilities,106 and mandated insurance benefit laws that encompass clinical trials or other research.107 These nonregulatory aspects of state law are beyond the scope of this paper.
About a dozen states have enacted “controlled substances therapeutic research acts,” which create an exception to narcotics control laws for marijuana use in certain cancer- or glaucoma-related research.108 Given the narrow scope of these laws, they warrant no further discussion.
States also facilitate research through their regulation of dead bodies. State anatomical gift acts uniformly allow cadaver or organ donation for research purposes, although variations from state to state may affect access to tissue under some circumstances. These laws, as they affect human stem cell research, were discussed in Chapter 3 of the Commission’s report (National Bioethics Advisory Commission 1999, 32) and in two commissioned papers (Andrews 2000; Kinner 2000).
Of greater interest as the Commission considers the regulatory system as a whole, and the focus of this portion of the paper, is the interaction of state privacy laws and research. In what respects do state laws affect the acquisition of information for research purposes or research information itself?
M-9
Access for Researchers to Otherwise Confidential Information
State-Held Information
States hold vast amounts of demographic and health data of potential value to researchers. Every state is a repository of birth, death, and similar demographic information. For public health reasons, every state mandates reporting of some communicable diseases. Largely to aid research, most states have created cancer or other disease-specific registries. For health planning purposes, some states require health care providers or third-party payers to report details of patient encounters. Other collections of state records (for example, files on state employees) might yield important information for researchers.
Whenever a state assembles a pool of personally identifiable and highly sensitive information, the state recognizes its obligation to protect the privacy of the individuals. Although the protective measures vary widely, and some are open to criticism as inadequate (Pritts, Goldman, Hudson et al. 1999), virtually every law that mandates or authorizes the collection of the information also requires that personally identifiable information be kept confidential.
Yet, confidentiality requirements could vitiate the very benefit to research that is a primary or secondary goal of these data collection efforts. To avoid this effect, the laws that mandate confidentiality invariably contain an authorization for access by researchers if the researchers bind themselves not to redisclose personally identifiable data. Vital records laws typically have a broadly phrased research exception to their confidentiality requirements.109 Disease registry and mandatory reporting laws allow access for research purposes.110 To cite but one example, the Illinois law that mandates the reporting of Reye’s Syndrome cases allows individually identifiable data to be made available for “health-related research” to a researcher who agrees not to redisclose the data.111 Finally, public records laws, which always exempt individual health and other sensitive information from mandatory disclosure, usually allow researchers access to this information.112 For example, all government records in Georgia that by law are “confidential, classified, or restricted may be used for research purposes by private researchers” if a researcher is deemed qualified, the research topic “is designed to produce a study that would be of potential benefit to the state or its citizens,” and the researcher agrees to protect the confidentiality of the records.113 Apart from its confidentiality requirement, this broad authorization for research access, like comparable laws elsewhere, does not impose any requirements related to the protection of human subjects.
For some records, a research exception to state confidentiality laws derives from a federal model. The federal law that requires substance abuse patient records to be kept confidential, for example, permits certain research-related disclosures without patient consent.114 So, too, do the federal regulations governing the confidentiality of child abuse and neglect records.115 Many state laws parallel the federal exceptions.116
Medical Records
Much research would be foreclosed if investigators were unable to obtain baseline clinical data about the subjects. Of course, subject consent for access to medical records should be obtained if possible. State lawmakers, however, have by and large accepted that patient consent may not always be feasible and have accommodated researchers by building into medical records privacy statutes an exception for researchers’ access without consent. Sometimes the exception is just that open-ended. Rhode Island law, for example, permits a health care provider to release confidential health care information without a patient’s consent to qualified personnel for the purpose of conducting scientific research, provided that information about individual patients is never disclosed by the researchers.117 Other laws impose the useful check that the research be approved by an IRB. Maryland’s medical records act, for example, permits disclosure without patient consent “for educational or research purposes, subject to the applicable requirements of an institutional review board” and to an agreement not to redisclose any patient identifying information.118
M-10
Exempting Research from HIV Testing Requirements
State laws governing HIV testing typically provide that testing is to be done only with informed consent and after appropriate counseling. Some states have enacted an exception for anonymous testing done for research purposes.119 Maryland exempts certain research projects from mandatory reporting by unique patient identifying numbers, but only if, among other requirements, the research is either related to HIV vaccine development or “is not primarily intended to provide medical treatment to participants” and has been approved by an IRB.120
Protecting the Confidentiality of Research Data
The “certificate of confidentiality” procedure under federal law allows for sensitive, individually identifiable research data to be protected against compelled disclosure.121 A few states have enacted their own comparable protections. The Georgia legislature, for example, has found that “protecting the confidentiality of research data is essential to safeguarding the integrity of research in this state, guaranteeing the privacy of individuals who participate in research projects, and ensuring the continuation of research in science, medicine, and other fields that benefits [sic] the citizens of Georgia and other states.”122 To that end, this law protects “confidential raw research data” against compelled disclosure.123 New Hampshire law also deems “personal medical and /or other scientific data of any kind whatsoever obtained for the purpose of medical or scientific research” by the state health commissioner or authorized researchers to be confidential, not admissible in evidence and to be used “solely for medical or scientific purposes.”124
State-Conducted Research
Hundreds of state laws authorize or direct state agencies to conduct epidemiologic or other public health research. Most common are the ubiquitous laws that require reporting to state health authorities of various communicable diseases. These laws then authorize the use of the information for public health purposes. The statutory language is usually capacious enough to encompass not only classic epidemiologic investigations, aimed at protecting the public health by identifying the source of a disease that has recently spread quickly among a group of people, but also epidemiologic research in the regulatory sense, aimed at the acquisition of “generalizable knowledge” about the causes and transmission of a disease within a population.125 In Illinois, for example, AIDS cases are to be reported to an AIDS Registry, and the Illinois Department of Public Health may collect “such information concerning those cases as it deems necessary or appropriate in order to conduct thorough and complete epidemiological surveys of AIDS...in Illinois, and to evaluate existing control and prevention measures.”126 Depending on their purpose and design, these “surveys” and “evaluations” might or might not be “research” under the Common Rule’s definition.
Other state laws, when describing the overall public health responsibilities of state officials, use language that either explicitly authorizes “research” among other permitted activities or is phrased broadly enough to encompass it. An example of the former is this Georgia authorization: “The Department of Human Resources and county boards of health are empowered to conduct studies, research, and training appropriate to the prevention of diseases and accidents, the use and control of toxic materials, and the prevention of environmental conditions which, if permitted to develop or continue, would likely endanger the health of individuals or communities.”127 An example from Maryland illustrates the way in which a broadly worded provision can authorize research implicitly. The Maryland health secretary has a statutory duty to “investigate...[t]he causes of disease and, particularly, the causes of epidemics.”128 To investigate the cause of an epidemic may well not be “research,” because its design may not constitute “a systematic investigation...designed to develop or contribute to generalizable knowledge;”129 to investigate the causes of disease may well be. The statutory authorization covers both.
M-11
None of the three laws cited here is linked to any requirements for human subject protection. This omission is not atypical; the laws that authorize state-conducted research usually do not specify a procedure for review of the research or impose a requirement for informed consent. Presumably, state researchers seek to follow prevailing ethical norms, but state law ordinarily does not mandate any specific procedures.
One notable exception is Florida’s approach to research conducted under the authority of the Florida Department of Health. Human subject research is subject to review and approval by the Review Council for Biomedical and Social Research, a nine-member body appointed by senior state officials (the Governor, the President of the Senate, and the Speaker of the House) and consisting of three members “knowledgeable in biomedical research,” three members “knowledgeable in behavioral research,” and three members “from the client advocacy community.”130 The Review Council is to be “guided by the ethical standards for human research set forth in the [Belmont Report].”131
Case Law
The earliest case law on “research” practices involved physicians’ ad hoc experimentation in the course of clinical care. As a New York court observed more than a century ago, when standard therapy exists, “there should be no departure from it, unless the surgeon who does it is prepared to take the risk of establishing by his success the propriety and safety of his experiment.”132 These cases are based on the premise that, in the clinical setting, the patient is entitled to expect the skillful application of potentially efficacious standard therapy. “Use of an unproven method of treatment which damages the patient has generally been considered negligence, even if carried out with the highest possible degree of care, unless it is clear the patient knew that it was research or innovation” (Holder 1978, 738; Goldner 1993).
With informed consent, the use of experimental procedures is permissible.133 Conversely, “A physician who uses a patient as the subject of an experiment of any sort without a full disclosure of all of the risks involved and the nature and purpose of the investigation, especially in a situation in which consent might be obtained by duress, express or implied, may be in serious legal and disciplinary difficulties” (Holder 1978, 742).134 Professor Holder’s reference to “disciplinary difficulties” is illustrated by the aftermath of the research scandal at the Jewish Chronic Disease Hospital. The physician-investigators’ decision not to inform patients of the injection of live cancer cells was the equivalent of a deliberate misrepresentation of a material fact, the kind of unprofessional conduct that can result in discipline (Katz 1972, 60–63).
Indeed, when research is subject to federal or state informed consent regulations, the breach of those regulations is likely to be viewed as itself a basis for liability. In Daum v. Spinecare Medical Group, Inc., a recent California case, the plaintiff alleged that his physicians had failed to obtain his informed consent for implantation of a experimental spinal fixation device. According to his version of events, he was not informed orally that “the surgery was part of an FDA-approved clinical investigation” of the device.135 The investigational status of the device, however, together with an account of risks and alternatives, was set out in a written consent document, presented for signature on the morning of surgery. In the trial court, experts testified for both the plaintiff and the defendants on the issue whether the informed consent process fell below the standard of care. After the judge instructed the jury that they should decide the case based solely on their assessment of the expert testimony, the jury found for the defendants.
The appellate court reversed, holding that the jury should have been allowed to consider all of the evidence, not just expert testimony, in deciding whether the standard of care had been breached. In particular, the jury should have considered the effect of federal and California informed consent requirements. Because the physician-investigators had agreed to comply with FDA regulations, the regulations themselves established the standard of care. The plaintiff, the appellate court held, “presented sufficient evidence that his injury resulted
M-12
from the kind of occurrence the statutes and regulations...were intended to prevent: participation in a clinical trial without the subject’s fully informed consent in writing, with a copy for the subject and under circumstances permitting a free and deliberate choice.”136 Under this analysis, failure to adhere to regulatory standards for informed consent amounts to the tort of “research negligence,” the holding in a well-known Canadian case.137 Another California decision, Moore v. Regents of the University of California,138 suggests that some courts will view the informed consent doctrine as encompassing disclosure not only of the protocol-related information specified in the Common Rule but also of material economic incentives affecting the researcher. Moore, arising in a clinical setting, held that a physician who used a patient’s surgically removed spleen to establish a patented cell line should have disclosed his research and economic interests to the patient prior to removing the spleen.
One can also speculate (to be sure, without any case law on point) that an aggrieved research subject might under some circumstances bring a breach of contract action against a researcher. This kind of claim could arise if a research protocol were described one way in an informed consent document but carried out another way in practice,139 or, under a third-party beneficiary theory, if research were conducted in a manner inconsistent with a single or multiple project assurance. Whether a plaintiff would have the kind of economic damages that are cognizable in a contract action, however, is open to doubt.
The T.D. case,140 discussed in the Commission’s Capacity report (National Bioethics Advisory Commission 1998, 71–72), may prove to be the forerunner of another branch of research-related case law: claims that the civil rights of specially vulnerable subjects were violated. With respect to involuntarily hospitalized adult patients and children in state psychiatric facilities, New York’s intermediate appellate court held, in a later-vacated aspect of its opinion, that the federal and state constitutions and state common law guaranteed certain protections for no-direct-benefit research, including adequate notice to potential subjects, review procedures for determinations of incapacity, and judicial approval of surrogate consent (Hoffmann and Schwartz 1998; Oldham, Haimowitz, and Delano 1998). An earlier federal case suggested that certain research practices with prisoners as subjects might violate the constitutional proscription of “cruel and unusual punishment.”141
Conclusion: Issues for the Commission
The singular title of this paper, “The Role of the States,” might better have been phrased in the plural. The regulation of human subject research varies markedly from state to state, from efforts at comprehensive regulation in a few to the imposition of limited protections in others. Their regulatory styles differ as well. Some codify informed consent and independent review requirements without detailed specification of methods; others implement these protections through incorporation into state law of federal standards, sometimes augmented by useful elaboration or specification; and still others add requirements or restrictions that reflect a state’s own policy choices. Indeed, in this last category are enactments that may be subject to constitutional objection—for example, restrictions on fetal research that may be worded so vaguely as to offend due process standards (Andrews 2000; Gelfand and Levin 1993).142 This diversity is the product of a policy environment in which an incomplete federal regulatory scheme encourages a focus on state-level agendas, be it advocates pursuing restrictive regulation in particular areas or research enterprises seeking to block undesired regulation. Whether in state legislatures or the courts, and whether they turn out to be mild or draconian, sporadic state responses to controversial research practices can be anticipated—unless, of course, the Commission were to recommend, and Congress were to pass, a broadly preemptive federal law.
Thus, a fundamental policy issue for the Commission is whether it is content with the prevailing relationship between federal and state law, in which federal standards are a floor, requiring compliance with both the federal standards and state law, if the state law is more protective of subjects; or whether the Commission
M-13
believes that federal law should be both a floor and a ceiling, reflecting a conclusive judgment about the extent to which the research enterprise is to be subject to regulation, a judgment that should not be diminished by state-to-state variation. Were the Commission to adopt the latter position, it would need to consider carefully a federal constitutional issue beyond the scope of this paper: the extent to which Congress, exercising its power over interstate commerce, or, by delegation, an Executive Branch agency, may preempt state law in this area (Andrews 1997).
This paper assumes, however, that the Commission will accept as inevitable, and perhaps even desirable, a continuing role for the states in an overall regulatory scheme. If so, the Commission may wish to consider the following policy issues:
Should the Commission encourage states to adopt comprehensive laws, like New York’s and Virginia’s, that apply the protections of informed consent and independent review to research generally, with an exception for research already covered by federal regulations? Should the Commission encourage the National Conference of Commissioners on Uniform State Laws to develop a model law on the regulation of research? Alternatively, should the Commission encourage states to adopt federal standards for discrete areas of regulated research?
A comprehensive solution to the problem of unregulated research is for Congress to enact a law that extends the protections of informed consent and independent review to all research in this country, whatever its funding source or site. Of course, Congress has not done so, and it is difficult to predict when, if ever, it will. Therefore, the Commission may wish to consider endorsing an alternative, albeit far more cumbersome, means of extending these protections: state-by-state enactment of general regulatory laws, like New York’s or Virginia’s. If these laws contained appropriate exemptions, they could help fill the regulatory gap while avoiding duplication of existing federal regulation. Such an initiative might be furthered if the National Conference of Commissioners on Uniform State Laws, a well-respected source for state legislative initiatives, developed a model research regulatory act. Alternatively, states might be encouraged, when they regulate discrete areas of research or research involving vulnerable subjects, to adopt federal standards, a strategy that has the salutary effect of applying known standards, with a history of implementation, to privately funded research. Examples include the New York and Oregon laws on recombinant DNA research143 and the California law on research in facilities for those with developmental disabilities.144
Should the Commission encourage states to require IRB approval prior to granting researchers access to confidential information? Should the Commission encourage the National Conference of Commissioners on Uniform State Laws to develop a uniform law on researchers’ access to confidential information?
Presumably, the Commission views favorably the basic policy decision, made by the states in a myriad of contexts, allowing researchers to gain access to otherwise confidential health information, whether maintained by state agencies or private health care providers. Nevertheless, overly permissive state laws not only risk compromising individual privacy but also reflect a lost opportunity to gain better protections for subjects. If more states followed the example of some and established as a prerequisite for access that the research be approved by an IRB, the policy goal of extending independent review to all research would be furthered. The Commission may wish to consider recommending this approach to the state legislatures. It is noteworthy that the National Conference of Commissioners on Uniform State Laws has included an IRB review requirement as part its Uniform Health-Care Information Act.145
M-14
Should the Commission recommend that DHHS establish a clearinghouse on state regulation of research?
Assuming that the federal policy decision about state regulation remains as permissive as it now is, the highly diverse landscape of state regulation surveyed in this paper will persist. The Commission may wish to consider encouraging DHHS to establish a clearinghouse on state regulation of research. The clearinghouse would be a source of information for state legislators considering new proposals, for federal research managers considering the implications of state regulation on their priorities, and for researchers themselves. The existence of a central source of information might itself promote greater consistency among the states.
Should the Commission encourage pilot programs for state involvement in the enforcement of federal standards for research?
Some federal regulatory systems incorporate shared enforcement responsibilities between federal and state (or local) agencies. Perhaps the most notable example is in the enforcement of equal employment opportunity laws, which involves “work-sharing” agreements between the Equal Employment Opportunity Commission and designated state and local agencies (Lindemann and Grossman 1996, 1221–1223). Other examples may be found in federal environmental laws.146 Analogies to these highly detailed regulatory schemes are imperfect at best, but they suggest the merit of exploring cooperative agreements to augment federal oversight of research. For example, it is not difficult to imagine a contract between DHHS and a state legislative audit agency under which the auditor’s performance audit of a state research university or a state health department would include a special focus on compliance with standards for human subject protection. While many practical problems would have to be addressed to make such an arrangement feasible, the Commission may wish to consider recommending that approaches of this kind be explored.
Notes
1 Crosby v. National Foreign Trade Council, 68 U.S.L.W. 4545, 4547 (U.S. June 19, 2000).
2 45 CFR § 46.101(f). 3 45 CFR § 46.116(e). 4 21 CFR § 50.26(c).
5 Panhandle Eastern Pipe Line Co. v. State Highway Commission, 294 U.S. 613 (1935).
6 Letter to Assistant Attorney General Jack Schwartz from J. William Pitcher and Deron A. Johnson. November 10, 1999.
7 NBAC Summary of Preliminary Findings: Adequacy of Federal Protections for Human Subjects in Research, appended to letter to the President from Harold T. Shapiro, Chair, National Bioethics Advisory Commission. May 4, 1999.
8 A full consideration of this question is beyond the scope of this paper. Although the Supreme Court has recently voided congressional enactments on the grounds that they exceeded the scope of the Commerce Clause, federal regulation of privately funded research would likely be upheld because, as the United States Court of Appeals for the Fourth Circuit (considered by many the most conservative of all the federal appellate courts) recently held, scientific research is “an interstate market.” Gibbs v. Babbitt, 2000 WL 793941 (4th Cir. June 20, 2000).
9 United States v. Morrison, 120 S. Ct. 1740, 1748 (2000).
10 Pine Grove Township v. Talcott, 86 U.S. 666 (1874).
11 The Territory of Guam imposes requirements comparable to those in the Common Rule for all research conducted at the University of Guam, which one supposes is the sole locus of biomedical research on the island. Interestingly, if research is conducted in violation of the law, the investigator is subject to both a $1,000 fine for each violation “and shall be prohibited from continuing and conducting human research studies for not less than two...years.” Guam Code Title 17, § 24109.
M-15
12 New York Public Health Law § 2441(2).
13 New York Public Health Law § 2441(2) and 2445.
14 New York Public Health Law § 2442.
15 New York Public Health Law § 2441(5).
16 45 CFR §46.116.
17 New York Public Health Law § 2441(5). 18 New York Public Health Law § 2444(1). 19 New York Public Health Law § 2444(2). 20 New York Public Health Law § 2444(2). 21 New York Public Health Law § 2444(2). 22 New York Public Health Law § 2444(2). 23 Virginia Code § 32.1-162.16.
24 Virginia Code § 32.1-162.17(2), (3), and (5), comparable to 45 CFR § 46.101(b)(2). In a slight variation, whereas the Common Rule exemption is lost if disclosure “could reasonably...be damaging to the subjects’...reputation,” the Virginia provision omits the reference to “reputation” but specifies instead that research is not exempt if it “deals with sensitive aspects of the subject’s own behavior, such as sexual behavior, drug or alcohol use, or illegal conduct.”
25 Virginia Code § 32.1-162.17(4), comparable to 45 CFR § 46.101(b)(3)(i). 26 Virginia Code § 32.1-162.17(6), comparable to 45 CFR § 46.101(b)(4). 27 Virginia Code § 32.1-162.17(1).
28 Virginia Code § 32.1-162.20.
29 Virginia Code §§ 32.1-162.18A and 32.1-162.19B.
30 Virginia Code § 32.1-162.18A.
31 Virginia Code § 32.1-162.18B.
32 45 CFR § 46.406(a).
33 Virginia Code § 32.1-162.16(1)-(5), comparable to 45 CFR § 46.116(a). 34 Virginia Code § 32.1-162.19B, comparable to 45 CFR § 46.111(a). 35 45 CFR § 46.111(b).
36 Virginia Code § 32.162.19B.
37 45 CFR § 46.109(e).
38 Virginia Code § 54.1-2407.
39 California Health & Safety Code § 24174. 40 California Health & Safety Code § 24174. 41 California Health & Safety Code § 24178.
42 California Health & Safety Code § 24177.5, adopting language from 21 CFR § 50.24(a)(1), (2), (3), and (5).
43 California Health & Safety Code § 24175.
44 California Health & Safety Code § 24175(e). 45 California Health & Safety Code § 24176. 46 California Health & Safety Code § 24173(c)(9).
M-16
47 California Health & Safety Code § 24173(c)(10). 48 California Health & Safety Code § 24185. 49 Louisiana Statutes § 40:1299.36.2.
50 Michigan Laws § 750.430a.
51 Rhode Island Laws § 23-16.4-1.
52 In addition, Missouri has prohibited the use of state funds for human cloning. Missouri Statutes § 1.217. Michigan has as well (superfluously, given its prohibition of the procedure itself). Michigan Laws § 33.26403.
53 New York Public Health Law § 3222.
54 Oregon Statutes § 431.810.
55 45 CFR § 46.201(b).
56 45 CFR § 46.207(a).
57 New Mexico Statutes § 24-9A-2.
58 45 CFR § 46.301(b).
59 Maryland Administrative Code § 12.14.04.02.D; Massachusetts Administrative Code Title 103, § 80.07; New Jersey Administrative Code §§ 10A:1-10.1 and 10A:16-2.20; New York Administrative Code § 7651.27.
60 Oregon Statutes § 421.085(2).
61 Oregon Statutes § 421.085(1).
62 Arizona Statutes §§ 31-321 to 31-323. 63 Georgia Administrative Code 125-4-4.12. 64 California Penal Code §§ 3502 and 3502.5. 65 California Penal Code § 3505.
66 45 CFR § 46.306(a)(2)(A) and (B).
67 45 CFR § 46.306(a)(2)(C) and (D).
68 Virginia Administrative Code Title 6, § 15-26-30C.
69 45 CFR § 46.402(a).
70 45 CFR §§ 46.404, 46.405, 46.406, and 46.407. 71 Oregon Statutes § 444.230. 72 Massachusetts Administrative Code Title 110, § 11.23. 73 Illinois Administrative Code Title 89, § 432.5(a)(1). 74 Illinois Administrative Code Title 89, § 432.7.
75 Illinois Administrative Code Title 89, § 432.5(c).
76 Virginia Code § 32.1-162.18A(iii).
77 Maryland Health-General Code § 20-102(a).
78 See, e.g., Maryland Health-General Code § 20-102(c); Oklahoma Statutes Title 63, § 2602A3.
79 Oklahoma Statutes Title 63, § 2601(c).
80 Protection of Human Subjects, 38 Fed. Reg. 31,740 (1973). 81 Protection of Human Subjects, 38 Fed. Reg. 31,745 (1973). 82 Missouri Statutes § 630.192.
M-17
83 Montana Code § 53-21-147(1). 84 Montana Code § 53-21-147(1). 85 Montana Code § 53-21-147(2). 86 Michigan Laws § 330.1919(1)(2).
87 South Dakota Codified Laws § 27A-12-3.21.
88 Delaware Code Title 16, § 5175(a).
89 Delaware Code Title 16, § 5175(g). 90 Delaware Code Title 16, § 5175(d). 91 Delaware Code Title 16, § 5176(2). 92 Delaware Code Title 16, § 5176(6). 93 Delaware Code Title 16, § 5174(2).
94 Delaware Code Title 16, § 5172(b)(2) and (4).
95 Delaware Code Title 16, § 5173(b)(1).
96 Texas Health and Safety Code § 592.053.
97 D.C. Code § 6-1969.
98 Montana Statutes § 53-20-147.
99 California Administrative Code Title 17, §§ 50413 and 50417.
100 Arkansas Administrative Code § 12.890(a)(12); Connecticut Statutes § 19a-550(b)(3); South Carolina Code § 44-81-40(B)(5); Vermont Statutes Title 33, § 7301.
101 Delaware Code Title 16, § 1121(4); Illinois Statutes Chapter 210, § 45/2-104(a); Maryland Health-General Code § 19-344(f)(2)(i); Rhode Island Laws § 23-17.5-7; Washington Code § 74.42.040.
102 Rhode Island Laws § 23-17.16.2(16); Wisconsin Administrative Code § HFS133.08.
103 Maryland Administrative Code § 10.05.01.10C(3).
104 New York Administrative Code § 794.1(a)(5).
105 See, e.g., Nebraska Statutes § 85-804 (cancer research grants).
106 See, e.g., New Jersey Statutes § 34-1B (New Jersey Economic Development Authority Act).
107 See, e.g., Maryland Insurance Code § 15-827 (clinical trials related to cancer and other life-threatening conditions).
108 See, e.g., Alabama Code § 20-2-114.
109 See, e.g., Arkansas Code § 18.50.310(b); Arizona Code § 36-341F; Kentucky Statutes § 213.131(4); Utah Code § 26-2-22.
110 See, e.g., Arizona Code § 36-664B(4) (communicable disease related information); California Health and Safety Code § 103850 (birth defects information); Idaho Code § 7-1706 (cancer registry); Illinois Administrative Code Title 77, § 663.90 (Reye’s Syndrome information); Iowa Code § 7-1706 (brain and spinal cord injury registry); Massachusetts Laws Title 111, § 111B (“malignant disease” registry); Nebraska Statutes § 81-647 (cancer registry); Washington Code § 70.54.250 (cancer registry).
111 Illinois Statutes Chapter 410, § 245/4(3).
112 See, e.g., Maryland State Government Code § 10-624(c).
113 Georgia Code § 50-18-101.
114 42 USC § 290dd-2(b)(2)(B); 42 CFR § 2.52.
115 45 CFR § 1340.14(i)(2)(xi).
116 See, e.g., Colorado Code § 19-1-307(2)(o) (child abuse and neglect records); Delaware Code Title 16, § 2214 (alcohol treatment records).
M-18
117 Rhode Island Statutes § 5-37.3-4(b)(3).
118 Maryland Health-General Code § 4-305(b)(2)(i).
119 Louisiana Statutes § 40:1300.13F(2); Maine Statutes Title 5, § 19203(5); New York Public Health Law § 2781; Pennsylvania Statutes Title 35, § 7605(f).
120 Maryland Administrative Code Title 10, § 10.52.09.01-1A.
121 42 USC. § 241(d).
122 Georgia Code § 24-9-40.2(a).
123 Georgia Code § 24-9-40.2(c).
124 New Hampshire Statutes § 126-A:11. 125 45 CFR § 46.102(d). 126 Illinois Statutes Chapter 410, § 316/4(a). 127 Georgia Code § 31-12-1.
128 Maryland Health-General Code § 18-101(1).
129 45 CFR § 46.102(d).
130 Florida Statutes § 381.85(3)(a) and (4).
131 Florida Statutes § 381.85(1)(c)3.
132 Carpenter v. Blake, 60 Barb. 488, 523 (N.Y. Sup. Ct. 1871). To like effect is Sawdey v. Spokane Falls & Northern Railway Co.,
70 P. 972, 973 (Wash. 1902), commenting that a physician “must not experiment in his treatment of [an] injury.”
133 Karp v. Cooley, 493 F.2d 408 (5th Cir.), cert. denied, 419 U.S. 845 (1974) (applying Texas law).
134 See, e.g., Ahern v. Veterans Administration, 537 F.2d 1098 (10th Cir. 1976) (applying New Mexico law).
135 61 Cal. Rptr. 2d 260, 263 (Cal. App. 1997).
136 61 Cal. Rptr. 2d at 273.
137 Halushka v. University of Saskatchewan, 52 W.W.R. 608 (Sask. Ct. App. 1965).
138 793 P.2d 479 (Cal. 1990), cert. denied, 499 U.S. 936 (1991).
139 Dingle v. Belin, 749 A.2d 157 (Md. 2000) discusses, in a clinical context, breach of contract theories related to informed consent.
140 T.D. v. New York State Office of Mental Health, 626 N.Y.S.2d 1015 (N.Y. Sup. Ct. 1995), aff’d 650 N.Y.S.2d 173 (N.Y. App. Div. 1996), aff’d in part and vacated in part 668 N.Y.S.2d 153 (N.Y. 1997).
141 Mackey v. Procunier, 477 F.2d 877 (9th Cir. 1973).
142 In assessing a proposed statutory ban on the use of tissue and organs from aborted fetuses for research, a recent opinion of the Nebraska Attorney General discussed the vagueness doctrine and a variety of other constitutional issues. Opinion to Senator Jim Jensen from Attorney General Don Stenberg. February 2, 2000. Available at http://www.nol.org/home/ago/opinions/00006.htm.
143 New York Public Health Law § 3222; Oregon Statutes § 431.810.
144 California Administrative Code Title 17, §§ 50413 and 50417.
145 National Conference of Commissioners on Uniform State Laws, Uniform Health-Care Information Act § 2-104(a)(7) (1985),
9 Uniform Laws Annotated 183, 201 (1999) (adopted in Montana and Washington).
146 See, e.g., 33 USC § 1342(b) (state programs, including enforcement responsibilities, for water pollutant discharge permits).
M-19
References
Andrews, L.R. 1997. The Current and Future Legal Status of Cloning. In Cloning Human Beings, Volume II, Commissioned Papers. National Bioethics Advisory Commission. Rockville, MD: U.S. Government Printing Office.
Andrews, L.R. 2000. State Regulation of Embryo Stem Cell Research. In Ethical Issues in Human Stem Cell Research, Volume II, Commissioned Papers. National Bioethics Advisory Commission. Rockville, MD: U.S. Government Printing Office.
Gelfand, G. and T.R. Levin. 1993. “Fetal Tissue Research: Legal Regulation of Human Fetal Tissue Transplantation.” Washington and Lee Law Review 50:647–694.
Goldner, J.A. 1993. “An Overview of Legal Controls on Human Experimentation and the Regulatory Implications of Taking Professor Katz Seriously.” Saint Louis University Law Journal 38:63–134.
Hershey, N. and R.D. Miller. 1976. Human Experimentation and the Law. Germantown, Maryland: Aspen Systems Corporation.
Hoffmann, D.E. and J. Schwartz. 1998. “Proxy Consent to Participation of the Decisionally Impaired in Medical Research: Maryland’s Policy Initiative.” Journal of Health Care Law and Policy 1:123–153.
Hoffmann, D.E., J. Schwartz, and E.G. DeRenzo. 2000. “Regulating Research with Decisionally Impaired Individuals: Are We Making Progress?” DePaul Journal of Health Care Law 3:547–608.
Holder, A.R. 1978. Physician’s Failure to Obtain Informed Consent to Innovative Practice or Medical Research. In American Jurisprudence Proof of Facts Second Series, Vol. 15, 711–761. Rochester and San Francisco: The Lawyers Co-Operative Publishing Company and Bancroft-Whitney Co.
Katz, J. 1972. Experimentation with Human Beings. New York: Russell Sage Foundation.
Kinner, J.K. 2000. Bioethical Regulation of Human Fetal Tissue and Embryonic Germ Cellular Material: Legal Survey and Analysis. In Ethical Issues in Human Stem Cell Research, Volume II, Commissioned Papers. National Bioethics Advisory Commission. Rockville, MD: U.S. Government Printing Office.
Lindemann, B. and P. Grossman. 1996. Employment Discrimination Law, Vol. II. Washington, D.C.: Bureau of National Affairs, Inc.
National Bioethics Advisory Commission. 1997. Cloning Human Beings, Volume I, Report and Recommendations. Rockville, MD: U.S. Government Printing Office.
National Bioethics Advisory Commission. 1998. Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity, Volume I, Report and Recommendations. Rockville, MD: U.S. Government Printing Office.
National Bioethics Advisory Commission. 1999. Ethical Issues in Human Stem Cell Research, Volume I, Report and Recommendations.
Rockville, MD: U.S. Government Printing Office.
Oldham, J.M., S. Haimowitz, and S.J. Delano. 1998. “Regulating Research with Vulnerable Populations: Litigation Gone Awry.”
Journal of Health Care Law and Policy 1:154–173.
Pritts, J., J. Goldman, Z. Hudson, A. Berenson, and E. Hadley. 1999. “The State of Health Privacy: An Uneven Terrain/A Comprehensive Survey of State Health Privacy Statutes.” Available at http://www.healthprivacy.org/resources/statereports.
M-20

PRIVACY AND CONFIDENTIALITY: AS RELATED TO HUMAN RESEARCH IN SOCIAL AND BEHAVIORAL SCIENCE
Commissioned Paper Joan E. Sieber
California State University, Hayward
N-1
Abstract
A s social and behavioral research expands to involve diverse populations, contexts, and sensitive topics, it raises many complex issues of privacy and confidentiality. These issues go beyond what is contained in textbooks or known to most researchers and Institutional Review Board (IRB) members. The literature on these issues is found in a variety of applied research publications in applied statistics, program evaluation, criminology, education, economics, and demography, but not in the mainstream social science literature. Understanding and solving some of these problems requires knowledge and resources that can be found via major research libraries, but only if one knows what to look for. IRBs cannot be expected to generate and disseminate the needed resources and body of knowledge on their own, though many have made a remarkable start in this direction.
The key recommendations of this report are that more useful definitions of privacy and confidentiality be given in the Common Rule and that two educational web sites be developed structured similarly to the Help menu of Microsoft Word. The small web site would guide IRBs in locating and structuring helpful materials and resources tailored to their specific location. The major web site would provide education and information needed by IRBs, researchers, and students, as well as teachers of research methodology who wish to use these materials for instructional purposes.
Only minor modifications of the Common Rule are needed, under definitions and informed consent requirements. No additional regulations or surveillance are needed except that IRBs would be required to take the steps specified in the small IRB web site to tailor the larger general web site to their specific setting. Enforcement of intelligent use of the web sites by researchers would be the role of IRBs when dealing with protocols, just as IRBs seek to foster intelligent ethical problem solving now.
The problem that needs to be solved is not lack of rules or lack of ethical concern on the part of researchers. The problem is lack of education—that is, lack of knowledge, problem solving skills, and resources to interpret the existing rules. Additional rules or greater specificity of rules would raise three serious problems (Evers, 2000):
| 1. |
Acceptance of many more detailed or specific rules across 17 agencies and diverse research contexts would be limited. |
| 2. |
Opportunities for intelligent interpretation, and deciding between principles or values in conflict would be diminished. |
| 3. |
Efforts required to follow a given rule may be disproportionally great relative to the expected gain or results. An intelligently developed and managed user-friendly web site in the hands of a capable scientific workforce |
and its managers creates a culture in which ignorance is no excuse and learning is easy. Even the occasional desperate individual, eager for a quick publication, would find it more difficult to skirt the rules. Three basic recommendations are proposed to the Commission:
| 1. |
Change the definitions of privacy and confidentiality contained in the Common Rule so that they are more generic and relate to a broader understanding of these two concepts, as opposed to the current definitions which seem to relate to biomedical research and medical records. |
| 2. |
Commission the development of educational information for the two web sites. Establish a small oversight committee that would edit the educational material, oversee the work of a web manager, and consider new ideas, submissions, or criticisms from users of the web site. Above all, this material and the roles connected with its development and management should be treated as educational and not as a regulatory requirement. |
N-3
| 3. |
Refer all readers of the Common Rule to the major web site for assistance with solving problems of privacy and confidentiality. Refer IRBs to the small web site for guidance in tailoring educational material to their context and location. |
Many specific recommendations are offered concerning the content of the two web sites. Most of these web-content recommendations concern concepts of privacy and confidentiality, relevant laws and regulations, approaches to learning what is private to individuals, and approaches to assuring privacy and confidentiality.
Introduction
The National Bioethics Advisory Commission (NBAC) has requested analysis and recommendations concerning issues of privacy and confidentiality that arise in social and behavioral research. In this response to NBAC’s request, relevant issues are raised and explored, and recommendations are offered. This introductory section provides an overview of the problem that is addressed and the general direction that is taken throughout this paper.
The Common Rule governing human research discusses privacy and confidentiality in ways more suited to biomedical research than to social and behavioral research (hereinafter referred to as social research). More useful definitions of privacy and confidentiality are offered herein. Even with more useful definitions, however, the interpretation of these ideas into an effective protocol sometimes requires kinds of knowledge, experience, and problem-solving skills that are absent from the training of most scientists, students, and IRB members. Hence, it is primarily education and easily accessed information, and not more rules or enforcement, that are needed by IRBs and their clientele.
The meaning of privacy and confidentiality in social research inheres in the culture and particular circumstances of the individual subject,1 the nature and context of the research, and the particular social and political environment in which the research and use of the data occur. Consequently, their definition, as well as the interpretation of requirements to respect privacy and assure confidentiality, is not a trivial or simple matter.
Informed consent is the mechanism through which subjects decide whether or not to allow access to themselves, and through which agreements are made concerning the handling of identifiable data. However, the regulations of human research, as currently written, give little hint of how finely the protocol and informed consent relationship must be crafted in response to the manifold aspects of privacy and confidentiality in social research. Worse, they do not allude to the background of concepts and plans that would underlie such an effective protocol and relationship. Remarkably, some IRBs function quite effectively despite these ambiguities, through wise interpretation by IRB members who are well schooled in ethical problem solving and whose scientific training has provided relevant research competencies. Such a fortunate confluence of education, competency, and effort is not the norm, however. Nor can such outstanding performance reasonably be expected of most IRBs, which are woefully overworked and under-budgeted. An educational program is recommended herein that would provide a foundation for effective ethical problem solving by IRBs and researchers with respect to privacy and confidentiality.
At the core of any ethical problem solving in research is the meshing of a) valid scientific methods with b) relevant ethical considerations such as respect for privacy and assurance of confidentiality, in pursuit of answers to c) nontrivial research questions—with as little compromise as possible in each of these three dimensions. Skilled IRBs can help guide this process.
For example, in the interest of confidentiality and informed consent, parents recruited for a study of child-rearing practices should be warned of the limits of confidentiality (e.g., mandatory reporting of evidence of child abuse). While this might distort the researcher’s random sampling scheme and jeopardize generalizability by eliminating those who decline to participate, it provides a higher level of confidence in the candor of those
N-4
who choose to participate and suggests conducting a parallel study of parents who have been convicted of child abuse.
In response to a problem such as this, the IRB can put an inexperienced researcher in touch with local resources who can counsel the researcher about issues of mandatory reporting. They can remind the researcher to use appropriate skills of rapport and effective communication to ensure that the limits of confidentiality are clearly communicated to potential subjects. A range of research approaches can enable the project to “surround” the research question despite distortion of the random sampling scheme. In short, many institutional resources can be focused on solving this set of confidentiality-related problems. Some IRBs operate at this level of sophistication.
Unfortunately, issues of privacy and confidentiality may also be handled in naïve or bureaucratic ways due to lack of knowledge and relevant problem solving skills. It is not a required role of IRBs to rehabilitate deficient protocols, nor is the knowledge required to do so necessarily inherent in the repertoire of skills of most scientists. Research methodology textbooks remain woefully deficient in this area. This places the IRB in an awkward leadership position and strains the constructive and collegial atmosphere that an effective research organization requires.
In response either to a particular interpretation by the Office for Human Research Protections (OHRP) or on account of their own inexperience, IRBs may respond to avoid one risk and unwittingly create another risk. For example, one irate parent objected to an extremely imprudent survey question about parents (administered to college students). In response, OHRP (then the Office for Protection from Research Risk, or OPRR) announced an interpretation to the effect that when questions are asked of a subject about another person, the consent of that other person must also be obtained (OPRR, 1999). This unfortunate general interpretation led an IRB to require a researcher interviewing parents about their childrearing practices to obtain the assent of their four-year-old children—a meaningless and developmentally inappropriate request. If taken seriously, this interpretation would also lead to the demise of much epidemiological research. An on-line educational program, shared nationally, would enable IRBs and researchers to engage in more effective ethical problem solving and to authoritatively question an OHRP interpretation that did not fit their specific circumstances.
However, even the most capable, competent, and energetic IRB finds it difficult to educate every researcher and student within its domain. Individual researchers (especially students) may not know that their work falls within IRB purview or may decide to avoid his or her IRB because of the belief that it will impose requirements that will render the research impossible. Some privacy-related beliefs on the part of individual researchers (e.g., “You always have to get parental permission”) become urban myths that get disseminated among uninformed researchers and students. A user-friendly, web-based education within institutions would help prevent or dispel such urban myths. It will also create a culture in which ethical problem solving becomes a more integral part of the research curriculum and irresponsible faculty members will find it more difficult to transmit their scofflaw attitudes to their students.
Who needs to be reached through such an educational resource? The social sciences are usually considered to include psychology, sociology, anthropology, political science, and economics. However, for IRB purposes, the point is not which are and are not social sciences but rather who uses social science methods. One must look virtually everywhere within a university, clinic, prison, school, or hospital to find such researchers. An internet information and education approach, indexed and patterned somewhat after the Help feature of word-processing software, would make it easier for administrators to guide researchers and students to the information they need, with little ambiguity or inefficiency. That information could also be incorporated by individual instructors into the curriculum of courses on research methodology.
An adequate resource would introduce all major aspects of respecting privacy and assuring confidentiality and would explain how each is operationalized. It would provide information about all major social research
N-5
methods and present some illustrative cases to show how methods interact with privacy and confidentiality considerations. Each method brings with it specific problems of privacy and confidentiality depending on other related aspects of the research. Each method may be used in a range of contexts, e.g., survey research can be conducted in person, or via mail, phone, or internet. The data or research site and subjects may be shared with other scientists subsequently; the data may be used in secondary analysis and meta-analysis. The subjects may be anyone—students, gang members, prostitutes, business persons, people in therapy, families, infants, professionals, recovering patients, runaway children, kindergartners, or representatives of whatever population the research is focused on. Each method, context, and subject population has many variations, and each brings with it a range of privacy/confidentiality issues to be resolved. Consequently, an awareness of alternative methods and their scientific and ethical implications is vital to effective ethical problem solving.
An important aspect of respect for privacy involves giving people reasonable choices about participation, which is sometimes precluded under traditional ways of designing experiments with random assignment. One alternative is to use a “wait list” design in which the control group also receives the desired treatment—later. People who want to participate in an experiment because they consider the treatment highly desirable can choose to participate with the understanding that they might get the treatment they desire right away or later.
For example, in a randomized trial on the effects of monetary scholarships paid to high-achieving students from impoverished families (Spencer et al., 1999), the monetary payments were delayed for one year for equally eligible students in the control group. As this example illustrates, developing a protocol that respects subjects’ privacy and autonomy requires selecting the method(s), procedures, and context(s) that best meet both scientific and ethical criteria.
No IRB can be expected to embody all of the scientific competencies needed to help craft the best protocols for all research projects. However, an educational resource, including a list of consultants by research topic, can be developed that would make it relatively easy for IRBs and investigators to locate the information they need in order to develop effective protocols. This would remove the IRB from the hostile role of “ethics police” and place it in a more professional and constructive role.
The Difficulty of Defining Privacy and Confidentiality
With the rise of concern for personal privacy, many confusing definitions have emerged, adding to the difficulty of specifying what it means to respect privacy or to avoid invading subjects’ privacy. The difficulty of defining invasion of one’s own privacy is compellingly expressed by Melton (1992, p. 66):
‘I know it when I feel it.’ A gut sense of personal violation may be the tie that binds such disparate events as being subjected to a body search, being the subject of gossip, having one’s mail read, being asked one’s income, or having one’s house entered without permission. It should come as no surprise that such an intensely personal construct is difficult to define.
Even more difficult to fathom, define, understand, or respect is the privacy of other persons situated differently from ourselves with respect to age, ethnicity, locale, socioeconomic status, gender, or the context in which the issue of privacy arises. Privacy is an aspect of respect for persons that can be difficult to translate into respectful behavior in cultures and contexts in which one does not understand the relevant norms and beliefs.
Without a useful definition or theory of privacy to guide them, or at least guidelines to remind them of possible threats to privacy, researchers and IRBs must depend on their own culture-bound notions of what people consider as private. They are left to invoke their personal and idiosyncratic definitions, resulting in a capricious standard of protection. Examples of inappropriate notions of privacy are provided herein, and guidelines are proposed for discovering what is private to others.
N-6
There are many definitions and concepts of privacy and confidentiality, some of which use the two words interchangeably. For purposes of guiding social research, it is important to distinguish clearly between these two concepts in precise terms that a researcher can act upon intelligently in any research context (e.g., internet, workplace, schools, hospitals, families, neighborhoods) and in the diverse ethnic and social cultures where social research is performed.
Privacy
One useful, though simple definition of privacy, following Boruch and Cecil (1979), is as follows:
Privacy refers to persons and to their interest in controlling the access of others to themselves. (Confidentiality refers to data, as discussed subsequently.)
This definition of privacy recognizes that control and autonomy, rather than isolation, are at issue. As
Beauchamp and Childress (1994) explain, rights of privacy are valid claims against unauthorized access; such claims have their basis in the right to authorize or refuse access. Accordingly, the above definition recognizes the vital role of informed consent (properly formulated and administered) in giving subjects control over whether they will allow the researcher access to themselves and to their attitudes, behavior, beliefs, and opinions. It alludes to the two directions of access: a) information that is given to one or rejected by one, e.g., pornography that a researcher wishes to show male subjects to study its effect on subsequent response to scenarios of violence towards women, and b) information one reveals to, or withholds from, others, e.g., a subject’s willingness or unwillingness to disclose personal details about his or her own life.
Informed Consent and Privacy
There are many verbal and nonverbal dimensions of informed consent that impact a subject’s decision of whether or how much to reveal to the researcher. The informed consent statement that fulfills the elements required by the Common Rule provides the objective factual information needed to control access satisfactorily. But there is a nonverbal dimension that is at least as important to individuals as they seek to manage their privacy.
Suppose, for example, that a disheveled researcher administered informed consent without establishing rapport and with body language that is “closed” and hostile. He or she would be regarded as having invaded the subject’s privacy before even uttering a word. Such a researcher’s manner would belie any “respectful” informed consent language. An unconcerned or ritualistic recitation of the informed consent conveys that it is merely a legal maneuver designed to protect the institution—that whatever happens to the hapless subject is of no personal concern to the researcher.
The nature, timing, and delivery of informed consent has important implications for subjects’ sense of privacy. For example, a longitudinal or ethnographic study or any research that involves repeated contact between researcher and subject should treat informed consent as an ongoing process of communication in which the nature of the relationship is repeatedly clarified and discussed in an informal, friendly way. Research in which it is easy for the subject to end the relationship (e.g., a survey that is conducted by mail or phone) should not require a signed consent, as this is unnecessary, inconvenient, and regarded by some to mean that they are making an irrevocable commitment to participate. Moreover, it reduces response rate and distorts random sampling by eliminating the least literate of potential subjects. Worst of all, it risks, unnecessarily, a breach of confidentiality and harm to subjects if a signature is somehow attached to sensitive data that could as well have been gathered anonymously.
N-7
Cultural and Developmental Determiners of Privacy
The boundaries between wanted and unwanted information proffered to us by others, as well as between information we will and will not share, are partially defined by sociocultural values. For example, current efforts to survey people’s safe-sex practices are made difficult because the very mention of some specific sexual practices (quite apart from responding to the survey) is taboo or offensive in some subcultures in the United States, but acceptable in others. On the response side, providing information about one’s sexual practices is acceptable to some respondents and highly offensive to others.
Some ethnic differences in sense of personal privacy are counter-intuitive to most Americans. For example, most Americans consider eye contact a sign of respect and honesty, but ethnic Japanese (especially in Hawaii) consider a researcher who establishes eye contact as disrespectful. Guessing at cultural nuances of privacy is a poor practice for social researchers.
How can researchers learn what is private in cultures and contexts that are foreign to their own personal experience? One solution is to employ research assistants from the same cultural community as the subjects. However, this sometimes risks a breach of confidentiality (disclosure of personal secrets) within a close-knit community. Correspondingly, subjects sometimes disclose more sensitive information to a stranger who has established appropriate rapport and promised confidentiality than to a member of their own group. The optimal solution to this problem rests on judicious selection and training of research assistants—training that is designed to produce the most culturally sensitive behavior possible without any actual or apparent risk to confidentiality within the community.
Everyone wants to have some choice about those with whom they will interact and what the conditions of that interaction will be. Early in life, people learn a variety of ways to avoid or evade unwanted interactions. The researcher who is not mindful of the privacy interests of subjects will be lied to, stood up, or complained about. In contrast, the researcher who understands or respects the privacy interests of subjects may find them overwhelmingly forthcoming, and may even find it difficult to end the session.
The researcher who understands the sociocultural aspects of privacy looks beyond the immediate scientific need to “get data.” He or she does some ethnographic work before designing recruitment and research approaches. By learning about the norms, beliefs, and culture of the subjects, the researcher can then appropriately express respect, establish trust, and create rapport that makes interaction acceptable.
The recruitment of subjects and design of research should be culturally appropriate and should instill sufficient trust that subjects will want to participate candidly. Much of the local ethnography needed to conduct social research effectively cannot be found in textbooks. However, networks of local researchers, educators, and outreach workers can share valuable information about the most appropriate ways to approach members of various cultures. Individual IRBs would do well to add to their educational web site suggestions of useful contact persons (e.g., AIDS outreach workers, social workers, farm agents, public health nurses) and to even sponsor local conferences on culturally sensitive approaches to research and develop proceedings for future use in designing appropriate protocols.
To respect privacy is to let subjects control the access of others to themselves: to provide the conditions under which the researcher’s inquiries are welcome, and to provide adequate opportunity for people to decline to participate. To breach privacy is to violate people’s space, to intrude where not welcome or not trusted, or to seek to control access to people against their wishes—for example, organizational research on employees to which management gives the consent and deceptive research in which the researcher induces the subject to behave in a way the subject would not wish to be observed are invasions of privacy. An educational resource for IRBs and researchers should discuss practices of recruitment, consent, timing of procedures, research methods, and debriefing in terms of whether they respect privacy or are likely to constitute breaches of privacy.
N-8
The definition of privacy developed so far works well with theories borrowed from the social sciences that describe the conditions under which access by others is welcome or aversive. Such theories describe the controls people use to limit the access of others to themselves and the conditions under which those controls are available and likely to be employed.
Theories of Privacy
Various theories of privacy are instructive to researchers and need to be incorporated into the education of researchers and IRB members. The following two are offered as examples.
Laufer and Wolfe (1977) describe how self-ego, environmental, interpersonal, and control-choice factors operate to regulate the experience of privacy. This elegant analytical framework indicates virtually every element one must consider to understand the privacy of another.
The self-ego dimension of privacy refers to the development of autonomy and personal dignity. For young children, being alone is aversive. By middle childhood, time alone is sought to establish a sense of self and autonomy and to nurture new ideas, creating a basis for self-esteem, personal strength, and dignity. Thus, children in middle childhood have a need and right to privacy not found in infants and younger children. Adults continue to need time alone and develop many means of protecting that privacy.
The environmental dimension includes cultural, sociophysical, and life-cycle dimensions. Cultural elements include norms for achieving privacy, e.g., one culture may permit lying while another may permit persons to have private rooms. Sociophysical elements refer to physical settings that offer privacy (e.g., indoor bathrooms, tree houses, automobiles, etc.) Life-cycle elements vary with age, occupation, available technology, and changing sociocultural patterns. The kinds of privacy one establishes at one age, under one set of responsibilities, constraints, and technological aids may be unsatisfactory or unavailable in another stage of one’s life.
The interpersonal dimension refers to how social interaction and information are managed. One’s social setting and its physical characteristics provide options for managing social interaction; physical and social boundaries can be used to control people’s access to one another.
The control/choice dimension develops out of one’s dimensions of self-ego, culture, and environment. Young children have no control over their privacy, except through hiding. Later, they learn to use personal, cultural, and physical resources to control their privacy. Events that would threaten one’s privacy early in the development of these mechanisms are later so easy to control that they are no longer considered a threat to privacy.
Thompson (1982) presents a developmental theory describing how the sense of privacy changes from early childhood through late adolescence, and how youngsters learn to control access by others. Thompson shows that popular ideas about vulnerability decreasing linearly with age are inaccurate: older children are more easily embarrassed, more concerned about personal and informational privacy, and more likely to feel upset if they reveal more than they intended. However, younger children’s sense of privacy is enhanced when their parent is present during a study, while an older child is confident of ability to manage access with the researcher, but wants the parent out of the room.
Theories of this nature would have an important role in any effort to foster greater skill in discovering and understanding the privacy interests and privacy management approaches of a given research population.
Places and Privacy
The concept of privacy is related to the notion of private places that can be used to control access. There are degrees of privacy in relation to places:
N-9
In research, only the first category is clearly without ethical concerns. Observational research or participant observation in private places is especially problematic.
Summary
Individuals, organizations, and cultures all have their own ways of wishing to be perceived, and one’s sense of privacy is intimately related to controlling the way one is perceived. However, researchers are often so focused on getting data that they forget even the norms of their own culture and are still more oblivious to the norms of other cultures. Moreover, researchers typically examine subjects from a critical perspective that may differ from the way their subjects wish to be perceived.
Researchers should be well informed and mindful of the ethnography of their subject population so that their approach is tactful and their critical analysis is objective. Yet, they will still function as outsiders, almost voyeurs, and should compensate for this by carefully masking the identity and location of those whose behavior they critically analyze in scientific publications. In summary, the concepts of privacy offered here do not presume that:
Rather, these concepts lead the researcher to look to theory, methods, and ethnographic information to learn how to respect the interests of a given subject population in controlling the access of others to themselves.
Should IRBs vary the degree of protection of privacy depending on the ethnographic standards of the research population? Clearly, some subgroups have particular concerns about privacy that are not shared by others, and that must be respected for scientific as well as ethical reasons. That is, respect for privacy is not only ethically required, it also encourages subjects to give truthful and unguarded responses. But what about populations that have diminished expectation of privacy in their everyday life, such as celebrities, prisoners, or citizens in a totalitarian state where privacy protections are few and confidentiality is always in doubt? Respect for privacy means giving people the privacy they would like to have, not the invasions of privacy that are regularly imposed on them. For example, a psychologist studying prisoners or workers in a company where the employer disrespects privacy should respect subjects’ privacy even if others do not. The IRB or researcher in doubt about the sense of privacy of a particular population can remove that doubt by asking surrogate subjects drawn from the target population. For more autonomous populations such as celebrities, the matter may be settled through pilot research and informed consent in which persons can participate or decline based on an accurate understanding of what will be asked of them.
N-10
Confidentiality
A useful, though simple definition of privacy, adapted from, and similar to, that developed by Boruch and Cecil (1979) is as follows:
Confidentiality is an extension of the concept of privacy; it refers to data (some identifiable information about a person, such as notes or a videotape of the person) and to agreements about how data are to be handled in keeping with subjects’ interest in controlling the access of others to information about themselves.
This definition provides a clear distinction from privacy, which is vital. For in addition to understanding what is private to given subjects and how that privacy may be respected, the researcher must be able to assure subjects that the access of others to information about themselves will be controlled in a way that is acceptable to them. Every detail of subsequent data management need not be communicated to subjects, though it should be worked out clearly in the protocol. Some matters such as required disclosure of child or elder abuse, plans for possibly sharing identified data with other scientists, or sharing archives of videotaped data should be part of the informed consent, if relevant.
This definition of confidentiality does not presume that:
This definition helps researchers and IRBs to recognize that there are various risks of unintended disclosure, e.g., through snooping by hackers or research assistants, theft by blackmailers, legally mandated disclosure, or careless construction of data tables that permit some readers to deduce the identity of individual subjects. It also reminds one that confidentiality is whatever arrangement about disclosure the researcher and subject agree upon, within the constraints of law and ethics. Confidentiality is more than just a promise or an intention on the part of the researcher. It is an arrangement to use certain techniques that are available for controlling the disclosure of identifiable information. There may be limits to what can be promised or guaranteed, and these must be discovered and stated at the outset.
Kinds of Confidentiality-Assuring Techniques
This definition of confidentiality leads naturally to the immense and growing literature on procedural, methodological, statistical, and legal approaches to assuring the confidentiality of research data. These methods are developed and described in various applied research literatures (e.g., Boruch and Cecil, 1979; Campbell et al., 1972; Jaro, 1989) and discussed subsequently. This definition also leads to recognition of the immense advantages of rendering data anonymous, where feasible.
The intelligent use of confidentiality-assuring techniques depends upon understanding what threats to confidentiality may exist or be perceived by subjects, what legitimate uses of the data may be anticipated including sharing with other scientists and agency audit of the data, and what costs and analytic disadvantages may accompany some of these techniques. There is considerable and ever-growing depth to this body of knowledge. When anticipating gathering of any data, and especially sensitive data, it is important to a) make early plans concerning the confidentiality-assuring techniques that will be used, b) incorporate these appropriately into any consent agreements with subjects and contractual arrangements with subsequent users (including funders who wish to audit the data), and c) include these details in the IRB protocol.
N-11
Emerging Issues of Privacy and Confidentiality
Within the last two decades, data collection and storage practices have been altered radically. New digital media support a wide range of social relationships such that social scientists, their colleagues, and their subjects need not meet face-to-face and may even reside in different parts of the world. The issues of confidentiality that are emerging are more varied and dangerous than current policy makers can easily anticipate. However, there are also hopeful solutions on the horizon. Within another decade or two, issues of confidentiality will be transformed in ways we cannot imagine today. There are now digital communication networks on a global scale and the possibility that hackers with a laptop computer and internet technology could download any electronic data stored on any server anywhere in the world. There are also emerging technologies for protecting communication and personal identity, and there is a whole new cohort of technology-sophisticated privacy activists. New laws that protect data are being developed and tested, and globalization of culture and policy processes is occurring.
These major shifts in technology have already begun to produce an immense literature on the accompanying threats and safeguards to privacy and confidentiality (e.g., Agre and Rotenberg, 1998). Much of this literature is esoteric. It needs to be tracked by specialists who are concerned with the confidentiality of social research data and translated into a body of knowledge that is useful to researchers, research administrators, and IRBs.
One relatively old, low-technology form of data collection with which most researchers are familiar illustrates a few of the kinds of problems that will arise with the use of high-technology data gathering. When data are gathered on videotape, the distinction between persons and their data is blurred. If one observes another on a video monitor, in real time, is this direct observation or data? If the material that is seen in real time on the monitor is taped, does the same image then become data? Can anonymity be complete if there is any visually recognizable aspect of the persons’ identity? Risk of deductive disclosure (the possibility that someone who already knows the individual will see the tape and correctly deduce that he or she is the subject depicted) takes on new meaning with videotaped data. When data are in videotaped form, special attention must be given to consent and avoidance of incriminating or unnecessarily sensitive material. The researcher should disclose the projects’ plans for storage, sharing, and disposal of the tapes. Subjects should have the option of requesting tape erasure if they decide that they have just said or done something they do not want to be retained on tape.
Summary
The proposed definitions emphasize that privacy and confidentiality are quintessentially behavioral and social phenomena and appropriate topics of scholarship and investigation. Curiously, however, the federal regulations of human research and requirements of IRB review have tended to be treated by researchers as leading to last-minute paper work rather than to the creative use of current privacy and confidentiality-assuring techniques, or the development of new theory or methodology that would make for more ethical human research. Thus, a main principle developed in the Belmont Report, that research should be performed by competent scientists who apply ethical principles intelligently based on sound scientific knowledge and research skills, seems to be lost on many scientists and IRBs. Part of this problem may be due to the failure to frame the concepts of privacy and confidentiality in ways that would invite innovation and investigation.
Regulations and Statutes
A wide range of federal and state laws, as well as the Common Rule and other federal regulations, concern privacy and confidentiality in social research.
The Common Rule (Subpart A of 45 CFR 46)
The federal policy for the protection of human subjects, which formerly pertained only to research funded by the Department of Health and Human Services (DHHS) (45 CFR 46 Subpart A) has now become the Common
N-12
Rule and has been incorporated into the regulatory structure of 17 federal agencies, 8 of which have additional human subject protections beyond the Common Rule, most of which do not relate directly to privacy and confidentiality.
These agencies, their regulations that contain the Common Rule, and their additional regulations related to privacy and confidentiality are as follows: Housing and Urban Development (24 CFR 60), Justice (28 CFR 46 with additional protections in 28 CFR 512), Transportation (49 CFR 11), Veterans Affairs (38 CFR 16 with additional protections in 38 CFR 17.85, M-3, Part 1, Chapters 9 and 15), Consumer Product Safety (16 CFR 1028), Environmental Protection (40, CFR 26), International Development (11 CFR 225), NASA (14 CFR 1230), NSF (46 CFR 690), Agriculture (7 CFR 16), Commerce (15 CFR 27), Defense (32 CFR 219, plus 12 additional regulatory protections), Education (with extensive additional protections to privacy and confidentiality as noted below), Energy (10 CFR 745), Health and Human Services (45 CFR 46 Subpart A), Social Security (P.I. 103-296), and CIA (Executive Order 12333); the last three agencies also employ Subparts B, C, and D of 45 CFR 46.
The Common Rule specifically requires that informed consent include a statement about how confidentiality will be maintained, but it leaves to the IRB and the researcher the subtle matter of understanding what confidentiality is and how it relates to privacy. Moreover, the Common Rule does not define privacy, but defines private information as follows:
Private information includes information about behavior that occurs in a context in which an individual can reasonably expect that no observation or recording is taking place, and information which has been provided for specific purposes by an individual and which the individual can reasonably expect will not be made public (for example, a medical record) (45 CFR 46.102(f)(2)).
This definition of private information confuses it with confidentiality and fails to convey the notions of personal privacy that are important to ethical research. It also implies that everyone has the same concerns about others’ access to themselves and to identifiable data about themselves.
Based upon this confusing set of definitions, 45 CFR 46.111 (Subpart A), the criteria for IRB approval of research, states:
(7) When appropriate, there are adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data.
Unfortunately, this requirement assumes a level of sophisticated knowledge concerning privacy and confidentiality that many IRBs and researchers do not possess.
Subparts B, C, and D of 45 CFR 46
Subpart B contains protections in research, development, and related activities involving fetuses, pregnant women, and human in vitro fertilization. Its relationship to privacy arises in connection with 46.209(d) which concerns consent of the mother and conditions under which the father’s consent is not required. Being biomedical in focus, it is not within the purview of this paper.
Subpart C pertains to research on prisoners that is conducted or supported by DHHS. It defines “prisoners” quite loosely to refer to any individual involuntarily confined or detained in a penal institution. This encompasses those sentenced under a criminal or civil statute, detained in other facilities by virtue of statutes or commitment procedures which provide alternatives to criminal prosecution or incarceration in a penal institution, and any individual detained pending arraignment, trial, or sentencing. (In contrast, the Department of Justice provides a more comprehensive set of protections, as outlined below, and pertains only to research conducted within the Bureau of Prisons.) Subpart C of the DHHS regulations of human research is responsive to
N-13
privacy/confidentiality issues in that it recognizes the limits to prisoners’ voluntary and uncoerced decision whether to participate in research. It limits the amount of risk to which prisoners may be exposed. It requires an IRB containing at least one prisoner or a prisoner representative and a majority of the members having no association with the prison apart from their membership on the IRB.
A shortcoming of Subpart C is its failure to address the special problems concerning research on incarcerated juveniles who deserve special attention due to issues of distrust, incomprehension, and their often unusual (strained or nonexistent) relationship with their parents. Neither Subpart C nor the Justice Department regulations described below (28 CFR 512.11) consider that issue. Moreover, Subpart D, which deals with research on children, does not offer special guidance regarding research on incarcerated juveniles.
Subpart D discusses additional DHHS protections for children involved as subjects in research. It is sensitive to children’s personal privacy interests in requiring the child’s active assent when assent would be meaningful, as well as parental permission, with either party having veto power. Unlike research on adults, Subpart D requires IRB approval when the research involves surveys, interviews, and observation of public behavior when the investigator participates in the activities being observed. It sets forth limits to the amount of risk to which children may be exposed in research, and places strict requirements for consent and benefit when any more than minimal risk is involved except where there is no other way to benefit the subject or to study a serious problem affecting children in general. Subpart D recognizes that there are contexts or circumstances in which parental permission is not a reasonable requirement to protect the subjects (e.g., in the case of neglected or abused children), and that the IRB needs to designate other ways of protecting the interests of such children. Procedures for selecting guardians or advocates who function in loco parentis are discussed.
Additional Protections: Department of Education
The Department of Education provides additional protections regarding respect for privacy and assurance of confidentiality under 34 CFR 99. For example, Part 99 concerns “Family Educational Rights and Privacy” and sets out requirements for the protection of privacy of parents and students under section 444 of the General Education Provisions Act, known as the Family Educational Rights and Privacy Act of 1974 (FERPA). FERPA and other statutes governing research in educational settings are discussed below under “Federal Statutes.”
Additional Protections: Department of Justice
The Department of Justice has developed an excellent set of additional protections of privacy and confidentiality pertaining to prisoners (28 CFR 512). These are most noteworthy in their emphasis on a) creating an impermeable firewall between research data and prison administration, b) requiring an IRB membership of at least one prisoner and a majority who are not prison personnel, and c) giving prisoners a high degree of control over their identifiable data.
Protections specifically pertaining to privacy and confidentiality are many: The researcher must not provide research information which identifies a subject to any person without that subject’s prior written consent to release of the information. Identifiable data cannot be admitted as evidence or used for any purpose in any action without the written consent of the person to whom the data pertains. Except for computerized data kept at a Department of Justice site, records containing identifiable data may not be stored in, or introduced into, an electronic retrieval system.
Access to Bureau of Prison records by researchers is limited by law. No one conducting research may have access to records relating to the subject which are necessary to the purpose of the research without the subject’s consent. A nonemployee of the Bureau of Prisons is limited in access to information available under the Freedom of Information Act (5 USC 552). A nonemployee of the Bureau may receive records in a form not individually identifiable when advance adequate written assurance is given that the record will be used solely as a statistical research or reporting record.
N-14
Federal Statutes
The rise of concern about the amount of governmental record keeping and the potential for abuse through inappropriate and unauthorized use of such records led to enactment of the Privacy Act of 1974. This omnibus law governs record keeping and record use in all federal agencies. (See in this volume paper by Goldman and Choy for discussion of the Privacy Act of 1974.) The Privacy Act authorized the examination of privacy issues in diverse environments via the Privacy Protection Study Commission, whose findings have continued to influence concern to balance the interests of individuals, record keeping institutions, and society as a whole. One outcome has been increased attention to the individual’s role in controlling information about himself. In keeping with the overall philosophy of the Privacy Act of 1974, and as an alternative to the omnibus approach to regulation of record keeping taken by the Privacy Act, various laws have been passed that are tailored specifically to privacy in educational settings and to research in educational settings. The Family Educational Rights and Privacy Act of 1974 is designed to protect educational records from disclosure without consent of parents or students over 18. The Protection of Pupil Rights Amendment (PPRA) gives parents the right to inspect innovative curriculum materials and requires parental consent for students to participate in sensitive research. An interesting aspect of FERPA and PPRA is that, like the federal regulations of human research, they allow the participating institutions to develop and implement local substantive and procedural requirements that meet minimum requirements established by law. We turn now to FERPA and PPRA, and then to the National Center for Educational Statistics (NCES) Confidentiality Statute that protects the confidentiality of identifiable data collected by the NCES. Finally, we discuss legislation that protects identifiable data from subpoena.
The Family Educational Rights and Privacy Act of 1974 (FERPA)
FERPA (also commonly known as the Buckley Amendment) states that “An educational agency or institution shall obtain the written consent of the parent of a student or of the eligible student (if 18 or older) before disclosing personally identifiable information from educational records of a student, other than the directory information.” Directory information includes information contained in an educational record of a student which would not generally be considered harmful or an invasion of privacy if disclosed. It includes, but is not limited to, the student’s name, address, telephone listing, date and place of birth, major field of study, participation in officially recognized activities and sports, weight and height of members of athletic teams, dates of attendance, degrees and awards received, and the most recent previous educational agency or institution attended.
Educational records refers to records directly related to a student and that are maintained by an educational agency or institution or by a party acting for the agency or institution. Educational records does not refer to records of instructional, supervisory, and administrative and educational personnel that are kept in the sole possession of the maker of the record and are not generally accessible to others. It also does not include information gathered after the student has ceased attending that institution.
FERPA applies to educational agencies or institutions that receive federal funds under any program administered by the Department of Education. This includes all public elementary and secondary schools and virtually all post-secondary institutions.
In contrast to the Common Rule, FERPA provides post-violation remedy rather than prior approval of a research protocol. However, IRBs typically impose prior approval based on FERPA requirements. Some degree of IRB discretion exists at the fringes of FERPA’s definitions, but generally the Buckley amendment prevents a researcher from inspecting educational records without permission of the parent or of the student (after age 18). The Department of Education reports that while it receives quite a few complaints from parents, most contain no specific allegations of fact that would indicate that a violation had actually occurred. Consequently, most of the effect of FERPA on research arises from restrictions that school administrators and IRBs place on researchers.
N-15
The Protection of Pupil Rights Amendment (PPRA)
PPRA is also commonly known as the Hatch Amendment. It affects research in various ways by giving parents certain rights regarding Department of Education-funded activities. When first introduced in 1974, it gave parents the right to inspect instructional material used in connection with any research designed to explore or develop new or unproven teaching methods or techniques. A later addition in 1978 (the Hatch Amendment) requires parental permission for certain types of surveys administered to minor students that seek information about the student’s private life, specifically children’s attitudes, beliefs, or habits in the following seven areas: 1) political affiliation; 2) mental and psychological problems potentially embarrassing to the student and his/her family; 3) sexual behavior and attitudes; 4) illegal, antisocial, self-incriminating, and demeaning behavior; 5) critical appraisals of other individuals with whom respondents have close family relationships; 6) legally recognized privileged or analogous relationships such as those of lawyers, physicians, and ministers; and 7) income (other than required by law to establish eligibility for a program).
PPRA was further amended in 1994 (Grassley Amendment) to remove the terms “psychiatric and psychological examination, testing and treatment,” and to clarify PPRA to refer to any survey, analysis, or evaluation that elicits information in the above seven areas.
Like FERPA, PPRA provides post-violation remedy rather than prior approval of a research protocol. However, IRBs typically impose prior review following PPRA requirements, irrespective of whether Department of Education funding is sought.
Issues Concerning Regulation of Research on School Children
School children are a convenient captive subject pool, especially in the case of schools located near universities. Laws providing special protections for school children have been enacted largely because overzealous, insensitive, or culturally inappropriate researchers have offended parents. These laws make it easy for school administrators and IRBs to rein in intrusive research on children. Thus, they are highly effective, even though the Department of Education rarely acts on them.
Signed Parental Permission
Unfortunately, however, there is a problem with FERPA and PPRA that begs for resolution. Among the children who most need to be served through research and development programs are those whose parents are unlikely to be able to provide their written permission. Parents who are illiterate, do not speak English, are irresponsible or largely absent from the home, or are unfamiliar with American culture are unlikely to respond to a request for written parental permission. The requirement of a parent’s signature invites forgery of parents’ signatures by youngsters who want to participate with their friends, and who may be accustomed to signing for illiterate or non-English speaking parents in any case. It also invites poor solutions such as having a youngster do the translation of the consent statement, mentioning such matters as confidentiality to a parent who does not understand the concept. Most damaging of all, it results in invalid sampling of populations most in need of the kinds of understanding and support that research can help to provide.
Parental permission for school-based research often is handled poorly unless the IRB helps by providing useful, well-tested models of good communication—model letters, town meetings, use of adult community members trained to communicate effectively with their peers, tape recorded messages, and so on. Once the IRB has established such mechanisms, their adaptation to new situations may be relatively easy. However, signed parental permission may remain elusive in some populations even though the parents would not object to their child’s participation. It may be appropriate for legislators to consider whether there should be exceptional circumstances (e.g., when parents are functionally illiterate) in which the usual requirements for written parental consent can be modified and so that the informed consent communication process can be conducted in a way that is more conducive to comprehension, such as by a community leader or gatekeeper.
N-16
What Research Is Covered by FERPA and PPRA?
While the regulations and statutes reviewed above may pertain only to research that is intended to provide generalizable knowledge, intended to be published, or funded by a given agency, in fact most IRBs act on their prerogative to impose more stringent requirements. For example, most IRBs review master’s thesis research which is not funded, does not produce generalizable findings (perhaps merely assesses what clients in some kind of treatment program think of the program), and may not be done with publication as a goal. Arguably, some of these projects need not be reviewed at all. However in most contexts such a student research project that involved school children would be subjected to the same stringent requirements (e.g., FERPA and PPRA) as would a project funded by the Department of Education. The willingness of IRBs to apply FERPA and PPRA requirements, even in cases when not federally mandated, mirrors societal and school concerns for family privacy. It expresses respect for local schools and the standards they would also be likely to apply and teaches researchers to do likewise. Moreover, as a practical matter, it is awkward for an IRB to impose stricter regulations on similar projects that happen to vary with respect to funding.
Most IRBs act to protect human research subjects, not simply to follow the letter of the law. Some IRBs describe their more stringent requirements in their single or multiple assurances. Other IRBs, at their lawyers’ insistence, describe their requirements as the minimum requirements set forth under the law, but, as an internal policy, impose a broader and more stringent set of requirements. However, with ever-increasing workloads, IRBs are increasingly tempted to discontinue such extra responsibilities as reviewing proposals that do not even qualify as “research” and imposing strict standards such as FERPA and PPRA when this may make the research much more difficult to carry out and evoke complaints from researchers. Thus, in their laudable attempts to educate researchers and to respect family privacy in research on school children, IRBs are placing themselves in somewhat of a no-win situation by requiring, with some research populations, a standard that may be very difficult to meet and that is not federally mandated. (These observations are based on many years of general observation, as well as on a recent discussion on McWirb.)
The National Center for Educational Statistics (NCES) Confidentiality Statute
The NCES Confidentiality Statute was enacted in 1988 to protect research subjects in several ways, as follows:
Legal Protections of Data
Statutory protection of research data enables researchers to arrange to assure the confidentiality of research records on identifiable individuals from subpoena. Subpoena of social research data is rare. However, if vulnerable data could not be protected from subpoena, there would be a chilling effect, especially on criminological and delinquency research.
N-17
Certificates of Confidentiality
The Public Health Service Act (PHSA) was amended (1970) authorizing the researcher’s withholding of information concerning the identity of people who are subjects of research on use and effect of drugs. This withholding authority occurs through the issuance of certificates of confidentiality by the Secretary of the DHHS. A 1988 amendment broadened its scope to include mental health, biomedical, clinical, behavioral, and social research. Under this amendment, the Secretary of DHHS may
Authorize persons engaged in biomedical, behavioral, clinical, or other research (including research on mental health, including research on the use and effect of alcohol and other psychoactive drugs), to protect the privacy of individuals who are the subject of such research by withholding from all persons not connected with the conduct of such research the names or other identifying characteristics of such individuals. Persons so authorized to protect the privacy of such individuals may not be compelled in any federal, state, or local civil, criminal, administrative, legislative, or other other proceedings to identify such individuals (42 USC 242a(b)(1989)).
Various institutes within DHHS are authorized to issue certificates. Since 1993, certificates have become obtainable for research that is not federally funded. DHHS regards a certificate’s protection to supercede state law; this position has been challenged and upheld in the New York Court of Appeals (People v. Newman 32 N.Y.2d 379, 298 N.E.2d 651, 345 N.Y.S.2d 502, 1973) (Boikess, 2000).
A certificate does not protect identifiable data of “nonsubjects,” that is, other people about whom the subject provides information to the researcher, a point which researchers may fail to clarify in the informed consent. The certificates only protect against compelled disclosure of subjects’ names or other identifiers, coupled with their data. It does not protect a subject who voluntarily consents to disclosure of his or her research record, nor preclude a researcher from reporting the identity of subjects who disclose intentions to harm themselves or others. Moreover, the language of PHSA is rather imprecise, which gives rise to uncertainty. It offers protection to “names and other identifying characteristics,” but the data of a known subject may not necessarily be protected. Melton (1992, p. 81) provides an example of this possible loophole:
[I]n one of my own studies, all of the children in a particular county who are involved in criminal child abuse prosecutions are invited to participate. Knowing that fact, a defense attorney might seek the data of a particular child (not the names of participants) as a fishing expedition for information intended to impeach the child’s testimony. A literal interpretation of the statute would suggest that the subpoena might be enforceable if the data could be shown in some way to be relevant to the proceeding. Although it is also possible—perhaps even probable—that a court would interpret the statute more broadly in keeping with congressional intent, the uncertainty prevents unequivocal offers of confidentiality to participants and, therefore, should be eliminated by a technical amendment.
It is also unclear whether child abuse reporting laws are abrogated by certificates of confidentiality. Is such reporting a “legal proceeding” that cannot be mandated under a certificate of confidentiality? Finally, the certificate of confidentiality must be requested in advance of each research undertaking. Some researchers are unfamiliar with this protection or lack the diligence to make the request. Moreover, subpoenas typically occur for reasons unrelated to the study itself and therefore are not reasonably foreseeable by either the subjects or the investigator. In short, the protections offered by certificates of confidentiality may be unavailable when needed. It is perhaps unrealistic to expect that legislation will be enacted in the near future that would make all research data privileged information. However, it is reasonable to urge that the language of
N-18
the PHSA be clarified to indicate exactly what is and is not covered. It is also appropriate that IRBs be provided guidance as to when certificates of confidentiality should be considered. Given that the Secretary of DHHS may issue certificates for “biomedical, behavioral, clinical or other research,” there apparently are no reasonable grounds for refusing a certificate prior to the time that data collection is initiated, unless perhaps it were obvious that some kind of nonresearch work was being recast as research to obtain a privilege against subpoena.
Placing Data in a Foreign Country and Laws Governing Foreign Discovery2
Many researchers assume that sending confidential data to a foreign country (e.g., to a colleague in Canada) will protect data from subpoena. However, the relevant laws are complex, offering only a deterrent from subpoena, not a guarantee of protection. The Federal Rules of Civil Procedure govern the procedures for discovery, including foreign discovery, in federal cases. Rule 26(b) states that parties may obtain discovery of anything that is relevant, not privileged, and admissible or “reasonably calculated to lead to the discovery of admissible evidence.” Rule 34 states:
| (a) |
Scope. Any party may serve on any other party a request (1) to produce and permit the party making the request (2) to inspect and copy, any designated documents, or (3) to inspect and copy, test or sample any tangible things which constitute or contain matters within the scope of rule 26(b) and which are in the possession, custody, or control of the party upon whom the request is served.… |
| (c) |
Persons Not Parties. A person not a party to the action may be compelled to produce documents and things or to submit to an inspection. |
The courts cannot compel a party to produce data if the party does not have “possession, custody or control” of the documents, but it is unclear what constitutes “control” in the kinds of situations discussed here. If a researcher sends data out of the country for the express purpose of preventing subpoena, would this qualify as loss of control in the eyes of a court? Jason Gilbert (2000), a legal intern at the Federal Judicial Center, offers the following analysis of this question:
While the courts seem to have settled on defining control as when a party has a legal right to obtain something, questions remain for the researcher seeking to give up control of research data to a foreign colleague in an attempt to protect it from being disclosed. Legal rights to possession can come from a variety of sources, particularly when one is considering intellectual property such as research data. If a researcher were to create a set of data, when exactly would he or she no longer have a legal right to that set of data? What if the researcher gave one part of the data to a colleague? What if the researcher only gave up a small “key” to the data that allowed the individuals who participated in the study to be identified? What if the researcher gave part, or even all, of the data to a colleague but still continued to collaborate with that colleague to perform analysis on the data even though it was not in the researcher’s possession? Would that researcher still have a legal right to get back what he or she had surrendered? While the concept of giving away the legal right of possession is relatively straightforward, the mechanics of how exactly a researcher can give away the legal right to possess his own data (particularly if one does not allow for a sale or some type of contract) remains unclear.
Gilbert also reminds us of some other implications of “loss of control” of data: 1) Transfer of data out of the country would mean loss of all electronic or hard copies in the researcher’s possession. 2) Transfer of data must never be done after a subpoena has been received. Even if it is done as a safeguard beforehand, the researcher may still be found to have acted not in good faith and be cited for contempt of court. 3) If the research is done under a contract requiring that the researcher maintain control of the data, relinquishing control to a foreign colleague would constitute a breach of that contract. 4) The researcher’s professional code of ethics or the requirements of a future journal editor may require that the researcher maintain control of the data.
N-19
If the researcher can be said to have “lost control” of data by sending it to a colleague in a foreign country, the researcher then puts that colleague at risk of having to respond to a request for the data and having to seek legal means of protecting confidentiality. However, the rules and procedures of foreign discovery are so complex as to deter discovery. If the person who controls the subpoenaed information resides in a foreign country and is not a national or resident of the United States, the party seeking production must follow certain procedures for foreign production. The United States has ratified various treaties concerning the obtaining of evidence from foreign countries, each having its own procedures. Discovery in a foreign country is a lengthy process. It involves the sending of a formal “letter of request” by the court where the action is pending to a court in the foreign country, requesting that court to take a deposition or request documents of the person in possession of the desired information. There are various diplomatic and legal approaches to delivering such a request and accomplishing the discovery. These are time consuming, difficult, and costly and may make discovery of the information too unattractive to pursue.
A further protection may come from the courts themselves. Over the years judges have shown sensitivity to researchers’ need to protect sensitive data from disclosure. Their concern has been not so much to protect individual subjects as it has been to protect researchers who promised confidentiality and to prevent the chilling effects that excessive subpoena power would have on research in general. However, the decision to quash a subpoena that would require disclosure of confidential research data is left to the discretion of a judge who must balance conflicting interests.
Freedom of Information Act (FOIA)
The FOIA concerns the responsibility of government agencies to maintain, organize, and disclose agency records on request by members of the public. There has been much concern about whether this pertains to identifiable research data (see, for example, Morris, Sales and Berman, 1981). Fraud in government sponsored research has stimulated interest in full disclosure of research data. The Supreme Court, in Forsham v. Harris (445 US 1699) (1979), held that data developed by independent researchers funded by a government agency need not be disclosed. However, Congress recently passed the “Shelby Amendment” (Public Law No. 105-277 (1999)) requiring federal agencies to make available via FOIA request at least some raw research data. The Shelby Amendment pertains to researchers in institutions of higher education, hospitals, and other nonprofit organizations. Research data are defined as “the recorded factual material commonly accepted in the scientific community as necessary to validate research findings, but not preliminary analyses, drafts of scientific papers, plans for future research, peer reviews, or communications with colleagues” (Office of Management and Budget, 1999). Moreover, research data do not include materials necessary to be held confidential by a researcher until they are published or similar information which is protected under the law, or information that could be used to identify a particular research subject.
The Shelby Amendment appears to protect research data, but some precautions should be kept in mind. The pressure on the federal government to ensure the integrity of critical research that it sponsors is likely to remain high. Researchers may one day be required to release raw, identifiable data to the sponsoring agency, and those data may be vulnerable to FOIA disclosure. Moreover, the Shelby Amendment pertains only to research grants to nonprofit organizations. The legal standing of research performed by for-profit organizations remains unclear. In the case of research contracts that require access by the sponsoring agency, it is sometimes possible for researchers to guard against such breach of confidentiality by requesting that audits be performed at the research site and not transferred to a federal agency where they might be obtained by some other party under the FOIA. It seems unlikely that most IRBs would be sensitive to these risks of disclosure through FOIA and be prepared to advise the researcher about possible future FOIA threats to confidentiality.
N-20
State Laws
The Common Rule does not diminish any protections offered by state laws, nor may state law diminish any protections of federal regulations. This interplay of state and federal requirements of human research is set forth elegantly in the commissioned paper “Oversight of Human Subject Research: The Role of the States” (Schwartz, 2000). While the Schwartz paper focuses primarily on medically related research, the sections on the consent of minors to research participation, the certificate of confidentiality procedure, and the conclusion are especially pertinent to this paper as well. Of particular importance are Schwartz’ points about the state-to-state variability of laws and unpredictability of court decisions, the desirability of creating a DHHS clearinghouse on state regulation of research, and the desirability of seeking greater uniformity of laws governing human research. The fact that many multisite research projects cut across state boundaries increases the importance of these issues. In any event, if NBAC recommends the development of a web-based information and education program for IRBs and researchers, such a project would be enhanced if Schwartz’ recommendations were also acted upon.
Mandatory Reporting Laws
State laws relevant to privacy and confidentiality in social research include those that mandate reporting and hence require that the informed consent state the limitation to confidentiality that the mandate implies. All states have mandatory reporting of evidence from identifiable subjects of child abuse or neglect and many require reporting of elder abuse or neglect. These state laws are in response to the federal Child Abuse Prevention and Treatment Act of 1974, which required that child protective services be established and that states mandate reporting laws. By 1978, state laws were in place, along with federally reimbursable child protective services. The history of the literature on child protection and the evolution of these laws may be found in Levine and Levine (1993).
Who must report and to whom? In most states, it is only helping professionals (e.g., teachers, therapists, physicians, nurses, social workers), not necessarily the typical researcher, who must report abuse, neglect, or intention to harm. Researchers might claim that they are not helping professionals, and not bound by the mandate. However, that might not be a winning argument in court. Most researchers are also teachers (university professors) and may be perceived by troubled subjects as an understanding professional to whom one might reach out for help. Moreover, IRBs would not permit researchers to ignore reportable evidence, and hence would require that they include in their informed consent a statement such as the following:
What is discussed during our session will be kept confidential with two exceptions: I am compelled by law to inform an appropriate other person if I hear and believe that you are in danger of hurting yourself or someone else, or if there is reasonable suspicion that a child, elder or dependent adult has been abused. [This statement was adapted from a statement developed by David H. Ruja and is discussed in Gil (1982).]
The same sort of warning must appear in the parental permission for research on one’s child. Such a warning is certain to muddle the sampling efforts and reduce candor in research on family processes.
The exact wording of reporting laws varies from state to state, though each state basically requires helping professionals to report any cases in which there is reason to believe or suspect child abuse—past or present.3 The variation in wording does not impact helping professionals, researchers (or IRBs) nearly as much as does the vagueness of every state’s laws. It is not clear whether “reason to believe” refers to a clinical hunch or to firm evidence, nor do these laws define what constitutes abuse. This leaves researchers to consider cultural differences, e.g., in the harshness of childrearing practices, and to weigh these against the possibility that the legal bureaucracy may be more harmful to the child or elder than are their seemingly abusive relatives. The
N-21
difficulties of defining abuse are many: Estimates of the amount of child abuse run from 1 percent to 30 percent of the U.S. child populations depending on one’s definition (Weis, 1989). How is the act perceived by the child—as done to teach an important lesson (Corbin, 1987), to cure a disease (Gray and Cosgrove, 1985), or out of malice? Thus, added to the costs of breaching confidentiality is the possibility that both the “victim” and the “perpetrators” will be wronged. Reporting laws vary from state to state with respect to how the professional learns about the suspected abuse. Some state statutes are limited to the child seen by the professional, while in other states a report is required even if the professional learns of it through a third party. Most statutes require the reporting professional to testify in court proceedings, include a criminal penalty for failure to report, and permit civil action against a professional whose failure to report was followed by injury to the child. However, all statutes provide immunity from a suit when a report made in good faith turns out to be unfounded (Levine, 1992).
What is ethically responsible research behavior with respect to reporting? Should the researcher stop and warn the subject who starts to mention abuse? Should the researcher listen to what is reported and follow the law…or ignore the law? Should the researcher actively seek and report evidence of abuse and neglect? How much discretion should the researcher use in deciding what should trigger reporting, in relation to the likely outcomes of reporting for the researcher, the project, the institutions involved, the child, the parents, and so on? How should the likelihood of inept handling by the Child Protective Services influence this decision?
An inexpensive, simple, safe, and legally acceptable way to study child abuse is through retrospective study of reported cases. This allows the researcher to trace abuse backward in time from its discovery to its apparent antecedents without accompanying reporting requirements. However, comparison of retrospective versus prospective research approaches on other problems of human behavior show that this is unlikely to lead to valid and useful findings. By proceeding in the opposite direction (selecting a random sample of children and collecting repeated-measure multivariate data with appropriate controls) one is likely to find a different set of conditions associated with emerging cases of abuse or neglect (Weis, 1989; Sieber, 1994). Given the popular media interest in the topic of child abuse, it is doubly crucial that scientists report valid findings. But the legal barriers to such an approach make it unworkable.
This is an area in which IRBs and researchers need wise guidance. Some IRBs may not recognize when there is risk of uncovering evidence of child abuse. Or if risk is recognized, the ambiguity of state laws concerning reporting can lead to capricious IRB decisions such as rejecting the protocol out of hand or suggesting poor solutions. If the IRB does not have a knowledgeable clinician among its members, it should call upon such a person for advice as needed. Clinically trained practitioners know how to interpret verbal or behavioral communications, and are able to determine the appropriate action to take. They probably are acquainted with the Child Protective Services agency in their area and with the strengths and weaknesses of its professional staff. They will know how to report suspected abuse in a way that maximizes the likelihood of a beneficial outcome. Researchers who do not have clinical training and who plan research on subjects who might be at risk of harming themselves or others or of being harmed need to plan ahead. They and their IRB should arrange to have access to a licensed health care practitioner in advance and have a plan for responding to events that seem indicative of relevant harms perpetrated or likely to occur.
Since most IRBs frequently review protocols for research that might happen upon evidence of abuse, most IRBs should arrange permanent institutional resources to advise and support researchers in this area in their decision-making. Without a trained clinician to advise on what constitutes “reasonable evidence,” a risk-averse researcher or IRB may over-report to protect themselves from possible prosecution. Both the IRB and the researcher need to be clear that their duty is to make a considered decision in consultation with others qualified to advise. It is not their duty to jump to conclusions and report without consultation or without good advice on the agency to which they should report. IRBs and researchers would also benefit from having carefully
N-22
developed guidelines concerning the duty to report. The guidelines should be tailored to the specific state and local situation, and to the particular institutional resources available for consultation.
In the short run, it is important that investigators at risk of discovering abuse understand the manifold significance of warning respondents of their duty to report. Federal regulations regarding confidentiality require that subjects be warned of mandatory reporting requirements, and researchers must be ready to respond appropriately to signs of abuse. Realistically, however, this requirement protects researchers, primarily, and not abused children or elders; worse, it hinders efforts to understand the dynamics of abuse. The message that researchers are required to deliver does not evoke appreciation that society cares about abuse; rather it may be interpreted as something like: “If I discuss (such and such) they’re going to put me in jail and take my kid away from me.” Such a message skews the sample by eliminating those subjects who have committed abuse or eliminating their candid admission of so doing.
In the long run, it might benefit society if reporting requirements for funded research on family processes deemed of great national importance could be altered. For example, the modified requirement might mandate training in effective parenting or elder care with follow-up supervision and built-in provision for such training and supervision. A warning to this effect might produce better outcomes for science, society, and the families involved.
Spreading of Legal Principles from State to State
Beyond these somewhat uniform mandatory reporting state laws, the task of informing oneself about the possibly relevant laws in any given state is daunting. Moreover, some high-profile legal principles “spread” from state to state. The case of Tarasoff v. UC Regents is instructive. A UC Berkeley graduate student, Prosenjit Poddar, revealed to a campus psychologist his pathological intent to kill Tatiana Tarasoff, who had spurned his affections. The psychiatrist notified the police, who found the man rational. Poddar understandably did not return to therapy, and stabbed Tarasoff to death. Through a series of appeals, the Tarasoff family persuaded the California Supreme Court (1976) that professionals have a duty to intervene effectively in such cases.
Depending upon the case, this might mean warning the intended victim, notifying authorities, or securing an involuntary commitment. Although some therapists and researchers consider this an unacceptable infringement on their duty to hold professional information confidential, the Tarasoff law in California holds that there is a duty to intervene effectively when the subject of therapy (including those in research on the therapeutic process) reveals an intent to harm another. The Tarasoff law is now widely embraced in other states, in one form or another. Even if one does not live in a state that has a “Tarasoff law,” it is reasonable to consider whether victims or their families might seek, as the Tarasoff family did successfully, to apply the Tarasoff principle if a subject indicates intent to harm and then commits a violent act.
N-23
Threats to Privacy and Confidentiality
Threats to privacy of subjects and confidentiality of research data may be viewed from the perspective of subjects, researchers, or IRBs.
Perspective of Subjects
Viewed from the perspective of subjects, one might begin with the specific harms that may occur to them and work backward to the source and nature of those harms. The basic harms and some examples of sources are shown in the following table:
Harm Example
Inconvenience
Psychological
A bothersome intrusion, e.g., phone surveys conducted at dinnertime. A decision to lie due to mistrust of researcher’s promise of confidentiality.
Stress, abhorrence, or embarrassment, personal harm from invasion of privacy; e.g., subjects view pornographic pictures and suffer self-blame, loss of dignity and self-respect, and loss of their sense of
or control of personal boundaries. Emotional Harm
Worry that responses will not be kept confidential
Note: Psychological or emotional harm may arise even if the researcher has taken appropriate steps to prevent harm but has not adequately assured the subject of this. The researcher has a dual duty— to prevent risk of harm and to effectively assure subjects that this has been done. In cross-cultural contexts this requires extra efforts to communicate in a way that is understandable and believable.
| Physical | A battered wife is observed by her husband while being interviewed. |
| Harm | Identifiable data from research on gay students is stolen by “gay bashers.” |
| Social | The above example of research on gay students also illustrates how the mere presence of the identified |
| Harm | researcher or the disclosure of identifiable data could lead to stigma, rejection, or other forms of social harm. |
| Economic | There may be significant economic costs to the battered wife or the identified gay students due either to |
| Harm | their efforts to evade their persecutors or to recover from resulting harm or stigma. |
| Legal | The battered wife and the identified gay students again suffice to illustrate the risk of being involved in an |
| Harm | arrest and interrogation and legal costs. |
A protocol recently discussed on McWirb illustrates all six of these risks of harm:
In a proposal to study response to homosexual invitation, a researcher would approach a same-sex stranger, express admiration, proposition the person, observe the response, then announce that it was just an experiment. This is not the sort of exposure to others that most people would welcome. This invasion of privacy and concern about what will be done with the findings may cause any of the following harms:
N-24
Perspective of Researchers
The ideal researcher is mindful of the possible harms to subjects and to the sources of, and solutions to, those harms. That ideal researcher is also mindful of the many procedural, methodological, statistical, and legal approaches to respecting privacy and assuring confidentiality. Unfortunately, that ideal is rarely realized because most researchers do not have the education, information, or support needed to develop and fully apply this ideal perspective to their research. Even today, most textbooks and teachers of research methodology do not include this material. IRBs cannot be expected to provide the tutelage and knowledge that researchers lack, nor would their efforts be appreciated in many instances. Moreover, most researchers have other perspectives that become countervailing pressures when they lack the resources to function in the ideal mode described above.
Above all, researchers have career concerns. Some researchers are at primarily teaching institutions where they must do research in order to be promoted, but are quite overwhelmed with teaching and student advisement responsibilities. Others are at primarily research institutions and must undertake major research responsibilities and publish extensively. In either case, time management and efficiency are major considerations. However, the way in which each type of career concern is expressed is influenced by whether the researcher has the resources of education, information, and support needed to approach the ideal described above. The following table was developed to suggest extreme opposite ways in which researchers might pursue career concerns, with and without the resources of education, information, and support.
| Concern | Resources Present | Resources Absent |
| Obtain Valid, | Researcher understands how to achieve rigor while | Researcher is trained in the tradition that overlooks |
| Publishable | gaining rapport and cooperation through respect for | the interests of subjects and considers subject |
| Scientific | privacy and confidentiality and other aspects of | autonomy a threat to rigor, e.g., to random |
| Data | subject autonomy. When in doubt, the researcher | assignment and response rate. Researcher does not |
| knows how to quickly seek relevant information and | know how to achieve rigorous research goals and | |
| skills to achieve research goals ethically. | follow the regulations. Perceives successful research | |
| as incompatible with following the Common Rule. | ||
| Publish as | Researcher gains in-depth knowledge of the culture | Researcher “wastes” no time relating to local |
| Much Research | of the target research population and develops | gatekeepers and other members of the research |
| as Possible | research sites in ways that fully demonstrate respect | population as they do not understand science and |
| for members’ privacy interests. Community members | may stand in the way of research if they know | |
| experience benefits of the research and welcome | what is going to be done. If necessary, the | |
| long-term, multifaceted R and D. | researcher sends others to handle these “public | |
| relations issues.” | ||
| Avoid Trouble | Researcher uses many avenues to discover risks to | Researcher perceives no risk, and tells subjects |
| Connected | subjects, is open and respectful with subjects about | nothing that would make them think about |
| with Harm to | possible risks and risk-prevention measures taken, | possible risks or assume that they had any say in |
| Subjects | reduces risks as much as possible. | the matter, as this would interfere with science |
| and with the researcher’s career. | ||
| Multitasking | Researcher integrates research, community and | Researcher has minimal time to “waste” on ethics |
| Between | university service, teaching, and scholarship under | in research planning and delegates it to students. |
| Research and | the umbrella of understanding and serving the | Does not get involved in activities that do not |
| Other Roles | subject population. | immediately produce publishable data. |
| Reduce Time | Researcher uses the educational and informational | Avoids, ignores, or placates the IRB as much as |
| Spent with IRB | tools available and consults with the IRB as research | possible. Sends the IRB a minimal protocol at the |
| plans develop. | last minute; complains if IRB delays the research. | |
The ideal researcher must at times be prepared to educate a nonideal IRB. Most of the researchers I spoke with in connection with this paper mentioned at least one instance in which the IRB wanted to take unnecessary precautions that would cause more harm than good. A better education program for the entire institution would reduce this problem. One example of such a complaint came from an eminent survey researcher who has been deeply involved in various aspects of social research ethics:
N-25
She and her colleagues proposed to study whether computer-assisted interviewing or audio-computer assisted interviewing yields better data on sensitive topics. Subjects would enter the lab and complete an interview including some sensitive questions and nonsensitive questions, in one mode or the other. In the consent form, they would be told that some of the questions asked would be sensitive, e.g., about their sexual behavior, alcohol usage, and drug usage. The IRB requested that the consent include examples of the questions. These highly experienced survey researchers responded that a “fair sample” is impossible, and that whatever examples were given would bias the respondent’s perception of what was to come. The IRB relented, but only after three tries.
Perspective of IRBs
The IRB must take into account the perspective of subjects and researchers in addition to the perspectives that are solely theirs. This is an onerous responsibility if there is not a good research ethics educational program available at that institution. To move beyond my own quarter century of experience as a researcher and IRB chair and administrator, I discussed with a number of my colleagues, who have been in roles similar to mine, the issues of privacy and confidentiality they had found particularly challenging. The issues they described and many issues from my own experience are organized below according to when they occur in relation to when the protocol is reviewed.
Privacy and confidentiality issues are prevalent at every stage of research and solutions are wide-ranging. Some of these problems and solutions are ones to which the federal regulations do not address. Some of the issues raised were (and remain) so sensitive that the actual cases related to me are not retold here; only the issues involved are described. Moreover, some of the issues raised by IRB members are critical of the decisions of their fellow IRB members.
The reader should bear in mind that most social and behavioral IRBs apply the same standards required of funded research to nonfunded research, unpublishable student “research” (such as evaluations of what clients in some local program think of the program), and research not necessarily intended to produce generalizable findings—kinds of projects that are not considered research under the federal regulations. Their concern is to protect human subjects and to teach their constituency to be ethical. They are also concerned to protect the institution from law suits and poor public relations. Hence many of the issues raised below may be typical of IRBs even though they do not flow strictly or exactly from government requirements.
Overarching Issues
The following general issues were raised in various forms by many respondents.
Ignorance About “How”
Overwhelmingly, researchers simply do not know how to carry out the regulations. They do not suffer from lack of morality or even lack of familiarity with the regulations, but from lack of the knowledge, preparation, and craftsmanship required to interpret regulations. For example, how does one know what is private or sensitive from a subject’s perspective? How can one anticipate when a subject might reveal sensitive information, e.g., concerning child or elder abuse. Relatedly, how does one recognize when data might be subpoenaed and how to block a subpoena? How is a child’s sense of privacy different from that of an adult?
Details of the Regulations
Some aspects of the Common Rule are like road signs or road maps—extremely easy to follow if you already know how, but quite confusing otherwise. For example, the issue of children’s privacy is intrinsically connected to parental permission, which intrudes on the child’s privacy in some contexts and protects it in others; there
N-26
are situations in which parental permission should be waived. However, to the incredulity of those who are well versed in 45 CFR 46 Subpart D, virtually no one else seems to find it easy to locate and understand the material on waiver of parental consent. Subpart D is buried behind Subpart B concerning fetuses, in vitro fertilization, pregnant women, etc., and Subpart C concerning prisoners. Having found Subpart D, one must then interpret the regulations pertaining to waiver of parental permission. Neither researchers nor most IRB members approach this interpretation without some sense of uncertainty about whether they are being perhaps too liberal or too restrictive in their interpretation.
Research the IRB Does Not See
The amount of research activity that is never overseen by an IRB is difficult to estimate, but it is a topic often raised by IRB members who express bewilderment that certain departments in their institution have never submitted a protocol. Institution-wide education and commitment would reduce the incidence of such violations.
Omission of Details to Which the IRB Might Object
Among researchers, “urban myths” grow about what the regulations allow and do not allow, e.g., “The IRB always requires signed parental consent.” One researcher sought to undertake important and useful research on inner-city teenagers’ drug use and sexual practices. Under the circumstances there was no way to obtain parental consent. The parents were variously absent, high on drugs, or dependent on their sons for drugs and drug money and hence not supportive of research designed to help kids get off of drugs. The researcher did not know about the legal exemptions for parental permission and did not reveal to the IRB that parental approval would not be sought. A researcher who was educated about waiver of parental permission could have worked with the IRB to solve this problem legally.
When Is It Human Subjects Research?
Among IRBs, tales are legion about particular departments that consider themselves not engaged in human subject research and therefore exempt from IRB review. For example, a professor of marketing believed that marketing research is not about human subjects hence need never go through the IRB. He undertook research on problems of introducing computer technology into the workplace. He had his students administer surveys to university staff to learn what problems they experienced with their supervisors in connection with computerization of offices. The survey was purportedly anonymous—only job title and department were requested! Needless to say, the IRB chair learned of this from outraged staff who felt politically vulnerable and could not imagine how the IRB could approve such a dangerous and invasive study. This was the first the IRB knew of the research. The researcher neither understood nor wished to understand the issues.
Late Protocols
Many researchers learn, part-way through their research, that they should have obtained IRB approval. A protocol is submitted as though the study had not begun. The innocent IRB raises issues and the researcher resubmits, correcting the mistakes. But the research already has been done.
Evasion in Response to Prior Rejection of Protocol
While most of the overarching problems of noncompliance are caused by ignorance, some represent blatant skirting of IRB requirements. In response to rejection of an extremely faulty protocol, some researchers find ways to get around IRB review. For example, a researcher wished to survey Chicano fifth graders about safe sex and AIDS. He did not plan to get parental approval as he judged that to be too difficult. The IRB indicated a number of deficiencies in the protocol, including the fact that no competent school administrator would permit such a study to be done, particularly without parental permission. It was later learned that he had assigned his student teacher to administer the questionnaire, as coursework, in a local school.
N-27
There may always be persons who would skirt the regulations. However, the more thorough and user-friendly the institution’s resources for education concerning human subjects research, the more one’s colleagues and students will exert pressure for adherence to the regulations.
Issues Preceding IRB Review
Nonresearch activities that turn into research, requiring special guidance. Example: A student started an in-class paper based on observation of a family with a recently aphasic member. She met with the extended family regularly, often over dinner, and not always to discuss her project. She gained enormous insight into the family and how it coped with the person’s gradual recovery from a stroke. She and they want the case turned into a book. However, family members do not realize that she is getting from each individual information that is devastatingly personal about the other family members also being interviewed. The risk is not that she is asking personal information, but that they are insisting on telling her personal information (not about the aphasic individual) and that the family members are at risk of learning harmful things about one another. The IRB is working with her on reasonable ways to omit damaging information.
When IRBs are asked to tell thesis students what they need to know. These meetings are useful, but just a first step. Each thesis is likely to raise different issues of privacy and confidentiality. Too much or too little detail is inappropriate for a one-hour seminar. The main accomplishment of such a meeting is to encourage subsequent consultation with the IRB on specific issues. While it takes time and commitment for an IRB member to meet with individual students who have questions, this is far easier and more satisfactory than dealing with poor protocols. However, the process of educating individual students would be made much easier through use of a comprehensive web site such as that proposed herein.
When approaching the IRB raises confidentiality concerns. Example: A historian obtained a list of the names and addresses of high-profile, historically important criminals in hiding, whom he planned to interview. The nature of the list and his research plans were so intriguing that he feared one or more IRB members would be unable to resist discussing it with others. He feared that ultimately the FBI might learn of the study, trace his movements, and arrest his subjects. Solution: The IRB chair agreed with this concern about possible breach of confidentiality by IRB members. He and an appropriate dean reviewed the protocol and approved it. An important book ultimately was published based on the interviews.
Issues Encountered in Reviews of Protocols
When research resembling investigative journalism raises special issues. What rules pertain regarding consent, privacy, confidentiality, anonymity, and risk? Example: A Yugoslavian student disbelieved Western analysis of events in Bosnia. He proposed interviewing survivors from two neighboring cities who had become involved in terrible atrocities against one another. He believed that only they could tell the real story. He planned to ask for the names of perpetrators. What standards of consent or confidentiality should pertain?
Deductive disclosure. Researchers often design studies that they claim to be anonymous although they plan to request (often unnecessary) information that would make it possible for persons to deduce the identity of individual subjects. Or, they may present tabular data with such small cell sizes that persons who know the people who participated in the research could deduce the identity of certain subjects. For example, suppose people in a convalescent home were interviewed about suicidal ideation and the data were broken down by age. Further, suppose that there were just one person over 90 in the institution and a table showed the number of person over 90 who had considered suicide (one). Anyone familiar with the institution could deduce the identity of that person. A skilled IRB can suggest changes in design or data display that eliminate risk of deductive disclosure.
N-28
Promises of confidentiality that preclude referring persons to appropriate help. Example: Researchers survey teens in school about whether they have contemplated suicide. If the survey is anonymous to respect privacy and foster candor, then the researchers cannot directly locate and assist respondents who report having suicidal ideation. In essence, the usual methods of respecting privacy could result in a subject’s death. Solution: The IRB recommended giving each participant a handout listing suicide-prevention services (e.g., counseling, hotlines, web sites, etc.) Moreover, surveys are coded by class. If there were one or more suicidal risks in a class, that entire class would receive a curriculum designed to help students cope with causal issues (e.g., stress, rejection, too-high expectations). Thus, privacy and confidentiality was assured, and suicidal students received the needed treatment along with their peers who received useful information about mental health issues relevant to themselves and their cohort.
Promises of anonymity that preclude treating persons in need of help. Many master’s degree students in counseling psychology have teen clients and want to do theses on how those teens perceive and cope with problems of their generation (e.g., premarital sex, STDs, HIV infection, birth control, pregnancy). They conduct the interviews anonymously. What if a subject reveals a serious problem (e.g., pregnancy, STDs or HIV infection, abusive relationships)? Solution: the interviewer, without knowing the name or other unique identifiers of the subject, simply encourages the subject to seek help that will be given at no cost. The graduate program, working with the IRB, arranged with a campus clinic to provide extensive free support and medical service for any teen who appears with an anonymous referral slip from one of the researchers. The clinic encourages such teens to keep coming back until the problem is handled appropriately.
Culturally insensitive researchers. Researchers may lack sensitivity to cultural norms of their subject population. They risk communicating in ways that are considered invasive of privacy, insulting, and unacceptable.
Example: A researcher planned to interview ethnic Chinese mothers in Chinatown who had received training in AIDS/STD education, who would then educate teenagers in their community. The researcher was planning to ask some very direct questions about how these women’s own children responded to the information, not realizing that one does not directly ask sensitive questions about family members in that subculture. Rather one asks about the responses of “other people’s teenagers.”
Applying rules overzealously. Example #1: A student spent the summer at home, about 3,000 miles from college, working in a prison. He chatted with the prisoners, most of whom were uneducated. He soon found the conversations so interesting that he decided to keep a diary of the conversations. That fall, faced with an opportunity to write a senior thesis, he decided to write up these summer experiences and insights as a senior paper (not for publication). He was required to obtain IRB approval. The IRB instructed him to get consent from all of his “subjects.” He considered this too impersonal and inappropriate to do by mail, especially given the literacy level of most of the prisoners. It was also too expensive to travel home and administer the consent in person; moreover he judged it too intrusive into the lives of people who had considered him just a pal and who were likely to misunderstand the point of his request. He finally persuaded the IRB to accept the careful use of pseudonyms.
Example #2: A graduate student worked for a State Renal Dialysis Registry and was responsible for the registry data. She had a relationship with the patients and wished to conduct a survey (in her student role) that was designed to help understand the needs of the renal patients. The project was enthusiastically endorsed by her supervisor at the Registry. The questions were not sensitive, and her access to the data was legitimate. She wished to write to the patients and ask if she might survey them. The IRB where she was in graduate school required that she have each patient’s primary physician contact that patient to ask if she could contact the patient. It is normal and understandable that physicians and their staffs broker the relationship between their
N-29
patients and a researcher who is unknown to them and who would not otherwise have legitimate access to their data. However, in this particular instance the requirement was unnecessary and made the research impossible to carry out. The researcher could not afford to pay the costs of hourly employment of physicians’ staffs who would contact her patients and get their permission for her to then contact them. Useful research was thus rendered impossible to carry out.
Insensitivity to the stage of intellectual or social development of subjects when trying to preserve privacy or confidentiality. Example: An autistic woman was to be tested by a clinical psychologist studying autism. Her elderly parents must make provision for her care when they die and need to know the results of the testing. In the usual interests of privacy and confidentiality, the report should go only to the subject. In this case, it was decided that the researcher should inform the subject, in the informed consent, that her results would be shared with her parents.
How subjects are paid: implications for confidentiality. Example #1: In street-based studies such as interviews of drug addicts, it is not advisable for the researcher to carry cash, but it would not be acceptable to pay via checks, as the subjects are anonymous and probably do not have bank accounts or convenient check cashing arrangements. A voucher payable at the institution’s bank (no identification required) solved this problem. A one-use, limited-amount debit card might work as well.
Example #2: Studies that pay subjects more than $600 must comply with IRS requirements and issue an IRS Form 1099 to subjects, thus making it possible to trace subjects through payroll and tax records. Participants in a study that stigmatizes them (e.g., studies that recruit HIV-positive persons, pregnant teens) have concerns about exposure of their identity. Timing the study to spread over two calendar years with half-payment in each may be the only legal solution to this problem.
Example #3: This problem is even more complicated when the subjects are illegal aliens and do not have Social Security numbers.
Respecting relationships between gatekeepers and subjects. A breach or perceived breach of confidentiality or invasion of privacy may destroy a gatekeeper’s relationship with subjects. Researchers are often dependent on the goodwill of members of communities or organizations who provide access to a particular population of subjects. Researchers who violate or appear to violate privacy or confidentiality may destroy important community relationships. Example #1: A researcher received permission to interview patients at an out-patient drug rehabilitation clinic. The clinic director placed many restrictions on the kinds of questions that may be asked, the times when the researcher may seek appointments with clinic patients, and the amount of time he may spend with them. It also restricted his access to clinic information. Though disappointed, the researcher realized that the clinic has worked hard to win the trust of its clients, and that both the rehabilitation and the research depend on the maintenance of that trust.
Example #2: A researcher interested in the problems and recovery processes of persons who have been sexually abused “hung out” in an internet chat room support group of victims of sexual abuse. The chat room was a public venue even if the subjects tended to forget that others were free to “lurk” there. He took careful notes on what he read, but did not print out messages or write down anything that would identify specific individuals, locations, or the identity of the on-line chat room. He decided not to seek consent from the participants or from the therapist who ran the chat room, as this might have a chilling effect on the therapeutic processes that were taking place. He realized that anything he published based on this observation should carefully disguise the identity of the chat room and its members so that none of the activities or individuals could ever be recognized.
N-30
Certificates of Confidentiality. It is not always obvious when there is a possibility that data might be subpoenaed. Example: Data from a study of employees’ occupational exposure to an environmental toxin (including data on their history of alcohol and drug abuse, and neuro-psychological battery test data) were subpoenaed by the manufacturer against which workers had filed a class action suit. The data were coded, but the number of subjects was small enough that their demographic and workplace data could be identified. The IRB had not required a certificate of confidentiality. The subpoena could not be squelched. The researcher was called as a witness. The study methodology was trashed by expert witnesses for the defense. The case was settled out of court in favor of the plaintiff subjects.
Waiver of parental permission. The question of when parental permission may be waived is usually cause for concern. Many researchers do not know that provision for this waiver exists (are unfamiliar with Subpart D), and IRBs are often troubled about when to apply it. Example: How does one handle consent with respect to emancipated or run-away teenagers who subsequently return home in the course of the research? Does one abandon the study of these teenagers? Does one disclose to a homophobic parent that his gay son was recruited into a study of safe sex practices while he was a run-away? Solution: Because of potential harm to the teen by the parent, parental permission was waived in this instance.
Issues Arising After IRB Approval
IRB audit of informed consent. Example: An IRB proposed to audit the signed consents of subjects who are HIV-positive. The subjects were local people and some were probably known by some of the IRB members who do not know their sero-positive status. Solution: The IRB chair decided to conduct the audit alone and report the findings to the rest of the committee. (Although the possibility of audit could have been mentioned in the informed consent, that might have muddled the researcher’s sampling scheme. Perhaps this was the best solution.)
Sharing of videotaped data. Videotaped data present new problems of privacy and confidentiality. Important studies, e.g., longitudinal studies of family interaction, are worth archiving for other scholars to use, but individuals might be recognized on the tapes. Researchers and archivists are only now developing consent language and other appropriate safeguards or restrictions on the use of such tapes. Pioneers in this area are the Murray Center Video Archive, and the Stanford and Silicon Valley Archive Project. Example: The Murray Center now has a videotape archive. Donors of videotape data must submit to the Murray Center the informed consent document showing that what was promised subjects is compatible with the intended archiving and sharing arrangement. It requires that scientists present a research proposal that must be approved before they may have the use of a videotape. They must indicate that they have never been located in neighborhoods where the subjects might be known to them. They must agree that they will immediately stop the video and not observe tape of anyone they think they recognize. Tapes must be returned to the archive by a specified time.
Site sharing. Site sharing sometimes occurs when a prior researcher has established a rich archive of information based on an interesting community of persons. For a new researcher to have access to the history and current lives of these people would constitute an invasion of their privacy if not handled courteously, with respect for their feelings and autonomy. Example: A secondary analyst wished to build on longitudinal data collected two years earlier by the data donor. To simply “show up” as though he were entitled to peer into the lives of these people would probably be considered an invasion of privacy and affront to dignity by at least some of the subjects. The initial researcher agreed to return to the research site and work through key community leaders to report what he learned in his study and to ask if people would volunteer to participate in a follow-up conducted by the new researcher.
N-31
Data management. This issue is especially important when data are not anonymous or when deductive disclosure is possible. Researchers promise confidentiality in good faith, but often overlook steps that should be taken to assure that theft or snooping cannot occur. Example: A researcher transported identifiable data (including data on sexual practices, diagnoses of STDs, etc.) from the research site to his home overnight before bringing them to the office. The car was broken into at his home and the records stolen. The IRB had failed to require a data management plan. Subjects whose records were stolen were immediately informed and given contact information for a police detective assigned to the case. No harm to subjects occurred, but the data were never recovered, thus damaging the project.
Institutional use of research data. A firewall between scientific and administrative use must be established and guarded so that subjects remain in control of access to themselves and to their data. Thus, this is both a privacy and confidentiality issue. Example: A state department of health examined the rate of illness in employees in a local laboratory. A high rate of illness was noted in certain employees, and the lab was instructed to provide better shielding in some areas. Strict confidentiality was maintained so that credit rating, insurance rates, and employability of affected employees would not be jeopardized. The laboratory later requested the data (with identifiers) ostensibly to further investigate the effectiveness of the shielding. It proposed to safeguard privacy and confidentiality by getting employees’ (subjects’) consent to obtain their records from the agency. An IRB rejected the protocol because the employees could not autonomously refuse, and might be laid off to protect the lab from law suits and insurance claims.
Issues Requiring Special Competence
There are many issues touched upon in the survey concerning the need for special resources of information and expertise. Some have already been mentioned. A few deserve special mention here.
Plethora of state laws. Several IRB chairs mentioned bewilderment at the many state laws that might pertain to privacy and confidentiality in social research. Even an IRB chair who is an attorney felt that the state law situation was hopelessly complex, especially in the case of multi-state research projects. A well-organized web site that could be accessed by state and topic would serve IRBs and researchers well.
Specialists. Some IRBs have worked hard to provide information to researchers, as needed, on specialists they might consult on various issues. Other IRBs have not recognized how useful a service this might be or what areas of special skill would be useful, periodically, to their clientele. While the particular kinds of specialists would vary by location, IRBs would benefit by having guidelines for developing appropriate informational resources of this kind tailored to their location and situation.
Methodological sophistication. Several related issues were raised that call for sophistication in statistical and research design. Some IRBs are troubled by research designs that appear to use more or fewer subjects than are necessary, or to employ an overly long set of questions. Unfortunately most investigators and IRB members are fairly parochial in their understanding of what might comprise adequate designs and analyses for given types of research. Does the IRB understand the methods of disciplines other than their own? Can it assist researchers in recognizing and rehabilitating poor designs?
| a. |
Methods such as grounded theory, historiography, use of focus groups, and even interview and survey research are sometimes rejected as inappropriate by some who were trained that experimental designs are the only valid research designs. |
| b. |
What is the appropriate number of subjects? Single subject designs, and approaches to reducing variance between subjects are important but often poorly understood methods of producing useful data with one or a small number of subjects. |
N-32
| c. |
There is now a literature of virtually hundreds of approaches to protecting privacy or assuring confidentiality. This literature is rarely sought out by IRBs, researchers, or teachers of research methods. Most are not even aware that it exists. It needs to be available in a user-friendly format. |
Federal Interpretations of the Regulations
There was a startling amount of spontaneously generated expression of concern among those surveyed that two recent governmental interpretations of the regulations are unworkable:
Requiring the consent of persons about whom a survey inquires. This virtually destroys the possibility of epidemiological research. It also gets applied foolishly to such contexts as interviews of parents about their young children. It is an example of a bad rule growing out of an extreme case.
Requiring written consent of participants in Centers for Disease Control and Prevention health surveys.
Concern was expressed that this requirement would decrease response rate, bias samples, increase time and cost required to do the research, and add a risk to confidentiality.
IRB Practices Regarding Privacy and Confidentiality
As the above examples illustrate, the range of privacy and confidentiality issues confronting IRBs is immense, and the regulations are too general to offer useful guidance in dealing with these issues. This is probably as it should be, for if the regulations were too specific they would leave no room for interpretation and flexibility. However, without intelligent interpretation, the regulations tempt IRBs to engage in overzealous, heavy-handed, or capricious enforcement, resulting in more harm than good. Some researchers can anticipate issues inherent in their research plans and argue effectively with IRBs that propose inappropriate applications of the regulations. Other researchers are not so fortunate, and may not know where to turn when faced with requirements they cannot translate into workable research procedures. IRBs may seek to educate their members and clients, but this is not always welcomed or practical and requires much initiative and commitment.
Many IRBs have little sense of the range of privacy issues they should consider or of the range of solutions that might be employed in the service of valid and ethical research. Many IRB chairs, members, and staff persons are not in a position to effectively guide or teach their clientele, or to gain the respect of their clientele. Other IRBs are able to do a remarkably effective job of teaching and guiding their clientele. This section will be devoted to discussion of current practices of highly effective IRBs.
Effective IRB Practices
Some IRBs have members and staff persons who have cultivated the knowledge and resources needed to engage effectively in creative ethical problem solving. Moreover, some IRB chairs have shown considerable wisdom and courage in their willingness to bend rules when necessary to protect human subjects. My colleagues, the IRB members who shared relevant experiences with me, have participated regularly as faculty in IRB workshops and other related professional activities at a national level. Their level of awareness and creativity is probably far above average. Several characteristics emerge concerning effective IRB practices.
Proactive Problem Solving
Effective IRBs identify key areas of difficulty and develop a range of resources to handle these issues. For example, some IRBs review many sensitive protocols involving adolescents. A key issue is finding a way to help or treat teens in trouble once their problems have been disclosed to the researcher. Confidentiality and teen/parent conflict are usually at the heart of the problem. A range of ready solutions can be created in consultation with knowledgeable persons within the institution and community. Extensive referral lists can be kept on disk and
N-33
can be revised as needed for each relevant project. Competent professionals can be identified who can be called upon for advice and assistance. Arrangements can be established with departments that are in a position to help.
Long-Term Professional Membership
Key IRB members have a long-term commitment to keep abreast of the literature and are involved professionally at a national level. Through personal contacts and involvement with IRB organizations, they can seek advice and information as needed. Thus emerging issues having relevance for privacy and confidentiality and the myriad approaches to respecting privacy and assuring confidentiality are quickly integrated into the IRB’s collective consciousness, policy manual, evolving guidelines, web site, etc. New members find a treasure trove of experience and resources at hand when they join the IRB. The researchers served by the IRB learn to respect the helpful intelligent human beings who serve on the IRB. Deans and department chairs learn to look to the IRB as a prudent and diplomatic collaborator who can help them deal with the “lost sheep” or “black sheep” in their flock.
Availability as Educators and Advisors
Effective IRB members, and especially chairs and key staff personnel, seek to be user-friendly problem solvers who are readily available by phone, e-mail, and in person. They give or arrange for workshops tailored to the current needs of specific groups of students and researchers. They are available on a consultative basis. They realize that there are as many privacy issues as there are kinds of human interaction, and as many confidentiality issues as there are conduits for data. Hence they see their own role as that of listening and helping researchers think through privacy and confidentiality problems and solutions. They work patiently with students, and regard themselves as teachers. As one IRB chair/researcher said:
I get students to stay in a confidentiality mode by insisting that they use pseudonyms and aliases even with me. I make a big fuss if I accidentally find out who somebody is and insist that the student be much more careful from then on. I ask to look through their field notes, and act really annoyed if I spot real names or identifiers. I ask ‘What if you lost the notes and the respondent found them. Or, if their worst enemy found them?’
Relevant Competencies
The IRB membership includes persons who have statistical and methodological sophistication, quantitative and qualitative skills, clinical skills, and legal skills. Alternatively, it has ties with key persons within the institution and community so that needed expertise is always at hand and individual members never hesitate to consult with these experts on problems. With this range of competencies they are quick to detect privacy and confidentiality issues and to help devise solutions that respect subjects and the integrity of the research.
Referral of Issues Outside of IRB Purview
One effective IRB has developed a chart listing all the kinds of borderline issues they cannot afford to ignore but that do not fall quite within their purview. The chart gives examples of each kind of issue and indicates the office or individual to whom each such issue might be referred. Thus, when presented with a sticky problem—judging whether a piece of research apparatus is safe, quashing a subpoena of data, or responding to a whistle-blower’s allegations of research fraud—the IRB has a ready process to engage and need not waste time and energy re-inventing solutions to each case. For each kind of case, the chart also indicates whether the office to which they refer must report its decision back to the IRB or whether the matter is settled once the referral is made. This IRB devoted much time developing this ever-evolving document, and got approval from those who would be involved in the referrals. Thus, those who used to complain that the IRB over-reached now have no complaint!
N-34
Web Site and E-Mail Efficiency
Some IRBs conduct much of their routine communication and education on line. Their web site contains their guidelines, protocol application forms (to be used either as hard copy or transmitted back to the IRB via e-mail), sample materials (e.g., consent forms) for a variety of purposes, etc. Many protocols are transmitted as e-mail attachments. Preliminary issues can be discussed and disposed of by e-mail; expedited reviews can be handled in a day or two. Some materials can be sent out instantaneously.
Time saved in this manner is then available for serious discussion of policy issues and proactive problem solving. Problems of privacy and confidentiality are often procedural ones with tidy solutions. A web-site reference to such problems and solutions coupled with on-line advising of researchers can make for much more efficient solving of privacy and confidentiality problems.
Discovering What Is Private to Research Subjects
The difficulty of understanding what is private to others and how that privacy might be invaded has been the underlying theme of this discussion. We have seen that experience, dedication, and wisdom builds something akin to intuition about issues of privacy and confidentiality. How can one directly nurture this ability? There are literatures, exercises, and other sources of information and expertise that are available to assist researchers, IRBs, and teachers of research methodology. This section discusses concepts and resources that can provide insight into the privacy needs of diverse subject populations.
Privacy as a Developmental Phenomenon
Thompson (1982) and Melton (1992) have summarized literature on child development to show how the sense of privacy grows and changes through the years of childhood and what this implies for research on children. Studies by Wolfe (1978), Rivlin and Wolfe (1985), and Parke and Sawin (1979) indicate the privacy is salient even for elementary school children. Children evaluate the quality of living situations by the degree of invasion of privacy and infringement of liberty present in them. Being alone is an important element of privacy to children. By adolescence, maintenance of control over personal information becomes important to the development of intimate relationships, development of self-esteem, and sense of personhood. Melton (1992) has opined that the range of inherently abhorrent intrusions is broader for children and adolescents than for adults because of the acute significance of privacy to personality development. Thus, the privacy interests of children and families are to be respected even if there is no risk of disclosure of intimate information. Many researchers fail to focus on this fact of human experience. IRBs should enlist members who are competent to advise in this area and be prepared to provide an annotated bibliography on developmental aspects of privacy and confidentiality.
Privacy as Situational
“I don’t feel comfortable discussing that here.” An important aspect of privacy is the freedom to pick where one says what to whom, what aspects of one’s life are observed, what degree of control one has over one’s personal space, and how fully one can control access to personal information. However, that generalization is too abstract to guide research practices; researchers situated differently from the subject are not good judges of what subjects might consider private. An important element of planning sensitive research is to ask gatekeepers or surrogate subjects what contexts would be most conducive to candid discussion of the specific topics of the research, what space is considered personal, and what one would not want others to observe—even if the data would be kept confidential.
N-35
Privacy as Culturally Determined
As Laufer and Wolfe (1977) have shown, persons’ privacy interests are idiosyncratic to their culture. To learn about the privacy interests of one’s research population, it is useful to a) ask someone who works with that population regularly (e.g., ask a social worker about the privacy interests of low socioeconomic status parents); b) ask an investigator who has had much experience working with that population, or read ethnographic literature on that population, provided that work was recent and in about the same geographical area; c) ask members of that population what they think other members of their group might consider private in relation to the intended study; or d) employ research associates from the subject population and work with those individuals to develop procedures that are sensitive to the group’s privacy interests.
Privacy as Self-Protection
What is it about the subject that might create a special interest in controlling the access of others? Is the subject engaged in illegal activities, lacking in resources or autonomy, visible, public or famous, stigmatized, institutionalized, etc? Each role carries its own special interests in privacy, which the researcher needs to understand in order to gather valid data and respect subjects. Experience with the target population, use of focus groups and surrogate subjects, knowledge of the ethnographic literature on that population, and consultation with expert professionals who have conducted research similar to one’s planned project—all can be useful sources of information on the privacy needs and interests of the given population. There are many national and local sources of this kind of information which a given IRB might help researchers locate and use.
Subject Population Beliefs About Research
Researchers and IRB members who have conducted community consultations have been astonished at the perspectives of some community members and at the obvious impact of those perspectives upon candor and beliefs about researchers’ promises of confidentiality. Time spent in “town meetings” or focus groups with members of the target population, parents of potential child subjects, or gatekeepers of the target population is well spent. It invariably yields useful insight regarding the privacy interests of subjects, and the implications for organizing the research to respect those interests and provide appropriate assurances of confidentiality.
Privacy as a Personal Perspective
One’s sense of privacy grows out of one’s system of values, morals, norms, experiences, beliefs, concepts, and language. The following table is a useful heuristic for research teams seeking to integrate what they have learned about their subjects into a viewpoint that will generate good solutions to the problem of respecting privacy appropriately:
| Subject Knows | Subject Does Not Know | |
| Researcher Knows | Shared culture or understanding of subject’s | What the subject does not understand about the |
| culture and beliefs. | researcher’s culture and beliefs. | |
| Researcher Does | What the researcher does not understand about | Shared ignorance. |
| Not Know | the subject’s culture and beliefs. | |
This table concerns differences in perception, knowledge and communication. After consulting with members of the target population, it is useful to fill in this matrix to better understand the perspectives of the researcher and subjects with regard to their respective interests in access to personal information. When there is much shared culture, the researcher can readily negotiate a valid agreement concerning privacy and confidentiality. Otherwise special measures are needed.
N-36
Procedures for Assuring Confidentiality4
There is a major body of literature on approaches to assuring confidentiality. The most outstanding and comprehensive source is Boruch and Cecil (1979). Approaches to assuring confidentiality fall into nine categories:
Kinds of Confidentiality Assurances and their Consequences
Researchers’ pious promises of confidentiality are not always effective in producing trust and candor in research participants. Moreover, they are not always promises that can be kept due to faulty data management practices and other possible compulsory disclosures. What kinds of assurances of confidentiality are there and what are their consequences? There is considerable research and scholarship on this topic (e.g., National Academy of Sciences, Committee on Federal Statistics, 1979; Singer, Mathiowetz, and Couper, 1993; Singer, VonThurn, and Miller, 1995; Sieber and Saks, 1989.).
Procedures that Eliminate Linkage of Data to Unique Identifiers
Anonymity offers the best assurance that disclosure of subjects’ responses will not occur. Many dozens of techniques have been developed that are responsive both to the need for anonymity and to other research needs as well. Different kinds of data—cross-sectional, longitudinal, and data from multiple sources—bring with them different research requirements and different ways of meeting these without use of unique identifiers of subjects. The following brief summary is illustrative, not comprehensive. See Boruch and Cecil (1979) for a comprehensive review.
Cross-Sectional Research
Cross-sectional research in its simplest form requires just one data collection session. Anonymity, in which even the researcher is at all times ignorant to the identity of subjects, protects the respondent from legal prosecution, social embarrassment, and concern that the data may fall into corrupt hands. However, it may be desirable to have some form of follow-up to test for sampling validity, response validity, or to do further research on some or all subjects. These refinements are impossible with complete anonymity, but can be realized through temporarily identified responses with subsequent destruction of identifiers, or through use of brokers to provide anonymous data to the researcher after completing one or more of these refinements.
Longitudinal Research
Longitudinal research seeks to track change in individual subjects over time. This cannot be accomplished with strictly anonymous data. However, there are many ways in which aliases or arbitrary identifiers can be used as
N-37
a basis for linking observations over time while preserving the confidentiality of individual responses. The simplest of these involves having subjects choose an easily remembered alias and using it on repeated occasions. There is a considerable body of literature examining the success of variations of this procedure. Some approaches are quite complex. For example, in research by the American Council on Education (Astin and Boruch, 1970) on political activism among American college students, a three-file linkage system was used as follows:
Initial Data Collection:
Second Data Collection:
Data Analysis:
could be used to obtain some of the identifiable data before File C is destroyed, hence a certificate of confidentiality could be obtained to preclude that unlikely event.
Intersystem Linkage
It is sometimes necessary to link research records on subjects with other, independently stored records on the same individuals. In the case of highly sensitive data such as psychiatric or police records, a linkage strategy may be needed so that the researcher does not have access to any identified records. One such method is as follows:
| 1. |
Researcher wishes to link data on 50 subjects with information from their police records. |
| 2. |
Subjects each provide data and an alias (no name) to the researcher. |
| 3. |
Subject provides to the archive (e.g., police) his name and alias. |
| 4. |
Archive provides the requested police information with the aliases (not the names) attached. |
| 5. |
Researcher analyzes relationship between his research data and the police record data. This brief summary is merely illustrative of some of the many specific procedures for preserving anonymity |
or confidentiality and the problems they are intended to solve. The actual literature on this topic is immense.
N-38
Statistical Strategies
Various statistical strategies have been developed to eliminate any direct link between the respondent’s identity and his true answer. All of these methods involve the injection of a specified amount of random error into the data set so that no individual’s true condition can be ascertained but useful statistical analysis of the data is still possible. A very simple example (oversimplified for purposes of this exposition) is the randomized-response method which can be used in direct interview. Suppose the researcher wished to ask whether subjects had struck their child in anger this week, or cheated on their income tax this year—an obvious invasion of privacy. The subject is instructed to roll a die in his cupped hands and observe which side came up without showing it to the researcher. If (say) a “one” came up, the subject was to respond “yes,” irrespective of the true answer. By an algebraic removal of the expected number of false “yes” answers from the data, the researcher can determine the true proportion of yes responses. Neither the researcher nor anyone else besides the subject knows who gave a true “yes” response. As with procedural strategies, statistical strategies have been designed for use with longitudinal and multiple source data, as well.
Secondary Analysis or Audit of Data
Concern for the integrity of data and for extending the analyses of important data sets brings with it the need to do so without risk to privacy or confidentiality. The simplest solution is to render the data anonymous. However, anonymity is not always acceptable or useful from the perspective of the secondary user or auditor. There are many procedures that diminish a) outright breach of confidentiality, b) likelihood of deductive disclosure, c) the sensitivity of the information to which the secondary users have access, or d) the need for the secondary user to actually take possession of the data. Researchers and IRBs who have knowledge of the literature on these issues will know what agreements to make with funders who require audits about how confidentiality will be assured. They can also decide what to include in the informed consent so that potential subjects understand what will be done with the data subsequent to the initial project.
Legal Protections of Confidentiality
As discussed in a prior section of this paper, there are few legal protections of research data. While the courts have shown considerable respect for the need to keep promises of confidentiality made to subjects, they must weigh the importance of this against countervailing issues. However, there is growing use of certificates of confidentiality. Researchers and IRBs need to understand the uses and protections these provide, and their limitations.
Descriptive Statistics
Much work has been done by statisticians in governmental agencies in the United States (e.g., Bureau of Census), Great Britain, and Sweden to develop practices of adjusting tabular presentations so that deductive disclosure is not possible. The most common of these practices is to broaden categories so that data from unique individuals (e.g., top income earners) are not apparent.
Various procedures for preventing disclosure from presentations of quantitative data and descriptive statistics have been developed. The issue of deductive disclosure and the methods that will be employed to prevent published information from disclosing identities should be considered in the planning stages of research and discussed in the IRB protocol, but this is rarely done as most researchers and IRBs lack knowledge in this area. A web site describing main approaches to preventing deductive disclosure from statistical presentation, as well as an annotated bibliography of more complex methods, would be useful aids to researchers and to teachers of quantitative methods.
N-39
Qualitative Research
It is sometimes possible to deduce personal information about identifiable individuals from qualitative data, such as a cultural anthropologist’s “anonymous” account of life in community that is described, but in which persons and places are given fictitious names (Johnson, 1982). The same kinds of problems will probably arise as more studies are conducted in virtual communities of participants in on-line chat rooms (King, 1997). Any clue such as a description of the research site (e.g., a map or a web address) might permit deductive disclosure by anyone familiar with that territory. Johnson (1982) reviewed cases of well-known publications in cultural anthropology in which the identities of specific rural community members could be deduced. In some of these cases, anthropologists had written detailed accounts about the secret, illegal, immoral, or reprehensible deeds of these people, with no awareness that the actual identities would be discovered.
Those who do qualitative studies of the lives of others cannot ensure confidentiality; the subjects themselves, the research assistants, or even the sponsor of the research may inadvertently leak the identity of the research site. Since total confidentiality or anonymity cannot be guaranteed, the issue becomes one of ongoing communication and agreement with subjects (informed consent) and respectful communication of the findings. There is, by now, a growing literature on this issue (e.g., Cassell, 1982; Gallaher, 1964; Glazer, 1982; Johnson, 1982; Kelman, 1968; King, 1999; Wax, 1982).
For example, Johnson recommends guidelines for “ethical proofreading” of cultural anthropology manuscripts to diminish potential harm to subjects or communities as follows:
Internet Research
Internet research was mentioned above in relation to participant observation or field data from the virtual environment of chat rooms. However, there is now a rapidly emerging literature on various other kinds of internet research, associated methods of solving problems of privacy and confidentiality, and uncertainties or vulnerabilities connected with these “solutions.” Researchers’ insouciant claims that internet data are anonymous or that confidentiality will be protected are reminiscent of such promises regarding non-web research of two or three decades ago.
N-40
This area of research will grow rapidly since it enables researchers to reach far-flung subjects quickly, inexpensively, round-the-clock, and without a research staff. The problems and solutions to issues of privacy and confidentiality will change rapidly over the ensuing years as new technologies render old problems and old solutions obsolete. Some of the rapidly evolving issues include:
to recommend that assurances of confidentiality contain appropriate disclaimers. A detailed review of the current literature on this topic is not warranted since the details will change within a few months. However, for purposes of the recommendations of this paper, sources of current information (typically web sites) should be made available to IRBs and researchers with frequent updates and assurances of confidentiality should be limited appropriately.
Summary: Key Issues for IRBs and Researchers
A poor and misleading definition of privacy and confidentiality in the regulations. Specific definitional problems are discussed below, in “Improving the Regulations.”
Research topics and methods that do not fit within the federal regulations. Research that more closely resembles investigative journalism, historical research, or biography is subject to conflicting standards—those of its discipline and those of the federal regulations. If IRB review and ethical guidelines must be applied to such research, rather than making it conform to inappropriate standards, a better solution would be to ensure that subjects understood what standard would apply. For example, instead of trying to impose some inappropriate limit on disclosure of unique identifiers, one IRB simply requires that oral history researchers inform potential subjects of all the kinds of disclosures that will appear in the resulting material that is made public.
Lack of effective, ongoing education of IRBs and their clients. One of the most serious problems facing the research community is the lack of time and resources to educate IRB members. IRBs are drowning in paper work. They need resources to foster their own effective learning and problem solving and create appropriate learning contexts for their clientele.
Lack of time and resources for proactive problem solving. Proactive problem solving includes creating and continuing to update educational materials for members, researchers, and students. IRB members need released time, resources, and administrative support if they are to function effectively as proactive problem solvers.
Lack of relevant research skills among IRB members. The many contexts and methods of social/behavioral research are mysteries to some IRB members not trained in those disciplines. It is important to have IRB members who view research methods with intelligent skepticism and who can anticipate the concerns of subjects. However, it is not useful to have many IRB members who are ignorant of research methods. For example, a competent applied statistician, qualitative methodologist, survey researcher, and field experimenter can educate fellow IRB members and their clientele.
N-41
Students who are poorly trained in ethics and methodology. Most research methodology still focuses on the
“get data” approach and pays scant attention to learning the culture of subjects in their various contexts and to understanding how to effectively respect the privacy of subjects. The absurd notion that ethics is the enemy of methodology prevails and is reinforced throughout students’ training. Students are the scientists of the future. If user-friendly and science-friendly curriculum is available on the web, this material can be used in the classroom and in anticipation of student research. A recent symposium at the American Association for the Advancement of Science concluded that curriculum on research ethics needs to be taught integrally with methodology curriculum. Otherwise, it is seen as the study of the misdeeds of others, rather than as problems oneself might encounter.
Lack of awareness of the range of privacy and confidentiality issues extending from the conception of the project to data management, publication, and ultimate data archiving or sharing. This is the one problem that appears to lend itself to a checklist. To prevent the checklist from being used in a procrustean fashion, it might be part of an indexed web site that raises the issues and offers solutions.
Too many rules, not enough basis for good judgment. One last word about rules and checklists: More rules and procedures will only render overworked IRBs less effective at the complex, subtle task of responding to privacy and confidentiality issues. It is tempting to devise a checklist to help identify and solve problems, but that, too, is subject to misuse by poorly educated IRB members. Once an issue is raised in some quasi-official way, there is a tendency to make much ado of nothing, or at least to require researchers to respond to an endless set of questions. At most, a checklist could be part of an on-line protocol and researchers could simply indicate N/A to items that seemed irrelevant. The web site could include a brief explanation of each checklist item.
Recommendations to the Commission
Two basic recommendations are offered for consideration by the Commission:
| (1) |
Provide, in the Common Rule, clear, separate definitions of privacy and confidentiality that are broad enough that researchers and IRBs can apply them to diverse research activities in different disciplines, and |
| (2) |
Recommend to OHRP the development of web-based educational materials, formatted much like Microsoft Word’s “Help” menu, using the “Book,” “Index,” and “Find” methods of retrieval. Two web pages are recommended. A major web page would provide the knowledge needed to design research ethically and prepare an effective protocol. A smaller web page would guide IRBs in locating, organizing, and tailoring information to serve local needs (e.g., state and local laws, local informational resources, helpful professionals who might consult with researchers, useful institutional resources). |
Improving the Common Rule by Redefining Privacy and Confidentiality
The complexity of privacy and confidentiality issues that arise in social and behavioral research, especially in field contexts, cannot be directly embraced in the Common Rule. Rather, privacy and confidentiality, should be addressed in more comprehensive, useful ways both in the “Definitions” section and in the “Informed Consent” section of the Common Rule. The reader should also be referred to a web site that clarifies what it means to respect privacy and assure confidentiality.
Since the regulations are often the first and only things that new IRB members see and since most institutional assurances are copied out of the regulations, the actual content of the regulations is significant. Since the current regulations handle the definition of privacy and confidentiality in a most misleading way, it is essential that these be made more useful and less confusing.
N-42
Privacy
The Common Rule does not define privacy per se, but defines private information as follows:
Private information includes information about behavior that occurs in a context in which an individual can reasonably expect that no observation or recording is taking place, and information which has been provided for specific purposes by an individual and which the individual can reasonably expect will not be made public (for example, a medical record) (45 CFR 46.102(f)(2)).
This statement is embedded in a larger context as follows:
40.102 Definitions
(f) Human subject means a living individual about whom an investigator (whether professional or student) conducting research obtains
| (1) |
data through intervention or interaction with the individual, or |
| (2) |
identifiable private information. Intervention includes both physical procedures by which data are gathered (for example, venipuncture) and manipulations of the subject or the subject’s environment that are performed for research purposes. Interaction includes communication or interpersonal contact between investigator and subject. Private information includes information about behavior that occurs in a context in which an individual can reasonably expect that no observation or recording is taking place, and information which has been provided for specific purposes by an individual and which the individual can reasonably expect will not be made public (for example, a medical record). Private information must be individually identifiable (i.e., the identity of the subject is or may readily be ascertained by the investigator or associated with the information) in order for obtaining the information to constitute research involving human subjects. |
This is not a definition of privacy. Privacy should be defined separately, as 45 CFR 46. 102(g). A comprehensive and useful definition of privacy would be:
(g) Privacy refers to persons and to their interest in controlling the access of others to themselves (e.g., via informed consent).
It is widely recognized (e.g., Beauchamp and Childress, 1994) that informed consent (and the right to withdraw at any time) is a major way in which research subjects control access to themselves. However, 45 CFR 46.116 General Requirements for Informed Consent do not mention privacy. Those sensitive to privacy issues would recognize privacy as a concern under 46.116(2):
Because people are so accustomed to over-simplified notions of privacy, the recommended web-based educational document that OHRP might provide should include a range of well-chosen examples of privacy, such as the following:
N-43
The examples offered should convey that subjects’ ability to regulate the access of others to themselves depends on such factors as age, station in life, and culture. Populations vary with respect to their need for help in regulating access to themselves, and this should be taken into account in research planning, procedures, and formulation of the informed consent statement.
Many researchers think that privacy and associated consent requirements are an impediment to research. The kind of static definition currently found in the current regulations only reinforces this impression. The regulations and accompanying web site examples should convey the relationship between contextual factors and privacy interests, and convey that respect for privacy is good scientific practice. They should make it obvious that privacy concerns affect subjects’ willingness to participate in research and to give honest answers.
Confidentiality
The Common Rule does not define confidentiality and seems to refer to it somewhat interchangeably with privacy. This confusing and oversimplified language can be detrimental when it is focused on the conduct of research in which issues of personal privacy and access to data are vital to the protection of subjects and to the willingness of subjects to participate and to provide candid responses.
A separate definition of confidentiality (as CFR 46.102(h)5) should be added. Following Boruch and Cecil (1979) this definition might be as follows:
(h) Confidentiality is an extension of the concept of privacy; it refers to data (some identifiable information about a person, such as notes or a videotape of the person) and to agreements about how data are to be handled in keeping with subjects’ interest in controlling the access of others to information about themselves.
This definition should then be reflected in 45 CFR 46.116, the section on informed consent. Currently, in the presentation of the elements of informed consent, 45 CFR 46.116 (5) states:
(5) a statement describing the extent, if any, to which confidentiality of records identifying the subject will be maintained.
45 CFR 46.116 (5) does not distinguish between confidentiality and anonymity. In many cases data gathered in an identifiable form can and should be rendered anonymous. The procedures for doing so should be carefully planned and described, at least briefly, in the informed consent.
45 CFR 46.116 (5) also fails to distinguish between normal and extraordinary confidentiality concerns. With sensitive data, there may be unusual threats to confidentiality (e.g., subpoena, hacker break-in, theft, mandated reporting) and there may or may not be solutions to these problems. 45 CFR 46.116 (5) should be reworded to encompass these distinction, as follows:
(5) a statement of whether and how the data will be rendered anonymous; or, a statement describing the conditions of confidentiality of identifiable data: who will have access to such information, what safeguards will prevent or reduce the likelihood of unauthorized access, and what unavoidable risks of disclosure may exist.
N-44
This more comprehensive informed-consent requirement reminds the researcher and IRB that there are many methods that can be used to prevent or reduce unauthorized access, and that one should be mindful that some risks of disclosure are difficult to prevent entirely. It virtually leads the researcher and IRB to seek out some of the voluminous literature on this topic. It informs subjects about what they need to know concerning the confidentiality or their data.
The related web site material should emphasize that confidentiality is not an agreement not to disclose. Rather, it refers to any kind of agreement about disclosure.
Web-Based Educational Resources for IRBs, Researchers, and Teachers
A web-based educational resource is recommended that will guide the ethical problem solving in research. It should not be offered as an official regulation, or interpretation of regulations, but as a user-friendly educational resource that will challenge IRBs, researchers, teachers, and students to improve their ability to craft solutions to ethical and methodological problems. Much of the success of this resource will depend on the ability of the IRB to tailor it to the particular needs of their institution, and to present it effectively. While the general web site would be educational, not regulatory, a smaller IRB web site would contain required actions for the IRB to take, as appropriate to their locale. That second, much smaller web site would guide IRBs in the development of resources for handling issues of privacy and confidentiality.
The main goals of the recommended web resource are: a) to present the most current knowledge concerning protection of privacy and confidentiality, and b) to ensure that this information presented in the general (large) web site is perceived by IRBs and researchers as educational and informational resources, not as an interpretation or requirement of OHRP. The intelligent interpretation and use of the educational material in the general web sites would be required of researchers by their IRB, but would be regarded as guidelines and not as rules to be applied slavishly.
To reiterate the argument presented earlier, the reasons for making this an educational and not a regulatory document, and for making sure it is perceived thus by IRBs and researchers are as follows:
| 1. |
Acceptance of many more detailed or specific rules across 17 agencies and diverse research contexts would be limited. |
| 2. |
Opportunities for intelligent interpretation and deciding between principles or values in conflict would be diminished. |
| 3. |
Efforts required to follow a specific rule may be disproportionally great, costly, or even inappropriate, relative to the expected gain or results. |
Administration of the Web Resource
To ensure that the general web site is perceived as educational and not regulatory, its contents and possibly also the contents of the smaller IRB web site should be the result of work commissioned to subject matter specialists, though overseen by a standing committee including researchers, IRB specialists, and representatives of OHRP. The commissioned work, in turn, should be edited by this committee. The final draft of each set of elements prepared for incorporation into the two web sites should be put out (on the web) for IRBs and researchers to review and critique as they wish. The finally edited documents should be designed and developed into two web sites formatted much like the Help menu of Microsoft Word. The web sites should be managed by a professional web master employed by the standing committee of experts.
To function as an evolving resource, responsive to new problems and information, both web sites must be frequently updated by a consultant and reviewed by the standing committee. Moreover, all users should be invited to submit suggested additions and modifications for consideration by the committee.
N-45
Some Recommended Elements of the (Small) Web Site for IRBs
| 1. |
How to appraise the IRB’s need for expertise in its members and outside consultants. |
| 2. |
How to structure workshops and curriculum for the major segments of researchers, tailored to the needs of their institution. |
| 3. |
How to select and develop needed institutional resources (e.g., consultants, counselors, health center staff, subject matter specialists) who can satisfy typical needs of some major sectors of the IRB’s clientele. |
| 4. |
How to locate and enlist the cooperation of local specialists (e.g., school personnel, therapists, social workers, demographers, urban anthropologists, farm agents, etc.) who would be added to a local directory and serve typical needs of the IRB’s clientele. |
| 5. |
How to help faculty who teach research courses to adapt portions of the web site for instructional purposes. |
| 6. |
How to bookmark or extract materials especially pertinent to their institution and add them to an internal IRB web page or document, e.g., pertinent state and local laws. |
| 7. |
How to select and add new materials as they become relevant to the evolving goals of that IRB. |
| 8. |
How to communicate with the web master and consultants about issues with which they need assistance or clarification. |
The General (Larger) Web Site
The recommended elements of the general web site would include virtually everything that IRB members, researchers, teachers of methodology or student researchers might need to know about the nature of privacy and confidentiality, how to respect privacy and assure confidentiality, and how to handle unavoidable risks to confidentiality.
These elements should be presented at about the level of an upper-division methodology text, and should mirror and go beyond the material presented in this paper. There should be an annotated bibliography along with each topic, and those materials should be available at the institution’s library. It is beyond the scope of this paper to provide a detailed outline of all of the specific topics that might appear in the general web site.
However, a summary of the main topics appears in Appendix A.
Additional Points to Consider
Some other kinds of issues were raised in this paper which the Commissioners may wish to consider:
Legal Issues
In the section on Regulations and Statutes, some problems were raised which the Commissioners may wish to seek to resolve:
| 1. |
Signed parental permission is difficult to obtain in some populations (e.g., of non-English speaking or illiterate parents) even though the parents would not object to their child’s participation in research. It may be appropriate for legislators to consider permitting waiver of parental permission under these conditions, provided other mechanisms are in place to protect the children. |
46.408 states that parental permission may be waived for a subject population in which parental or guardian permission is not a reasonable requirement to protect the subjects (for example, neglected or abused children) provided there are appropriate other mechanisms for protecting the children, etc. In order to generalize this to permit research on children whose parents are functionally illiterate, perhaps a
N-46
community consultation model might be employed to explore the attitudes and concerns of the parent population, and to explore what safeguards are deemed appropriate. Particular attention would need to be given to the choice of community gatekeepers or leaders who would participate in this consultation, and who might then be responsible for confirming with parents that they understand what the research will entail and determining whether any of them object to their child’s participation. This alternative to written and signed informed consent might be tested in a variety of types of communities where most of the parents are functionally illiterate to determine its acceptability before such a provision becomes part of 48.408(c).
| 2. |
The language concerning protections offered by certificates of confidentiality is rather imprecise and should be clarified. Researchers and IRBs need clear guidance and rules regarding the issuance and use of certificates of confidentiality, an explanation of what they do and do not protect, and what the reporting requirements are for child and elder abuse and neglect, given a certificate of confidentiality. |
| 3. |
Neither Subparts C and D of 45 CFR 46 discuss special protections that should be given to incarcerated youths, many of whom have strained or nonexistent relationships with their parents. IRBs and researchers need to understand this population and protections that are needed. This is a complex topic that should be addressed by an expert in the field of research on juvenile justice. |
| 4. |
Because warning about mandatory reporting of child or elder abuse serves researchers but not children, the Commission may wish to recommend that reporting requirements for funded research on family processes deemed of great national importance be altered. For example, the modified requirement might mandate training in effective parenting or elder care with follow-up supervision and built-in provision for such training and supervision. A warning to this effect might produce better outcomes for science, society, and the families involved. |
Making the Regulations More Accessible
The Common Rule contains much that is not relevant to most social researchers, e.g., how to organize and conduct an IRB, how to do research on fetuses, pregnant women, etc., or on prisoners. The entire Common Rule should appear somewhere on the general web site. However, of most relevance to social researchers are the elements of a protocol and of informed consent (ideally with web links to explanatory material elsewhere in the general web site). Additionally, Subpart D of 45 CFR 46 is relevant to social research on children (ideally also with links to explanatory material). These should be presented separately along with examples of well-planned, well-designed protocols and consent statements, including well-designed explanations to parents and parental permission forms. Tips on how to plan the protocol should be included, along with discussion of how to make the informed consent process one of effective communication that promotes comprehension, trust, and good decision-making by the prospective subjects.
Acknowledgments
I am grateful to the many colleagues who reviewed earlier drafts of this paper and contributed to the collection of privacy/confidentiality problems and solutions discussed herein. They contributed immeasurably to the quality of this paper; its deficiencies are due entirely to me. Ironically, I cannot name these generous people because I promised confidentiality in return for their disclosure of the IRB foibles, problems, and solutions that enrich this paper. Some readers would inevitably deduce, correctly as well as incorrectly, who said what. Special thanks go to the competent and conscientious staff of NBAC for their guidance throughout the development of this paper.
N-47
Appendix A: Some Recommended General Web Site Topics
| 1. |
Explorations of the concepts of privacy, confidentiality, and anonymity and how they pertain to various specific contexts. |
| 2. |
Exploration of theories of privacy and how they guide researchers in asking the right questions about subjects’ privacy interests and suggest sources of answers. |
| 3. |
Exploration of kinds of confidentiality-assuring techniques; details of their use; exercises in tailoring confidentiality assurance techniques to specific problems. Techniques for preventing deductive disclosure from quantitative data. Techniques for masking identities of persons and places in qualitative data and for “ethical proof reading” of case study or ethnographic material so that accidental disclosure of identities is not damaging. |
| 4. |
Current updates on emerging issues of confidentiality, e.g., issues stemming from new modes of electronic communication, new safeguards, new privacy advocacy, policies and laws, and new electronic venues for conducting research. |
| 5. |
Research in public venues where people nevertheless interact with one another in an intimate or private way (e.g., some internet chat rooms) and where publication of research identifying the site could prove chilling to interaction or embarrassing to subjects. |
| 6. |
Summary and interpretation of regulations and laws (federal, state, and local) governing privacy and confidentiality of social research. |
| 7. |
Certificates of confidentiality and other protections of data: what they cover and how to use them (and any new emerging protections that become available). |
| 8. |
Responding to legal problems that actually arise in the course of one’s research, such as subpoena of one’s data or involvement in a situation that may mandate reporting. |
| 9. |
Approaches to evaluating what may be private to one’s subject population and to assessing actual or imagined threats to confidentiality. Designing research that satisfies subjects’ interest in controlling access to themselves and to safeguarding the confidentiality of data. |
| 10. |
Tips to researchers on organizing their careers so that they can readily generate the resources (information, networks, community ties, ethnographic knowledge, techniques for assuring confidentiality, etc.) required to conduct research successfully in ways fully respectful of privacy and confidentiality concerns. |
| 11. |
Issues of research on children, the elderly, and other vulnerable populations and their special privacy and confidentiality interests. |
| 12. |
Frequently asked questions and answers about 45 CFR 46, including Subparts A, B, C, and D, and about FERPA and PPRA. |
| 13. |
How to identify appropriate ethical standards for kinds of research that do not fit the usual social/behavioral research paradigms (e.g., oral history, ethnography, market research, research resembling investigative journalism). When does each kind of research require IRB review and how are conflicts reconciled between the usual standards of research (e.g., anonymity of subjects, masking of location) and the standards of that discipline (e.g., full disclosure of that information). |
| 14. |
How to resolve issues of responsibility to help subjects in dire need, when anonymity or promises of confidentiality constrain the ability of the researcher to locate or help those subjects. |
| 15. |
How to resolve issues of paying subjects when these are in conflict with the need to maintain confidentiality or anonymity. |
| 16. |
How to build mutually beneficial relationships with gatekeepers that are respectful of the privacy and confidentiality concerns of subjects, the organization, and the gatekeeper. |
| 17. |
How to plan for subsequent data sharing and audits of one’s data. |
| 18. |
Data management issues. |
| 19. |
Understanding how many subjects, how many trials, etc., are needed for valid research and how this depends on the particular design and goal of the research. |
| 20. |
Kinds of research methods, where they typically are used, and associated kinds of data analysis techniques. Ethical and other issues connected with each method and approaches to resolving those issues. |
| 21. |
Research on research ethics. When researchers are unsure what procedure best solves their ethical or scientific concerns, they can build a study within a study. In a given research context, they can then examine which of two or more different procedures produce the best results. For example, does anonymity produce more disclosure of undesirable behavior or a higher response rate than promises of confidentiality in a given kind of research project? Suggest topics of research on ethical issues from the perspective of the various relevant scientific disciplines. Mention journals that would be likely to publish these studies. |
N-48
Notes
1 Many argue that the term research participant is more respectful than the term subject. For some purposes I agree. For the purposes of this paper, I prefer to use a term that reminds the reader that the person being studied typically has less power than the researcher and must be accorded the protections that render this inequality ethically acceptable.
2 I am indebted to Dr. Joe Cecil and Jason Gilbert, Federal Judicial Center, for providing me with their detailed summary and analysis of these issues.
3 A copy of state reporting laws may be obtained by writing to Dr. Seth C. Kalichman, Psychology Department, University of Chicago, 6525 North Sheridan Road, Chicago, IL 60626.
4 I am indebted to Drs. Robert F. Boruch and Joe S. Cecil for their work in this area, and particularly for their seminal work Assuring the Confidentiality of Social Research Data (1979) which has been my main source for this part of the paper.
5 The current items in 45 CFR 46 10 (g-j) would be moved down and become items (i.) through (l.)
References
Agre, P.E., and Rotenberg, M. (Eds.) (1998). Technology and privacy: The new landscape. Cambridge, MA: MIT Press.
Astin, A.W., and Boruch, R. F. (1970). A “link file system” for assuring confidentiality of research data in longitudinal studies.
American Educational Research Journal, 7, 615–24.
Beauchamp, T.L., and Childress, J. F. (1994). Principles of biomedical ethics. 4th edition. New York: Oxford.
Boikess, O. (2000). Personal communication. “Privacy Protection for Research Subjects: Certificates of Confidentiality.”
Boruch, R.F., and Cecil, J.S. (1979). Assuring the confidentiality of social research data. Philadelphia: University of Pennsylvania Press.
Buckley Amendment (1985). 20 USCA. Section 1232 et seq. The accompanying regulations are set down in 34 CFR pt. 99.
Campbell, D.T., Boruch, R. F., Schwartz, R.D., and Steinberg, J. (1972) Confidentiality-preserving modes of access to files and to inter-file exchange of useful statistical analysis. Evaluation Quarterly, 1(2), 269–300.
Cassell, J. (1982). Harms, benefits, wrongs, and rights in fieldwork. In J. Sieber (Ed.), The ethics of social research: Fieldwork, regulation and publication. New York: Springer Verlag. Pp. 7–31.
Child Abuse Prevention and Treatment Act of 1974, Public Law 93-247.
Corbin, J.E. (1987). Child abuse and neglect: The cultural context. In R. E. Helfer and R. S. Kempe (Eds.), The battered child (4th ed., pp 239–255). Chicago: University of Chicago Press.
Evers, K. (2000). Formulating international ethical guidelines for science. Professional Ethics Report, 13(2), 1,7–8.
Forsham v. Harris (1979) (445 US 169) holds that raw data developed by scientists in government-sponsored research projects but not actually in the agency is not covered by FOIA, even though the agency has the right to obtain the data if it is desired.
Gallaher, A. (1964). Plainville: The twice-studied town. In A. Vidich, J. Bensman, and N. Stein (Eds.), Reflections on community studies. New York: Wiley.
Gil, E. (1982). The California Child Abuse Reporting Law: Issues and answers for professionals (Publication No. 132 [10/86]). (Available from the State of California Department of Social Services, Office of Child Abuse Prevention, 744 P Street, M.S. 9-100. Sacramento, CA 95815).
Gilbert, J. (2000). Federal Judicial Center Memo of July 7, 2000, entitled “Protecting Confidential Research Information from Disclosure in Litigation.”
Glazer, M. (1982). The threat of the stranger: Vulnerability, reciprocity and fieldwork. In J. Sieber (Ed.), The ethics of social research: Fieldwork, regulation and publication. New York: Springer Verlag. Pp. 49–70.
Gray, E., and Cosgrove, J. (1985). Ethnocentric perceptions of childrearing practices in protective services. Child Abuse and Neglect, 9, 389–396.
Hatch Amendment (1984). 20 USC Section 1232h (b). The accompanying regulations are set down in 34 CFR pt. 98.
Jaro, M.A. (1989). Advances in record-linkage methodology as applied to matching the 1985 census of Tampa, Florida. Journal of the American Statistical Association, 84, 414–420.
N-49
Johnson, C. (1982). Risks in the publication of fieldwork. In J. Sieber (Ed.), The ethics of social research: Fieldwork, regulation and publication. New York: Springer Verlag. Pp. 71–92.
Kelman, H. (1968). A time to speak: On human values and social research. San Francisco, CA: Jossey-Bass.
King, S. (1997). Researching internet communities: Proposed ethical guidelines for the reporting of results. The Information Society, 12,119–127.
Laufer, R.S., and Wolfe, M. (1977). Privacy as a concept and a social issue: A multidimensional developmental theory. Journal of Social Issues, 33, 44–87.
Levine, M. (1992). Child protection legislation: origins and evolution. Child, Youth and Family Services Quarterly, 15(1), 2–3.
Levine, M., and Levine, A. (1983). Helping children: A social history. New York: Oxford University Press.
Melton, G.B. (1992). Respecting boundaries: Minors, privacy and behavioral research. In B. Stanley and J. Sieber (Eds.) The ethics of research on children and adolescents. Newbury Park, CA: Sage. Pp. 65–87.
Morris, R.A., Sales, B.D., and Berman, J. J. (1981). Research and the Freedom of Information Act. American Psychologist, 36(8), 819–826.
National Academy of Sciences, Committee on Federal Statistics (1979). The influence of interviewer and behavioral variables on reporting in household interviews. Vital and Health Statistics: Public Health Service Publications, No. 1000-Series 2, No. 26. Washington, D.C.: U.S. Government Printing Office.
National Research Act (1974). Pub. L No. 93-348.
Office of Management and Budget (1999) Circular A-110 1,64 Fed. Reg. 54,926.
Office for Protection from Research Risks (OPRR). U.S. Department of Health and Human Services. Http://grants.nih.gov/grants/ oprr/library_human.htm.
Parke, R.D., and Sawin, D. B. (1979). Children’s privacy in the home: Developmental, ecological, and child-rearing determinants.
Environment and Behavior, 11, 84–104.
Rivlin, L.G., and Wolfe, M. (1985). Institutional settings in children’s lives. New York: John Wiley.
Schwartz, J. (2000). Oversight of human subject research: The role of the states. Paper commissioned by the National Bioethics Advisory Commission.
Singer, E. (1979). Informed consent: Consequences for response rate and response quality in social surveys. American Sociological Review, 43, 144–62.
Sieber, J.E. (1994). Issues presented by mandatory reporting requirements to researchers of child abuse and neglect. Ethics and Behavior, 4(1), 1–22.
Sieber, J., and Saks, M. (1989). A census of subject pool characteristics and policies. American Psychologist, 44(7), 1051–1063.
Singer, E., Mathiowetz, N., and Couper, M. (1993). The role of privacy and confidentiality as factors in response to the 1990 Census. Public Opinion Quarterly, 57, 465–82.
Singer, E., VonThurn, D., and Miller, E. (1995). Confidentiality assurances and survey response: A review of the experimental literature. Public Opinion Quarterly, 59, 266–77.
Spencer, M. (1997). Proposal for a randomized experiment on the effects of scholarship awards to high achieving students from low-income families. Philadelphia, PA: University of Pennsylvania, CHANGES.
Tarasoff v. Regents of the University of California, (1976) 17 Cal 3d 425, 131 Cal Rptr 14, 551 p 2d 334.
Thompson, R. (1982). Developmental changes in research risk and benefit: A changing calculus of concerns. In B. Stanley and J. Sieber (Eds.), Social research on children and adolescents: Ethical issues. Newbury Park, CA: Sage. Pp. 31–64.
Wax, M. (1982). Research reciprocity rather than informed consent in fieldwork. In J. Sieber (Ed.), The ethics of social research: Fieldwork, regulation and publication. New York: Springer Verlag. Pp. 33–48.
Weis, J.G. (1989). Family violence research methodology and design. In L. Ohlin and M. Tonry (Eds.), Family violence (pp. 296–321). Chicago: University of Chicago Press.
Wolfe, M. (1978). Childhood land privacy. In I. Altman and J. R. Wohwill (Eds.) Human behavior and environment: Advances in theory and research (Vol. 3, pp. 165–222). New York: Plenum.
N-50
UNFULFILLED PROMISE: HOW THE BELMONT REPORT CAN AMEND THE CODE OF FEDERAL REGULATIONS TITLE 45 PART 46—PROTECTION OF HUMAN SUBJECTS
Commissioned Paper Harold Y. Vanderpool
University of Texas Medical Branch, Galveston

O-1
The power of the Belmont Report to amend the Code of Federal Regulations Title 45 Part 46 has never been realized. This paper will indicate why and how an incorporation of the content and spirit of Belmont into the body of the Federal Regulations can rectify major problems in the Regulations, strengthen the protection of human subjects, and accent the inescapable roles of moral judgments for assessing when research involving human participants is permissible.
Signed into law on 12 July 1974, the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (hereafter called the National Commission or Commission) was charged by the U.S. Congress “to identify the basic ethical principles that should underlie the conduct of...research involving human subjects.”1 In response to this charge, the National Commission published the Belmont Report2 in the Federal Register on 18 April 1979. Belmont was then adopted in its entirety as a policy statement by U.S. Department of Health, Education, and Welfare (DHEW)—now called the Department for Health and Human Services (DHHS). Its principles of respect for persons, beneficence, and justice are regarded as “the three quintessential requirements for the ethical conduct of research involving human subjects” by the National Institutes of Health’s (NIH) Office for Protection from Research Risks (OPRR).3
I. The Federal Regulations and the Belmont Report: Shared Purposes and Different Approaches
The Federal Regulations and the Belmont Report share the overarching purposes of promoting research and protecting human subjects.4 The Federal Regulations assume that extensive research conducted within or sponsored by 14 federal agencies should be approved according to the conditions set forth in the Regulations. In the Belmont Report, the promotion of research serves as a pervasive, but de facto purpose. Belmont 1) begins with the sentence, “Scientific research has produced substantial social benefits,” 2) views research as a moral “obligation” in several places,5 and 3) justifies a number of research activities and practices in order to accomplish the goals of research.6 As indicated by their subtitles, the explicit purpose of both the Federal Regulations and the Belmont Report is the protection of the human subjects of research. These documents, however, adopt different approaches to this protection.
The protection of research subjects in the Federal Regulations includes rules about how subjects should be protected by risk/probability of benefit determinations [46.111 (a) (1) and (2)], by an equitable selection of subjects [46.111 (a) (3)], by informed consent [41.111 (a) (4) and 46.116], and by adding additional safeguards for vulnerable populations of prospective subjects [46.111 (b)].
Notably, the Regulations also protect human subjects by giving detailed attention to organizational and enforcement mechanisms—the oversight of federal agencies such as OPRR, rules pertaining to the necessity and structure of IRBs, the documentation of IRB deliberations and informed consent, record keeping, and so on. This is as it should be in an effective regulatory system. The fact that some of the greatest abuses and harms in history perpetrated on human beings in research experiments occurred in Germany after its Minister of Interior in 1931 promulgated laudable and visionary ethical guidelines for conducting research involving humans underscores the necessity of institutionalizing ethics through organizational and enforcement mechanisms.7 In contrast to the Regulations, the Belmont Report proposes to protect human subjects through its ethical principles and guidelines. The Report asserts that its principles will enable investigators and Institutional Review Boards (IRBs) to resolve “ethical problems arising from research,” and will enable researchers, reviewers, human subjects, and interested citizens “to understand the ethical issues inherent in research.”8 The Regulations are either silent or virtually silent about the protective value of ethics. This paper will show that the ethical purposes and content of Belmont should serve as a basis for amending the Federal Regulations in order to strengthen its subject protections.
O-3
The purposes of protecting human subjects and enhancing the benefits of biomedical and behavioral research are fully justifiable, although their balance should be critically and periodically reassessed. It is a mistake to assume that the relationships between these purposes have to be a zero sum game, whereby increased protections for subjects subtracts from the conducting of research. Over the last 25 years, efforts to protect human subjects have enhanced, not just curtailed, research initiatives. Ethical ferment and regulatory protections have eased the public’s anxiety and increased the public’s trust, the effects of which are greater public participation in and advocacy for new and expanding levels and types of research.9
II. Promise: The Belmont Report as a Critical Source for Rectifying Major Problems in the Regulations with Respect to Human Subject Protections
The four explicit purposes of the Belmont Report are set forth in its entangled second and third paragraphs. The report 1) identifies ethical principles that will 2) provide a foundation for formulating, criticizing, and interpreting the regulatory rules of research found in codes of research ethics such as the Nuremberg Code, the Declaration of Helsinki, and existing codes of Federal Regulation, and 3) enable scientists, research subjects, IRBs, and interested citizens to understand the ethical issues that are “inherent in research involving human subjects.” At the end of these paragraphs Belmont then says that its “objective” is 4) “to provide an analytical framework that will guide the resolution of ethical problems arising from research involving human subjects.” This analytical framework includes the report’s principles and their applications.10 Belmont focuses on the first and fourth of the above purposes, and its contents display that the third is inescapably true. Notably, however, the report does not discuss the second purpose just listed. This paper will focus on this unaddressed and unfulfilled purpose articulated in the report itself.
The ethical content of Belmont consists of principles and guidelines that are organized according to the following schema, or, to use the report’s terminology, “analytical framework:”11
| The Principle of: | Applies to | Guidelines/Applications/Requirements for: |
| Respect for Persons | Informed Consent | |
| n Information | ||
| n Comprehension | ||
| n Voluntariness | ||
| Beneficence | Risk/Benefit Assessment | |
| Justice | Selection of Subjects | |
The term “guidelines” in this schema is drawn from Belmont’s subtitle and is accented in the introductory letter composed by the members of the National Commission when the Belmont Report was delivered to the secretary of DHEW.12 The guidelines are entitled “applications” in the body of the report. These applications consist of “requirements” (a term used several times) or “moral requirements” regarding informed consent, risk/benefit analysis, and the selection of subjects of research.
Taken together, the purposes and schematized content of the Belmont Report rest on the assumption that ethical principles, reasoning, and guidelines should serve as an essential basis for protecting human subjects of research. This includes formulating, criticizing, and interpreting codified regulations, including the U.S. Code of Federal Regulations. Yet for over 20 years the Belmont Report has not been systematically used as a source of additional and essential protections in the Federal Regulations. Instead, the report has been by and large marginalized to the status of encouragement and oratory. Belmont has been and is being preached about, rather than plowed into the fabric of human subject protections.
O-4
From the vantage point of Belmont, the present Federal Regulations (“Common Rule”) contain a number of major problems, all of which can be rectified by using the Report to amend the Regulations. The Regulation’s problems include:
propose clear and concise ways to amend the Code of Federal Regulations.
III. The Belmont Report: Treasures in an Earthen Vessel
A. Principles
The meaning of Belmont’s principles is often misunderstood. Robert J. Levine, for example, claims that the report mandates a form of “ethical reasoning,” in which “abstract” principles are taken to be “an ultimate foundation” for “second-order” rules or norms. Levine contrasts this type of reasoning with the views of certain “philosophers” who argue that “the traditional and received norms of society” are logically prior to basic ethical principles.13 This view easily lends itself to a “principle-based approach to bioethics” (often called “principalism” and viewed with suspicion) that begins with a-theoretical, top-down, “ad hoc constructions” that are unconnected to one another.14 The nature and functions of the principles articulated in the Belmont Report should be identified by the text of the report, by the thinking of its primary author, Tom L. Beauchamp,15 and by the recollections of its Commissioners. The following four points describe how the principles in Belmont should be understood in relation to ethical reflection, judgments, and action.
First, Belmont’s principles are condensations or constitutive elements of morality related to research and derived from culture. Belmont itself says that its principles were chosen because they are “particularly relevant” to research and are “generally accepted in our cultural tradition.”16 In other words, rather than serving as abstract norms, they reflect “the fabric of morality and morally sensitive cultures” to such an extent that “no responsible research investigator could conduct research without reference to them.”17 “Principles are the common coin of moral discourse.”18 When the Belmont Report was composed, principles were used by ethicists and bioethicists to identify the basic and comprehensive elements of morality—akin to identifying the basic elements of nature in the periodic table.19 Principles summarized the right-making and wrong-making elements of human interaction that could serve as “an easily grasped set of moral standards” for persons with diverse backgrounds and training.20 Consider the first principle articulated by Belmont—respect for persons. Commissioner Jonsen could understand this principle as an action guide drawn from ethical theory. Commissioner Lebacqz could regard it as crediting all human beings and communities with measures of dignity and worth. Lawyers could identify it
O-5
with the U.S. Constitution’s rights of self-determination and privacy, theologians with the sacredness of human life and human dignity endowed by God, and philosophers with respect for the autonomy of the will from Immanual Kant and liberty of choice from John Stuart Mill. Belmont says that its “comprehensive” principles are “stated at a level of generalization that should assist scientists, subjects, reviewers and interested citizens to understand the ethical issues inherent in research involving human subjects.” The report’s principles were suited to diverse professional groups within the pluralism of American culture.22 Second, the report does not explicitly ground its principles in ethical theory, but this does not mean that theory is superfluous and, consequently, that its principles are the ultimate foundation of moral reasoning. The Belmont Report does not purport to offer a canonical understanding of philosophical ethics. Its purposes are practical and specific. Why, then, is Belmont silent about ethical theories? It is silent primarily because the Commissioners could not agree upon the theoretical justifications of the principles, even though they “had no difficulty in agreeing on the principles themselves.”23 Nevertheless, even though the Belmont Report does not display its indebtedness to ethical theory, that theory lurks behind both its principles and applications—the theories of Mill and the deontological ethics of Kant within the principle of respect of person, ethical consequentialism behind the principle and applications of beneficence, and so on. Beauchamp and others “ultimately relented” to Commissioners’ requests that the “bolder philosophical defenses” of theories and theorists in earlier drafts of Belmont should be stripped away from the final report.24 Third, the text of Belmont shows that its principles are advanced as free standing moral norms that are not linked together or prioritized by one or more overarching theories. How these principles are understood in Belmont is subject to two interpretations. First, they are sometimes regarded as equally and absolutely required: “they all must be satisfied before research is ethically acceptable.”25 Second, they are widely construed as non-absolute, prima facie ethical duties that are always morally binding unless they come into conflict.26 When prima facie principles conflict in particular situations, decision makers must determine how these principles ought to be “balanced” with one another or whether one of the principles appears to be overriding and, therefore, binding. The Belmont Report does not choose between these very different moral perspectives.27 And fourth, this discussion about the nature of Belmont’s principles partially defines what the report is promulgating. To say that according to Belmont, “the purpose of a regulatory system is to promulgate ethical principles” in order to protect human subjects and enhance science28 skims the surface of Belmont’s intent. Belmont promotes the ethics of research for the sake of protecting human subjects 1) through general principles that reflect basic, readily understood, and commonly shared moral values found in and advanced by philosophical ethics, law, and religious traditions and 2) through particular moral requirements and guidelines that resonate with these principles. This presses us to explore the protections provided by Belmont.
B. Protections
To apprehend the protections of human research subjects Belmont provides, its principles must be morally related to its “applications.” The language of the report says that its principles “lead to” “find expression in,” and serve as “a basic justification for...particular ethical prescriptions” (presumably its applications). These phrases and the word “applications” itself imply that Belmont’s applications are deduced from its principles.29 This should not, however, be taken to mean that its “applications” are morally secondary or inferior to its principles in terms of their ethical weight. Belmont’s principles represent “moral requirements,” “obligations,” and “imperatives” found in its applications. As such, its applications “require” actions and consist of “obligations.” Informed consent “requires conditions free of coercion and undue influence;” and the “assessment of risks and benefits requires a careful arrayal of relevant data.” The protections provided the Belmont Report are rooted in the moral requirements of both its principles and applications, which in the report’s subtitle are referred to as ethical “guidelines.”
O-6
While Belmont’s text primarily connotes that its applications are “derived from” its principles, the contents of the report display a reciprocal interplay between its principles and applications. Members of the National Commission did not merely discover and affirm a set of ethical principles from which they deduced all the applications of the report. By the time Belmont was published, the Commissioners had produced a series of reports on various aspects of biomedical and behavioral research that either focused upon or were filled with the “applications” found in Belmont. These included reports on research involving prisoners (1976), children (1977), disclosure of research information (1977), and IRBs (1978).30 The Commission’s extensive work enabled it to recognize which set of ethical principles (among other moral principles in the ethics literature)31 were “relevant” or “particularly relevant” to research.32 The equally strong moral requirements of its principles and applications and the interplay between them directly relates to the protections provided by Belmont.33
The most noteworthy feature about the protections promulgated by Belmont is that at critical points its protections are far greater in the “applications” section of the report than in its “basic ethical principles” section. The crucial place in which this occurs entails protections pertaining to respect for persons. According to Belmont, the principle of respect for persons “requires” that persons “should be treated as autonomous agents, which involves giving “weight” to the opinions and choices of individuals who are capable of deliberating about and acting in accord with their “personal goals.” Respect also requires refraining from heavy-handed disrespect, such as repudiating the considered judgments of prospective subjects or denying their freedom to act on these judgments. To “give weight” to a research subject’s opinions and choices, however, implies that the authority to weigh and judge resides with someone other than the subject. This phrase undercuts the ethical and legal understandings of respect for autonomy, namely that individuals are free and self-determining agents who have the final authority to decide what should happen to them.34 But what the principle (as stated in Belmont) denies, the applications supply. All prospective participants 1) must be granted “the opportunity to choose what shall or shall not happen to them;” 2) must be given all the information (much of it detailed in the report) that “reasonable volunteer[s]” would need to know to decide “whether they wish to participate;” 3) must comprehend this information (which involves the way the information is organized, the time needed to understand and ask questions, and communication suited to subjects’ language and levels of intelligence, maturity, rationality); and 4) must be situated in “conditions free of coercion, undue influence (due to excessive or improper rewards, overtures, or inducements), and “unjustifiable pressures” from “persons in positions of authority or commanding influence” over either the prospective subject or “through the controlling influence of a close relative.” These applications show that the subject’s choice should be free and final.
The principle of respect for persons in Belmont also includes “the requirement to protect those with diminished autonomy,” which may involve excluding some groups of subjects from research depending upon “the risk of harm and the likelihood of benefit” to these subjects. Belmont’s applications are far more explicit than this dimension of the principle of respect.35 The principle of beneficence encompasses the “obligation” to reduce social harms and increase social benefits through research—an obligation that can be viewed as nonprotective, if not outright threatening to human subjects of research. This obligation, however, is restrained by the other obligations included within the principle of beneficence, namely, the “imperatives” of “protecting [subjects] from harm” and “making efforts to secure their well-being” by maximizing possible benefits and minimizing possible harms. The applications of this principle offer further restraints. For example, they require “a careful arrayal of relevant data, “including, in some cases, alternative ways of obtaining the benefits sought in the research,” and they embrace a broad range of risks and foreseeable benefits—physical, psychological, social, legal, and economic.36 Belmont’s principle of justice charts the different ways justice is defined, but primarily focuses on injustice as occurring “when some benefit to which a person is entitled is denied without good reason or when some
O-7
burden is imposed unduly.” Within its discussion of justice as a principle, the report catalogues historical examples of excessively burdened and exploited patients and begins to deal with how justice and injustice apply to past policies of selecting subjects due to their “easy availability,” “compromised position,” or “manipulability.” The report extends this analysis in its applications section.37 Importantly, the principles of beneficence and its applications and the principle of justice and its applications serve as gate keeper functions. They are the ethical criteria which IRBs must use to determine which research projects and protocols are acceptable enough to move to the stage of subject enrollment. They serve, therefore, as essential, but nevertheless initial moral screens prior to the ethical bedrock of Belmont’s human subject protection—the vital protections surrounding informed consent.
The Belmont Report’s great reliance upon informed consent accords with the fundamental moral values of a free and democratic society. This requires a high bar for informed consent, which Belmont strongly upholds as its bedrock basis for subject protection. Without this bar, permissible research would have to reflect strict and delimited research risks.38 Without the protections inherent to informed consent, the resulting stricter controls of risks and benefits would have the effect of transmogrifying the relationships between subject protections and the research enterprise into a zero-sum game.
These points enable us to complete the description (begun at the end of section A above) of what the Belmont Report is promulgating for the purpose of protecting human subjects. Belmont promulgates the ethics of research through general principles 1) that reflect basic, readily understood, and commonly shared moral values found in and advanced by philosophical ethics, law, and religious traditions, and 2) that are strengthened and expanded by the ethical requirements and guidelines specified in its applications.
C. Flaws
In spite of its manifest strengths, the Belmont Report bears the flaws and cracks of an earthen vessel. First, in spite of its clear, schematized outline, it is not easily understood or fathomed. It contains the multilayered features of a document comprised by a committee with many agendas.39 Second, Belmont’s silence about how its three principles are related to each other has given rise to confusions and conflicting interpretations about their relationships. This is regrettable, because relating, prioritizing, or “balancing” Belmont’s principles is inevitable for the assessment of many protocols. Some level of guidance is needed for the sake of encouraging moral reflection and more responsible decision making by IRBs.40 Third, Belmont is conceptually flawed. By linking its principle of respect for persons with autonomy, the report exposes itself to cogent criticisms by bioethicists who argue that its principle of autonomy is anemic, if not wrong headed.41 Belmont’s principle of autonomy does not square with its applications. Bioethicists also point out that this principle should be distinguished from the principle of protecting persons from harm, which pertains to beneficence.42 But unless the Commissioners were hopelessly confused, which, given all the philosophical papers they heard and read, we have no pressing reasons to believe, Commissioner Lebacqz’s interpretation of what Belmont was meant to convey under its respect for persons principle is probably correct. The majority of Commissioners wanted the rather open-ended phrase “respect for persons” to denote respect for autonomy, respect of persons with diminished autonomy, respect of fetuses and infants, and possibly respect for communities of persons.43 A sentence or two could have clarified some of the confusion.
Fourth, the Belmont Report mentions (which has often gone unnoticed),44 but does not accent or expand upon the ways distributive justice or fairness requires extending the benefits of research to underserved populations of patients.45 The report also does not mention issues related to compensatory justice—the imperative of compensating at least some subjects in some circumstances for the injuries sustained in research.46 Fifth, Belmont focuses on the protection of individuals to the neglect of communities. Had the report discussed probable benefits and harms to communities, its protections could have been extended to other areas of research, including stem cell, xenotransplantation, and genetics research.47
O-8
And sixth, the report does not offer direct guidance or examples regarding one of its explicit purposes—the purpose of formulating, criticizing, and interpreting codified regulations. Belmont’s silence in this regard has likely contributed to its marginalization and neglect, as well as to its not yet being used as a basis for critical revisions of the Code of Federal Regulations.
D. Power
Do Belmont’s flaws undermine its power to effect changes in the Federal Regulations in order to enhance subject protections? No. The treasures of the Belmont Report are found in its legal, historic, and revered status, and its great intrinsic worth. Its intrinsic value encompasses:
Belmont’s flaws limit its ability to enlighten and resolve day-to-day decision making by IRBs and researchers, but they do not keep Belmont from serving as a powerful basis for correcting serious problems in the Federal Regulations.
IV. Changes in the Code of Federal Regulations Mandated by Belmont
This section has a clear and specific goal. It will show how coherent and systematic linkages between the Belmont Report and the Federal Regulations will rectify the serious problems in the Regulations identified in section II above. These changes will also make the Regulations clearer, better focused, easier to use, and less bureaucratic.
The subsections that follow are arranged in a problem-solution format. Each will first describe a major problem in the Federal Regulations, then suggest how the relevant section(s) of the Regulations should be reformed and reorganized.
A. Corrections of the Federal Regulations’ Disregard of Ethics
Unfortunately, the accent on the importance and roles of ethics in the Belmont Report is virtually absent from the Federal Regulations. Any mention of ethical standard or ethical principles appears only in later Subparts that deal with special populations of patients/subjects. Subpart B (Protections of Fetuses and Pregnant Women) 46.202 says that the activities under review should “conform to appropriate ethical standards.” And subpart D (Protections for Children) 46 (b) (1) (ii) states that “the research will be conducted in accordance with sound ethical
O-9
principles. Furthermore, the term “ethics” is mentioned only once in the main body of the Regulations—in 46.103 (b) (1) in connection with a “statement of principles” that should be adopted by institutions that are applying for assurance of compliance agreements with a federal department or agency.49 Specific recommendations for correcting this disregard are given in the sections that follow.
B. Rectification of the Regulations’ Irresponsible Standards Pertaining to the Sources that Define and Articulate Research Ethics
The one place where “ethics” is mentioned in the main body of the Federal Regulations reflects an irresponsible approach to the ethics of research. Section 46.103 (b)(1) says that “a statement of principles” for the purpose of “protecting the rights and welfare of human subjects of research” is required in an assurance of compliance agreement. But the actual content of such a statement is not taken seriously, and its uses are not addressed.
Here is the wording about the statement that is required of institutions: “This [statement] may include an appropriate existing code, declaration, or statement of ethical principles, or a statement formulated by the institution itself.”50 So stated, a document that deals with the “ethical principles” of research is 1) viewed as an option on equal par with some “existing code” (The Nuremberg Code?), or “declaration” (The Declaration of Helsinki?), or some “statement” formulated by an institution itself. This falsely assumes 2) that all of the above options will serve to protect the rights and welfare of human subjects. 3) This wording also apparently assumes that the statement that is submitted in an assurance of compliance application will inform IRB deliberations at the institution making the application. But how or when the statement is to be used is not mentioned. And 4) this directive conflicts with explicit statements in the first paragraphs of the Belmont Report that the often “inadequate” and conflicting codified rules in Nuremberg, Helsinki, and the U.S. Federal Regulations of 1974 need to be expanded, criticized, and interpreted by utilizing ethical principles.
Section 46.103 (b) (1) should be revised to convey the following [in which changes and additions are underlined]:
Assurances applicable to federally supported or conducted research shall at a minimum include:
(1) A statement of ethical principles and rules governing the institution in the discharge of its responsibilities for protecting the rights and welfare of human subjects of research conducted at or sponsored by the institution, regardless of whether the research is subject to Federal regulations. This statement of ethical principles should include, at minimum, the tenets of the Belmont Report. The statement should serve as an ongoing basis for training programs and protocol evaluation by the institution’s IRB members and investigators.
C. Correction of the Flawed Understanding of the Ethics of Research in the Federal Regulations
Regrettably and surprisingly, the Federal Regulations themselves incorporate a seriously flawed understanding of research ethics. This is found in the body of the Regulations 46.107 (a) under the heading of IRB membership. The third sentence of 46.107 (a) says:
In addition to possessing the professional competence necessary to review specific research activities, the IRB shall be able to ascertain the acceptability of proposed research in terms of institutional commitments and regulations, applicable law, and standards of professional conduct and practice. The IRB shall therefore include persons knowledgeable in these areas.51
O-10
In this directive the critical task of “ascertaining the acceptability of proposed research” 1) rests on the vague, unspecific category of “institutional commitments and regulations,” 2) does not even mention ethics or “sound ethical reasoning,” and 3) falsely assumes that “standards of professional conduct and practice” (presumably, professional codes of ethics) directly relate to the ethics of research. The import of this last and false assumption is highlighted in the discussions and recommendations of the Final Report of the Advisory Committee on Human Radiation Experiments. Recommendation 9 of this Final Report includes the following:
The historical record and the results of our contemporary projects indicate that the distinction between the ethics of research and the ethics of clinical medicine was, and is, unclear. It is possible that many of the problems of the past and some of the issues identified in the present stem from this failure to distinguish between the two.
The Committee suggests...the following:…Incorporating of research ethics, and the differences between the ethics of research involving human subjects and the ethics of clinical medical care, into curricula for medical student, house staff, and fellows.52
To correct this flawed understanding of research ethics in the body of the Federal Regulations, the wording quoted above from part 46.107 (a) should be revised to convey the following [in which the word changes are underlined]:
In addition to possessing the professional competence necessary to review specific research activities, the IRB shall be able to ascertain the acceptability of proposed research in terms of sound ethical reasoning that distinguishes the ethics of research from the ethics of clinical care, applicable law, and each institution’s statement of ethical principles and rules specified under 46.103 (b) (1).
D. Clarification of Organizational Confusions in the Regulations
To make the Regulations clearer, better focused, and easer to use, the criteria investigators and IRB members are to follow and utilize as they develop, approve, or disapprove of research protocols should be reorganized. The places where the Regulations require researchers and members of IRBs to make judgments or determinations regarding research protocols are separated from each other and are interspersed between IRB “house keeping” rules and enforcement powers. This results in an array of directives that obscures, rather than highlights, what investigators and IRBs are supposed to do.
The primary organizational confusion of the Regulations occurs between sections 46.111 (Criteria for IRB approval of research) and section 46.116 (General requirements for informed consent). Between these sections there are several others that deal with the powers and limits of non-IRB institutional officials (46.112), the power of IRB’s to suspend or terminate research (46.113), the roles of IRB’s respecting cooperative research projects (46.114), and management-and-federal-oversight concerns regarding IRB records (46.115). In short, the types of judgments that researchers and IRBs are to make are imbedded in lists of rules pertaining to management and enforcement.
In order to bring these judgments into focus and highlight their importance, the Federal Regulations should be reordered in a logical progression and, on occasion, display different titles. A cursory review of the items that follow indicates how they are scrambled and disorganized in the Common Rule. [Note: Changes in ordering, numbering, and wording are underlined in the suggested reorganization that follows, and the numbering of the Regulations as they now exist are placed in brackets.]
O-11
46.109 IRB review of research.
46 .110 [46.111] Criteria for IRB approval of research.
46 .111 [46.116] Criteria for informed consent.
46 .112 [46.117] Requirements for the Process and Documentation of informed consent.
46 .113 [46.110] Expedited review procedures for certain kinds of research
46 .114 [46.113] Suspension or termination of IRB approval of research.
46 .115 [46.114] Cooperative research.
46 .116 [46.115] IRB records.
46 .117 [46.112] Additional review by institutional officials.
46.118 Applications and proposals lacking definite plans for involvement of human subjects.
E. Emendations of Sections in the Federal Regulations that Deal with Criteria for IRB Determinations and Investigator Compliance
The rules and requirements IRBs should utilize to review and approve of research involving human subjects (other than special populations) and that investigators should follow are in sections 46.111 and 46.116 of the Regulations—reordered as sections 46.110 and 46.111 above.
These sections of the Regulations display four main problems:
O-12
Taken together, these four problems show that the Federal Regulations do not embody the purposes and power of the Belmont Report—its insistence on critical and organized reasoning, and a number of its recommendations regarding the protection of research subjects.
These serious problems should be corrected. The purposes, concepts, and language of the Belmont Report readily serve as a foundation for this correction.
The corrections/revisions suggested for 46.111 (Criteria for IRB approval of research) are given in Appendix A of this paper. The suggested corrections/revisions of 46.116 (General requirements for informed consent) and 46.117 (Documentation of informed consent) are given in Appendix B. The emendations suggested for 46.116 and 46.117 reflect the “applications” of informed consent in the Belmont Report, as well as the discussion in section III. B of this paper concerning the bedrock protective value of informed consent in a democratic society.
V. Putting Ethics into Practice: What are Researchers and IRB Members Supposed to Be Doing?
To be effective, protections of human subjects of research should be suited to the everyday concerns and practices of researchers and the reviewers of human subject research. This can be done by stepping into the shoes of investigators and IRB members who, upon wrestling with research regulations and working with regulatory bodies, regularly ask, “What are we supposed to be doing?” This question serves as a simple, straightforward test for what an effective regulatory system ought to be communicating through its codified regulations and the ethical principles and requirements upon which these regulations rest. This pragmatic test means that those who compose and/or amend and alter the Code of Federal Regulations should ask themselves, “Are we clearly communicating what those on the front lines of human subject protection need to be doing to protect human subjects?“ The foregoing sections of this paper show that at the present time the Federal Regulations tell investigators and regulators what they should be doing in a considerably disorganized, incomplete, confusing, and follow-the-rules fashion. For example, IRB members are told that risks to subjects should be minimized and “reasonable in relation to anticipated benefits” [46.111 (a) (1) and (2)] without being told how this can and should be done by means of a systematic assessment of risks and probable benefits. IRBs are also told that informed consent “will be sought...in accordance with, and to the extent required by 46.116.” Four sections later in the Regulations, 46.116 does not mention IRB oversight. In its initial paragraph, 46.116 tells investigators that they should seek to secure the consent of subjects or their representatives “only under circumstances...that minimize the possibility of coercion or undue influence,” and through giving information “in language understandable to the subject or the representative.” This paragraph is a prelude to the clearly outlined and listed “basic elements of informed consent” [46.116 (a)], the wording, outline, and contents of which clearly convey what investigators and IRB members should be doing to protect human subjects: They should make sure that consent forms contain all the information listed in the Regulations, and they should refine or “tweak” consent forms that cannot be understood or that lack any of the required information.57 The emendations advanced in this study retain the contents of above requirements, but they link these requirements together, outline what should be done more clearly, fill in blind spots, correct misunderstandings, and make them more thought provoking. These emendations give the following answers to the question, “What are researchers and IRB members supposed to be doing to protect human beings enrolled in research?”
O-13
The thesis of this paper is that the above actions and practices are necessary for the protection of human subjects. Each directly reflects the ethics of research promulgated in the Belmont Report. Each and all of these moral imperatives should, therefore, be clearly and adequately communicated through the Federal Regulations and suited to concerns and practices of researchers and IRB members.
Conclusions and Recommendations
This paper has explored the shared purposes and different approaches of the Belmont Report and the Code of Federal Regulations with respect to protecting human subjects of research. It has identified a number of major problems in the current Code of Federal Regulations, and has shown that the purposes and content of the Belmont Report can and should be used to rectify these problems. The presence of these problems prove that the protections of human subjects of research advanced within Belmont have not yet been adequately incorporated within the Federal Regulations. This paper also describes clear and specific ways to revise the actual wording and organization of the Regulations. And it argues that truly effective guarantees of subject protection must give clear and adequate answers to the practical concerns of and questions asked by investigators and reviewers of research protocols.
Predicated upon this study, the author makes two recommendations to the members of the National Bioethics Advisory Commission:
Note: I thank my colleagues Ronald A. Carson, Cheryl M. Chanaud, and William J. Winslade for their critical comments and suggestions.
O-14
Appendix A
Note: The word changes below are underlined. Deletions from the present Regulations are indicated by removing the bold typescript. And the numbering of the items have been changed, with the numbering as it now exists placed in brackets.
46.111 Criteria for IRB approval of research.
[a] In order to approve research covered by this policy the IRB shall determine that all of the following requirements pertaining to the welfare and rights of human research subjects are satisfied.
| (a) |
A systematic and rational assessment of the risks and probable benefits which shows: |
| (1) |
Risks to subjects are minimized: (i) by using procedures which are consistent with sound |
research design, (ii) by considering physical, psychological, and social risks, and (iii) [ii] whenever appropriate, by using procedures already being performed on the subjects for diagnostic or treatment purposes.
(2) Risks to subjects are reasonably balanced in relation to anticipated benefits, if any, to subjects, and the importance of the knowledge that may reasonably be expected to result from the research:
(i) by giving special weight to subjects’ risks over the probable social benefits of research, (ii) by considering only those risks and probable benefits that may result from the research (as distinguished from comparing these to the risks and probable benefits of therapies prospective subjects would receive as patients not participating in the research), and (iii) by giving particular consideration to the voluntariness and comprehension of subjects of research that offers fewer, if any, benefits and greater risks to the subjects, but significant benefits to society. The IRB should not consider possible long-range effects of applying knowledge gained in research (for example, the possible effects of the research on public policy) as among the research risks that fall within the purview of its responsibility.
(b) [3] Just and equitable procedures are used in the selection and recruitment of prospective subjects. IRBs should be particularly cognizant of the ethical problems of research involving vulnerable populations, such as children, prisoners, pregnant women, mentally disabled persons, or economically or educationally disadvantaged persons. They should also be cognizant of justly including women, ethnic minorities, and all age groups in research that will likely contribute to their health and well being.
| [ |
Keep the present wording of sections 46.111 (4) through (7), but renumber them as (c) through (f).] |
| (g) |
[b] When some or all of the subjects are likely to be vulnerable to coercion or undue influences, |
such as children, prisoners, pregnant women, mentally disabled persons, or economically or educationally disadvantage persons, (i) the appropriateness of involving them should be demonstrated, and (ii) additional safeguards have been included in the study to protect the rights and welfare of these subjects.
O-15
Appendix B
Note: The word changes below are underlined. Deletions from the present Regulations are indicated by removing the bold typescript. The numbering of the items has been changed, with the numbering as it now exists placed in brackets.
46.116 Criteria for informed consent.
Except as provided elsewhere in this policy, no investigator may involve a human being as a subject in research covered in this policy and IRBs may not approve of such research unless the investigator obtains the legally effective and ethically justifiable informed consent of the subject or the subject’s legally authorized representative. The consent process has the following three basic elements: (a) Voluntariness. An investigator shall seek such consent only under circumstances that provide the prospective subject or the representative the opportunity to freely choose whether or not to participate and that minimize the possibility of coercion, undue overtures, rewards, or inducements, and unjustifiable pressure from researchers or through relatives. No informed consent, whether oral or written, may include any exculpatory language through which the subject or the representative is made to waive or appear to waive any of the subject’s legal rights, or releases or appears to release the investigator, the sponsor, the institution or its agents from liability for negligence.
(b) Comprehension. The information that is given to the subject or the representative shall be organized and presented in language understandable to the subject or the representative. The subject’s comprehension of information and explanations should be assured through mutual communication and practical methods of comprehension assessment. Subjects or the representative should be given sufficient time to make an informed choice.
(c) [a] Understanding. Except as provided in paragraph (e) [c] and (f) [d] of this section, in seeking and evaluating informed consent, the following information shall be provided to each subject:
| (1) |
An explanation that the study involves research . . . |
| (5) |
An explanation describing the extent, if any, to which confidentiality of records identifying the |
subject will be maintained; . . .
(8) An explanation that participation is a voluntary choice the subject is free to make, that refusal to participate will involve no penalty or loss of benefits to which the subject is entitled and aware of, and that the subject may discontinue participation at any time without penalty or loss of benefits to which the subject is entitled and aware of.
| (d) |
[b] Additional elements of informed consent . . . |
| (1) |
An explanation that the particular treatment or procedure . . . |
| (5) |
An explanation that significant new findings . . . |
46.117 Requirements for the Process and Documentation of informed consent. . .
| (b) |
Except as provided in paragraph (c) of this section, the consent form may be either of the following: |
| (1) |
A written consent document that embodies the basic elements of informed consent required by |
46.116. This form may be read to the prospective subject or the subject’s legally authorized representative, but in any event, the investigator shall give either the subject or the representative adequate opportunity to read, discuss, ask questions, and comprehend it before it is signed.
O-16
Notes
1 Office of the Secretary, Department of Health, Education, and Welfare (DHEW) in The National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research, (OPRR Reports, 18 April 1979), p. 2.
2 The name “Belmont” was chosen because work on the report began at a four-day retreat (in February 1976) at the Smithsonian Institution’s Belmont Conference Center.
3 OPRR, Protecting Human Research Subjects: Institution Review Board Guidebook (Washington, D.C.: U. S. Government Printing Office, 1993), p. xxi.
4 Humans who become involved in research should be called human subjects, rather than participants. The term “participants” is too cozy and in part deceptive. The roles of human beings enrolled in research are portioned out, not commonly shared with investigators and investigator teams. Enrollees in research do not “participate” in the sense of contributing creatively or actively as do participants in discussion or activity groups. “Subjects,” on the other hand, connotes subjects of paintings, testees, persons opened to or exposed to danger, and conscious, thinking agents.
5 Belmont’s discussion of the principle of beneficence begins with two rules of moral “obligation” to human subjects: “1) do not harm and 2) maximize possible benefits and minimize possible harms.” It then says that “obligations of beneficence” “extend both to particular research projects and to the entire enterprise of research.” This is followed by the statement, “In the case of scientific research in general, members of society are obliged to recognize the longer term benefits and risks that may result from the improvement of knowledge and from the development of novel medical, psychotherapeutic, and social procedures.” The obligation to sustain biomedical research rests on the same rules that pertain to human subjects: the moral obligations to mitigate harm and to maximize benefits for the larger society.
6 On the assumption that medical research should be promoted and sustained, Belmont 1) justifies research involving children who do not directly benefit from the research; 2) justifies withholding information about research from patients under certain conditions when “incomplete disclosure is truly necessary to accomplish the goals of the research;” and (3) says that risks to subjects can be “outweighed by the sum of both the anticipated benefit to the subject, if any, and the anticipated benefit to society in the form of the knowledge to be gained from the research.”
7 Harold Y. Vanderpool, “Introduction and Overview: Ethics, Historical Case Studies, and the Research Enterprise,” in Vanderpool, ed., The Ethics of Research Involving Human Subjects (Frederick, MD: University Publishing Group, 1966), pp. 6 and 21.
8 The purpose of protecting human subjects in is the report’s subtitle, and its other purposes are stated in the report’s beginning paragraphs.
9 Vanderpool, ed., p. 40.
10 The first four paragraphs that discuss the report’s purposes are subtitled “Ethical Principles and Guidelines for Research Involving Human Subjects,” and the discussion that follows will show that the term “guidelines” refers to Belmont’s applications. The sentence before and the paragraph that follows the statement concerning Belmont’s “objective” show that the report’s framers viewed its “analytical framework” as inclusive of Belmont’s principles and their applications.
11 The arrangement of this schema is indebted to Tom L. Beauchamp, “How the Belmont Report Was Written,” (Unpublished MS presented at “Belmont Revisited,” a conference at the University of Virginia, 16–18 April, 1999), p. 8.
12 Office of the Secretary, p. 2.
13 Robert J. Levine, Ethics and Regulation of Clinical Research, second edition (Baltimore-Munich: Urban and Schwarzenberg, 1986), pp. 11–15.
14 K. Danner Clouser, “Common Morality as an Alternative to Principalism,” Kennedy Institute of Ethics Journal 5 (September 1995): 219–236, especially 221–224. Some authors argue that appeals to abstract, predetermined principles violate the particularity of concrete situations or that appeals to principles easily result in dogmatism and tyranny. (Seyla Benhabid, Situating the Self: Gender, Community and Postmodernism in Contemporary Ethics (New York: Routledge, 1992) and Stephen Toulmin, “The Tyranny of Principles,” The Hastings Center Report (December 1981): 31–39.) What the principles meant to each of the 11 members of the National Commission probably varied with each member. One commissioner, Albert R. Jonsen, viewed them as “action-guides” predicated upon the philosophical theory and reasoning of H. Tristram Engelhardt, Robert M. Veatch, and other bioethicists who presented scholarly papers to the Commission’s members. (Albert R. Jonsen, The Birth of Bioethics [New York: Oxford University Press, 1998], pp. 102–104, 333 and 357.) Another commissioner, Karen Lebacqz, says that Belmont’s principles represented broader categories that subsequently have been truncated by bioethicists. Lebacqz argues that the principle of “respect for persons” in
O-17
Belmont has been narrowly equated with “respect for autonomy” or freedom of choice, instead of denoting what the commissioners intended—a principle that encompasses respect for autonomous persons, for nonautonomous persons, and for the communities in which persons gain and express their identity. (Karen Lebacqz, “Twenty Years Older But Are We Wiser? (Unpublished MS Presented at “Belmont Revisited,” a Conference at the University of Virginia, 16–18 April, 1999, p. 1–10).
15 Beauchamp created the first drafts of the report, continued to revise the report in responses to feedback from the Commissioners and staff of the National Commission and participated in its final “touchup wordsmithing.” Beauchamp observes that Belmont ended as “a joint product.” Beauchamp, “How the Belmont Report Was Written,” pp. 1–11, quotations from pp. 9 and 10.
16 For a discussion of the interplay between Belmont’s principles and the practice and ethics of clinical medicine in the decades surrounding the writing of the Belmont Report, see Eric J. Cassell, “The Principles of the Belmont Report Revisited: How Have Respect for Persons, Beneficence, and Justice been Applied to Clinical Medicine?” (Unpublished MS Presented at “Belmont Revisited,” a Conference at the University of Virginia, 16–18 April, 1999), pp. 2–37.
17 Beauchamp, “How the Belmont Report Was Written,” p. 19.
18 Jonsen, p. 333.
19 See, e.g., Arthur J. Dyck, On Human Care (Nashville, TN: Abingdon, 1977), pp. 14–16, 52–54.
20 Tom L. Beauchamp, “Principalism and Its Alleged Competitors,” Kennedy Institute of Ethics Journal 5 (September 1995), p. 181.
21 Jonsen, p. 332–338.
22 Lebacqz, p. 9.
23 Beauchamp, “How the Belmont Report Was Written,” p. 7; see also Toulmin, p. 32.
24 Beauchamp, “How the Belmont Report Was Written,” p. 7. During the same time that Beauchamp was drafting versions of Belmont, he and James F. Childress were writing their first edition of Principles of Biomedical Ethics, which discussed how ethical theories 1) generate moral principles, 2) systematically link them together, and 3) defend their universal validity. Tom. L. Beauchamp and James F. Childress, Principles of Biomedical Ethics (New York: Oxford University Press, 1979), pp. xii–xiii; and Beauchamp, “How the Belmont Report Was Written,” pp. 11–14.
25 Robert M. Veatch, “From Nuremberg Through the 1990s: The Priority of Autonomy, ” in Vanderpool, ed., pp. 45–58, quotation from p. 52.
26 Levine, p. 12; and Albert R. Jonsen, “The Weight and Weighing of Ethical Principles,” in Vanderpool, ed., pp. 59–82, especially, p. 64.
27 In two notable places the report appears to avoid prima facie ethics. In the last sentences at the end of the report’s discussions of respect and beneficence, the text says that all or most “hard cases” of ethical conflict require “balancing” choices between the moral claims (or ingredients) within each principle, not, presumably, between them. On the other hand, in at least one place (identified in endnote 6, point 2 above), the report plays its principles off one another in prima facie fashion.
28 Donald Chalmers, “Alternatives to the Current Human Subjects Protection,” in Summary of Commissioned Papers for the Oversight Project (of the NBACP), 25 April 2000.
29 Levine, pp. 11–12.
30 Jonsen, p. 102.
31 Commonly recognized ethical principles that are not mentioned in Belmont include nonmaleficence, gratitude, fidelity, and veracity.
32 Beauchamp describes how the principles the commissioners invoked in their deliberations were “interpreted, modified, and specified by the force of examples and counterexamples.” Beauchamp, “How the Belmont Report Was Written,” p. 17. Belmont includes a number of cases that reflect the Commission’s work.
O-18
33 The report deals with human subjects of research who include competent adults, persons who are not fully competent or are incompetent (due to mental disability, terminal illness, or comatose status), humans in early stages of life (fetuses, infants, and children), and persons in life circumstances that include institutionalization, limited education, poverty (which includes persons dependent on public health care), and situations surrounded by cultural, racial, and gender bias. In the place where Belmont explicitly discusses subject “protection” (the 4th paragraph under Basic Ethics Principles: Respect for Persons), it asserts that, 1) some persons are in need of “extensive protection,” even to the point of excluding them from research that my cause harm, 2) some will be protected by making sure that they know about possible adverse consequences of the research and are free to chose whether to participate, and 3) the extent of protection that should be afforded to respective populations of prospective research subjects depends upon risks of harm and likelihood of benefit.
A general outline of the way the Belmont Report relates each of these levels of protection to the just-listed populations of prospective research subjects encompasses 1) special and ascending levels of protection (to the point of disallowing research) for persons who are vulnerable to harm because they are immature, incompetent, not fully competent, or are subject to coercion or abuse due to their life situations and 2) protection stemming from the principle of respect for persons and the requirements of informed consent (from subjects or their proxies) for all subjects of research, which should not be approved unless it accords with the principle of beneficence and the risk/benefit assessments that are required by beneficence.
34 Larry R. Churchill, “Disrespect for Autonomy: Why the Belmont Report Needs Revision,” (Unpublished MS Presented at “Belmont Revisited,” a Conference at the University of Virginia, 16–18 April, 1999), pp. 2–4; and Beauchamp and Childress, pp. 58–60.
35 The applications say that research for all such subjects 1) “should be considered on its own terms;” 2) should give all these prospective participants the opportunity to choose to the extent they are able, whether or not to participate in research; 3) requires seeking the permission of other parties “who are most likely to understand the incompetent subject’s situation and to act in that person’s best interest,” and 4) requires that these proxy decision makers are “given an opportunity to observe the research as it proceeds in order to be able to withdraw the subject from the research, if such action appears in the subject’s best interest.”
36 The applications also state that risks to subjects are permissible if they are “outweighed by the sum of both the anticipated benefit to the subject, if any” (which should “normally carry special weight”) and “the anticipated benefit to society” as long as “the subjects’ rights [through the process of informed consent] are protected.” They hold that harms and benefits of research pertaining to subjects must be determined by explicit, rational, systematic, nonarbitrary analysis. The “justifiability” of research is predicated on a set of additional considerations, which include determining “whether it is in fact necessary to use human subjects at all,” reducing risks to those that are necessary to achieve the research objective, and the need to demonstrate when it is appropriate to involve vulnerable populations in research.
37 These applications focus on subject selection: the injustice of “placing further burdens on already burdened persons,” the need to be aware of “social, racial, sexual and cultural biases institutionalized in society,” and the “injustice” of recruiting “vulnerable subjects” out of “administrative convenience.”
38 This is reflected in both of the subheadings under the Assessment of Risks and Benefits applications in Belmont, in which riskier research is deemed acceptable as long as “the subject’s rights have been protected” by, for example, “the manifest voluntariness of the participation.”
39 See the discussion in the first paragraph of section II above, and consider the following example of a logic-defying statement: “Applications of the general principles to the conduct of research leads to consideration of the following requirements” (the first sentence under C. Applications).
40 Robert M. Veatch, “Ranking or Balancing: Resolving Conflicts Among the Belmont Principles” (Unpublished MS Presented at “Belmont Revisited,” a conference at the University of Virginia, 16–18 April, 1999) 1–26.
41 Churchill, pp. 3–12; Beauchamp and Childress, pp. 58–60; and Robert M. Veatch, “Resolving Conflicts Among Principles: Ranking, Balancing, and Specifying,” Kennedy Institute of Ethics Journal 5 (September 1995), pp. 203–204.
42 Beauchamp, p. 13; and Churchill, pp. 12–13.
43 Lebacqz, pp. 1–10.
44 In its third sentence under its principle of Justice, Belmont says that “injustice occurs when some benefit to which a person is entitled is denied without good reason.” This point is not mentioned or expanded upon in Belmont’s applications.
45 Carol Levine, “Changing Views of Justice after Belmont,” in Vanderpool, ed., pp. 105–126; and Patricia A. King, “Justice Beyond Belmont,” (Unpublished MS Presented at “Belmont Revisited,” a Conference at the University of Virginia, 16–18 April, 1999), pp. 1–23.
46 See, e.g., Advisory Committee on Human Radiation Experiments, Final Report (Washington, D.C.: U.S. Government Printing Office, October 1995) pp. 827–828.
O-19
47 See, for example, Eric T. Juengst, “Respecting Human Subjects in Genome Research: A Preliminary Policy Agenda,” in Vanderpool, ed., pp. 401–429.
48 Harold T. Shapiro and Eric M. Meslin, “The Influence of the National Commission and the Belmont Report on the National Bioethics Advisory Commission,” (Unpublished MS Presented at “Belmont Revisited,” a conference at the University of Virginia,
16 –18 April, 1999), p. 3.
49 The terms “ethics” or “ethical” are mentioned six other times in the regulations: three times in Subpart B (Research Involving Fetuses, Pregnant Women, and Human In Vitro Fertilization) 46.204 respecting Ethical Advisory Boards that, upon producing two reports between 1978 and 1980, have never since been convened (Jonsen, Birth of Bioethics, pp. 54–55, 106–107, 310–311); and three times in relation to “ethics” as an area of expertise for expert evaluators (twice in 46.306 regarding research with prisoners, and once in 46.407 (b) regarding research with children).
50 The first, second, and fourth of these options are precisely those that are given in the DHEW/Public Health Service (PHS)/NIH
“Yellow Book,” entitled The Institutional Guide to DHEW policy on Protection of Human Subjects (Washington, D.C.: U.S. Government Printing Office, 1971), pp. 5 and 10.
51 This wording and the three bodies of information that are considered to be sufficient sources for determining the acceptability of research are directly drawn from the DHEW “Yellow Book.” DHEW/PBS/NIH, p. 4.
52 Advisory Committee on Human Radiation Experiments, pp. 817–818.
53 Consider how section 46.111 (a) (1) and (2) states that “the IRB shall determine that...risks to subjects are minimized” and that “risks to subjects are reasonable in relation to anticipated benefits” with no mention of Belmont’s accent on utilizing the standard of a careful, systematic, nonarbitrary assessment of the risks and benefits in question.
And consider how section 46.116 (renumbered and reworded above as “Criteria for informed consent”) urges simple-minded rule-following in its long sections 46.116 (a) and (b) that list the “basic” and “additional” elements of informed consent. Several of the rules in these sections deal with certain “statements” that must be made in consent forms. Two of these require that consent forms contain “a statement that the study involves research” (46.116 (a) (1)) and “a statement that participation is voluntary” (46.116 (a) (8)). The regulations indicate that these statements in consent forms satisfy two of the “basic elements of informed consent.” These statements, however, do not satisfy the standards pertaining to informed consent in the Belmont Report—the standards of assuring that subjects are fully informed (in this instance, informed about what “research” is) and of making sure that the circumstances in which research is conducted allow subjects to make voluntary choices, as opposed to merely telling subjects that their participation is “voluntary.” These and similar rules in the regulations encourage researchers to recommend and IRBs to approve of “boiler plate” language in consent forms. Examples of this language lifted word for word from section 46.116 (a) (8) of the federal regulations include the following: “Your participation is voluntary, and you may discontinue participation at any time without penalty of loss of benefits to which you are otherwise entitled.” (Advisory Committee on Human Radiation Experiments, pp. 707–709.) While the power and meaning of this language may not be grasped by many research participants, it is, in fact, precisely what the text of the regulations requires.
54 This part says that IRBs “shall determine” (46.111 (a)) that two requirements about risks should be “satisfied” (46.111 (a) and (b)), as well as other requirements regarding subject selection (46.111 (a) (3)), informed consent (46.111 (a) (4)), and other matters. The language pertaining to subject selection (“Selection of subjects is equitable”) in 46.111 (a) (3) is so cryptic that its moral import is easily lost.
55 With the exception of its last section, 46.116 addresses investigators instead of both IRB reviewers and investigators. The categories in its initial paragraph are not clearly organized around the Belmont Report’s cogent and easily understood and employed three “elements of consent.” The ways 46.116 distorts the meaning of Belmont is discussed in the next paragraph of this paper.
56 This identification of information respecting expected risks and benefits, explanation of procedures to be done, etc., with “the basic elements of informed consent” reflects the precise wording of DHEW’s “Yellow Book.” DHEW/PBS/NIH, p. 7.
57 Benjamin Freedman, “The Ethical Analysis of Clinical Trials: New Lessons for and from Cancer Research,” in Vanderpool, ed., pp. 325–326. Freedman rightly argues that this activity should not be denigrated; see also endnote 53 above, and Advisory Committee on Human Radiation Experiments, pp. 713–719, 817–818, and 874–856.
O-20

The Ethical Analysis of Risks and Potential Benefits in Human Subjects Research: History, Theory, and Implications for U.S. Regulation
Commissioned Paper Charles Weijer Dalhousie University
P-1
Summary
This paper addresses three questions central to the ethical analysis of risks and potential benefits in human subjects research:
1) How was the ethical analysis of risk understood by the members of the U.S. National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission)?
2) What conceptual framework should guide the ethical analysis of risk?
3) What changes to U.S. regulations would the implementation of such a framework require?
Three distinct views on the ethical analysis of risks and potential benefits in research can be found in the National Commission’s opus: analysis of entire protocols; analysis of protocols with particular components; and analysis of components. Basing the moral analysis of risk on the categorization of research into therapeutic or nontherapeutic research poses two problems: 1) “therapeutic research” is a contradiction in terms, and 2) research subjects are inadequately protected, as any number of procedures not for the benefit of subjects may be added to a therapeutic study. Recognizing these shortcomings, the National Commission adopted an analysis of risk that focused on whole protocols with particular components. New problems arise with this approach. Little guidance is given for the analysis of research that presents less than minimal risk; the concept of minimal risk is applied to both therapeutic and nontherapeutic procedures but sets a threshold for allowable risk only for nontherapeutic procedures; and, since clinical research often contains a mixture of procedures, differing rules for whole protocols may apply simultaneously, leading to confusion and conflict.
The ethical analysis of the various components in a research study seems to present a number of advantages:
1) It acknowledges that clinical research often contains a mixture of procedures, some administered with therapeutic intent and others solely to answer the research question.
2) Therapeutic procedures and nontherapeutic procedures are, by definition, administered with differing intent. This difference is morally relevant.
3) Therapeutic procedures are justified by their potential to benefit the subject, while nontherapeutic procedures are justified by their potential to generate knowledge. These two types of benefit are largely incommensurable.
4) Rigorous separate moral calculi for therapeutic and nontherapeutic procedures protect research subjects better than previous approaches. They prevent the justification of risky nontherapeutic procedures by the benefits that may flow from therapeutic procedures that are components of the same study.
5) It is a parsimonious model for analysis and thereby avoids confusion and conflict.
The model advocated in this paper establishes the separate ethical analysis of therapeutic and nontherapeutic procedures in research. Therapeutic procedures are those study interventions administered with therapeutic intent. The Institutional Review Board (IRB) must ensure that such procedures fulfill the requirements of clinical equipoise—that is, they must ensure that, at the start of the study, genuine uncertainty exists in the community of expert practitioners as to the preferred treatment. Nontherapeutic procedures are not administered with therapeutic warrant and are administered in the interest of answering the research question. The IRB must ensure that the risks associated with such procedures are 1) minimized and 2) reasonable in relation to the knowledge to be gained.
Minimal risk is a concept through which the risks of nontherapeutic procedures are compared to the risks of daily life. Minimal risk is used in regulation as a sorting mechanism and as a protection for the vulnerable. As a sorting mechanism, minimal risk is used to direct the attention of the IRB to risky research. Protection of the
P-3
vulnerable is, however, its most important role. Groups may be vulnerable for one or more of three reasons: they may be unduly susceptible to risk; they may be incapable of providing informed consent to study participation; or they may be in circumstances that throw the voluntariness of their consent into question. Protections for vulnerable groups include ensuring that the study hypothesis requires the inclusion of the vulnerable group; seeking consent from a proxy decisionmaker when subjects are incapable of giving consent; and limiting the amount of nontherapeutic risk to which subjects may be exposed to minimal risk or a minor increase over minimal risk.
A number of changes to the Common Rule and Department of Health and Human Services (DHHS) regulations are required if the regulations are to be consistent with this more comprehensive framework for the ethical analysis of risk:
1) Ambiguity in current regulations caused by a multiplicity of conceptual models must be eliminated. A single conceptual model should underlie all regulations for the protection of research subjects.
2) Definitions for therapeutic and nontherapeutic procedures should be added.
3) The IRB’s general obligations regarding the ethical analysis of potential benefits and risks of research participation must be stated more clearly.
4) The definition of minimal risk should be clarified.
5) Standards for expedited review must be more rigorous.
6) Regulations for the ethical analysis of risk in research on children should be greatly simplified.
7) A new subpart detailing protections for incapable adults should be added.
Introduction
The IRB is a social oversight mechanism charged with the mandate of protecting research subjects. Performing this task competently requires that the IRB scrutinize informed consent procedures, the balance of risks and potential benefits, and subject selection procedures in research protocols. It may be that IRBs spend too much time editing informed consent forms and too little time analyzing the risks and potential benefits posed by research.1 This imbalance is clearly reflected in the research ethics literature. A review of articles published between 1979 and 1990 in IRB: A Review of Human Subjects Research, for example, reveals a large number of articles on informed consent and confidentiality (142 articles), and considerably fewer on risk-benefit assessment (40), study design (20), and subject selection procedures (5).2 The obligation to ensure that study participation presents a favorable balance of potential benefits and risks to subjects is central to upholding the ethical principle of beneficence and fulfilling the IRB’s protective function.3 Some believe it to be the single most important determination made by the IRB. It ensures that potential research subjects—be they sick or well, young or old, capable or not—are presented with the option of entering a research study only when agreeing to study participation would be a reasonable choice.
Accordingly, the Common Rule requires that the IRB ensure that:
(1) Risks to subjects are minimized: (i) by using procedures which are consistent with sound research design and which do not unnecessarily expose subjects to risk, and (ii) whenever appropriate, by using procedures already being performed on the subjects for diagnostic or treatment purposes.
(2) Risks to subjects are reasonable in relation to anticipated benefits, if any, to subjects, and the importance of the knowledge that may reasonably be expected to result. In evaluating risks and benefits, the IRB
P-4
should consider only those risks and benefits that may result from the research (as distinguished from risks and benefits of therapies subjects would receive even if not participating in the research). The IRB should not consider possible long-range effects of applying knowledge gained in the research (for example, the possible effects of the research on public policy) as among those research risks that fall within the purview of its responsibility (45 CFR 46.111(a)).
The moral analysis of risk is neither obvious nor intuitive. Rules, including those of the Common Rule, are not self-interpreting. They must be situated within a conceptual framework that facilitates their interpretation by the IRB. The articulation of a conceptual framework for the ethical analysis of risk might therefore be a project assisting IRBs in fulfilling their mandate—the protection of research subjects.
Of the analysis of risk in research, the authors of the Belmont Report observe that “[i]t is commonly said that benefits and risks must be ‘balanced’ and shown to be ‘in a favorable ratio.’ The metaphorical character of these terms draws attention to the difficulty of making precise judgments.”3 Unpacking these metaphors will occupy the bulk of this paper. We will focus on three questions:
1) How was the ethical analysis of risk understood by the members of the National Commission?
2) What conceptual framework should guide the ethical analysis of risk?
3) What changes to U.S. regulations would the implementation of such a framework require?
The work of the National Commission receives special consideration in this paper. No other ethics body has had as much influence on the development of research ethics and regulation. As we shall see, pivotal conceptual advances in the moral analysis of risks and potential benefits can be traced back to work of the National Commission.
The last papers on the ethical analysis of risk written for a major U.S. ethics body were written almost 25 years ago by Robert J. Levine, a staff member and consultant to the National Commission. In “The Boundaries Between Biomedical or Behavioral Research and the Accepted and Routine Practice of Medicine,” Levine recognizes that many clinical studies are “complex activities,” involving both therapeutic and nontherapeutic procedures.4 In the second paper, “The Role of Assessment of Risk Benefit Criteria in the Determination of the Appropriateness of Research Involving Human Subjects,” he comprehensively describes the risks and benefits presented by research to subjects and society.5 He argues convincingly that quantitative approaches to risk analysis will, at best, be of limited use to the IRB.
As we shall see, there have been considerable refinements in our understanding of the ethical analysis of risk in the last 25 years. Nonetheless, this paper, solicited by yet another commission, the National Bioethics Advisory Commission (NBAC), relies heavily on the solid intellectual work that precedes it. All of the work of the National Commission is a source for learning; much of it should be preserved in our current understanding and regulation. Levine’s papers for the National Commission remain foundational in research ethics. This paper will assume familiarity with them.
Risks and Potential Benefits in Research Involving Human Subjects
Risk is a multidimensional concept involving both the probability and magnitude of harms to research participants.6 All too often, risk is equated with the magnitude of the outcome, e.g., death or serious disability. The proper ethical analysis of risk requires that both the magnitude of the harm and its probability of occurring be considered. A risk of death of one in one million is properly treated differently than a risk of death of one in ten. Benefit, on the other hand, is the magnitude of a positive outcome without reference to its probability.
P-5
In reference to the comparison of risks to benefits, reference is often made of the need to consider the “risk-benefit ratio” presented by study participation. This is not a parallel construction, and, hence, it is, strictly speaking, incorrect. One speaks accurately of “harms and benefits” or “risks and potential benefits.” Research subjects may be exposed to a broad array of risks and potential benefits as a result of study participation. Risk is not a concept exclusive to biomedical research; social science studies also present risks to participants. Indeed, there is a surprising degree of overlap between the kinds of risks presented in biomedical and social science research. As study methodologies continue to cross conventional disciplinary boundaries, we can expect increasing convergence in the risks and potential benefits in biomedical and social science studies. We will thus need to consider whether the moral calculi involved in risk assessment suffice for the assessment of risks of research in a variety of disciplines. Consider the risks to participants in the following four case studies:
Study A: Placebo controlled trial of a drug for people with acutely symptomatic schizophrenia. The study involves schizophrenic patients who are newly hospitalized with acute symptoms of their disease.7 Despite the existence of effective treatment for such symptoms, patients are randomized to a new antipsychotic drug, a standard drug, or placebo. Patients are treated in hospital for four weeks, and a variety of psychometric scales are measured. Risks to subjects include the possibility that the new medication may have serious adverse effects, some of which may be irreversible; patients assigned to placebo will be deprived of needed treatment for a month; patients may suffer from continuing hallucinations or paranoia; patients may be at increased risk of suicide; and, finally, patients may pose a risk to others. (The ethics of placebo controlled trials in schizophrenia is discussed in detail elsewhere.8)
Study B: Hypnotic induction of partial deafness to see whether paranoid symptoms result.
Hypnotically suggestible but otherwise healthy college students are randomly allocated to three different hypnotic suggestions: partial deafness without awareness of the cause; partial deafness with awareness of the cause; and no deafness but an ear itch.9 The hypothesis is that persons in the first group, compared to the other two groups, will demonstrate more symptoms of paranoia. Subjects are assessed with a variety of measures, including psychometric scales and scoring of observed behavior. After evaluation, subjects are hypnotized again, debriefed at the end of the study, and reassessed at one month. The study poses a variety of risks to participants, including distress associated with paranoia and hearing loss, suicide, the possibility of harm to others, and uncertain sequelae from hypnosis. (Some of the ethical issues raised by this study are discussed elsewhere.10)
Study C: Questionnaire examining adolescent sexual practices. The study involves the administration of a pencil and paper questionnaire to 400 Minneapolis high school students during regularly scheduled health classes.11 The survey seeks to document attitudes and behaviors related to HIV prevention. Accordingly, adolescent participants are asked whether they are sexually active, what types of sexual activity they have experienced (e.g., oral, vaginal, or anal intercourse), and the gender(s) of their partners. A variety of risks are presented by this study to participants: teachers or parents may become aware of undisclosed sexual activity; others may become aware of same-sex relationships; and participants might become aware that they are at risk of developing HIV. (The ethical issues raised by this study are thoroughly reviewed elsewhere.11)
P-6
Study D: Genetic epidemiology of BRCA1 and BRCA2 mutations in Ashkenazic Jews. The BRCA1 and BRCA2 mutations are known to be associated with an increased risk of breast and ovarian cancer. The study seeks to determine what proportion of Ashkenazic Jews carry the mutations in question and what risk is conferred by them in a nonhigh- risk population.12 Participants who respond to advertisements will be asked to give a blood sample and fill out an epidemio-logical survey including questions on health, family history of cancer, and family members who might also be willing to participate. Personal identifiers will be destroyed before genetic tests are conducted, and test results will not be disclosed to participants. Risks to participants are the risks of a venipuncture, the risk of anxiety provoked by answering questions related to family history of cancer, and risks of genetic testing, including unwanted disclosure of risk, discrimination, and stigmatization. (A review of ethical issues in genetic epidemiology studies may be found elsewhere.13)
As illustrated by these four examples, research participation may expose the study participant to a wide spectrum of risks. Levine classifies risks into four categories: physical, psychological, social, and economic.6 Let us consider each briefly:
So defined, risk is an inherently inclusive concept. As demonstrated by the above examples, a given study may present a variety of types of risk. For example, study C (sex questionnaire) posed both psychological and social risks. Furthermore, no category of risk is exclusive to medical or social science studies: study B (deafness and paranoia), a social science study, presented physical risks, and studies A (schizophrenia trial) and D (breast cancer genes), medical studies, generated psychological risks. Despite the various disciplinary backgrounds involved, all four of the study examples posed nontrivial risk to research subjects.
Levine provides a comprehensive description of particular potential benefits and risks presented to research subjects and society by biomedical and social science research, and the listing will not be repeated here.5
The Analysis of Risks and Potential Benefits in the Work of the National Commission
The first major question to be addressed regards how the ethical analysis of risk was understood by the members of the National Commission. The National Commission sat from 1975 to 1978 and issued a total of ten reports on differing aspects of human subjects research. The National Commission’s work represents the first sustained in-depth exploration of the moral analysis of risk in research. As such, it has had a lasting influence on research ethics scholarship and federal regulation. Little recognized is the fact that the National Commission’s views on risk analysis evolved over its four-year term. Three distinct views on the ethical analysis of risks and potential benefits in research can be found in the National Commission’s opus: analysis of entire protocols; analysis of protocols with particular components; and analysis of components.
P-7
Six reports of the National Commission were selected for this analysis based on their impact on public policy and the perception by National Commission staff of the overall success of the report.14 These reports are
Research on the Fetus (1975);15 Research Involving Prisoners (1976);16 Research Involving Children (1977);17 Research Involving Those Institutionalized as Mentally Infirm (1977);18 Institutional Review Boards (1977);19 and the Belmont Report (1978).3 What follows is a critical review of approaches to the ethical analysis of risks and potential benefits found in each of the reports. The interpretation is my own and is based on a review of the primary source documents.
The ethical analysis of risks and potential benefits presented by entire protocols.
Research on the Fetus was the first of the National Commission’s reports. It was produced under several constraints.15 Congress required the completion of the report in only four months, and it imposed a moratorium on fetal research pending the completion of the report. Thus, Levine observes that:
As a consequence of these time constraints, the Commission completed its report, Research on the Fetus, before it had the opportunity to address the general conceptual issues in its mandate. If the conceptual clarifications…had preceded the report, it is likely that the Commission would have developed substantially different recommendations.20
In the report, the National Commission defines research as “the systematic collection of data or observations in accordance with a designed protocol” (p. 6).15 The schema for risk analysis presented in Research on the Fetus relies on separating whole research proposals into two types: therapeutic research and nontherapeutic research. Therapeutic research is that which is “designed to improve the health condition of the research subject by prophylactic, diagnostic, or treatment methods that depart from standard medical practice but hold out a reasonable expectation of success” (p. 6).15 Nontherapeutic research, on the other hand, is “not designed to improve the health condition of the research subject by prophylactic, diagnostic, or treatment methods” (p. 6).15 Separate recommendations are presented for each type of study. Recommendation 1 addresses therapeutic research directed toward the fetus. Under this provision:
[t]herapeutic research directed toward the fetus may be conducted or supported, and should be encouraged, by the Secretary, DHEW, provided such research (a) conforms to appropriate medical standards, (b) has received the informed consent of the mother, the father not dissenting, and (c) has been approved by existing review procedures with adequate provision for the monitoring of the consent process (p. 73).15
Recommendation 4 outlines different ethical criteria for the assessment of nontherapeutic research. It states that:
[n]ontherapeutic research directed towards the fetus in utero (other than research in anticipation of, or during, abortion) may be conducted or supported by the Secretary, DHEW, provided (a) the purpose of such research is the development of important biomedical knowledge that cannot be obtained by alternative means, (b) investigation on pertinent animal models and non-pregnant humans has preceded such research, (c) minimal or no risk to the well-being of the fetus will be imposed by the research, (d) the research has been approved by existing review procedures with adequate provision for the monitoring of the consent process, (e) the informed consent of the mother has been obtained, and (f) the father has not objected to the research (p. 74).15
P-8
While there is intuitive appeal in categorizing studies as a whole, as either therapeutic or nontherapeutic, the validity of this approach has been criticized. Levine points out that this distinction invariably leads to deep conceptual problems. This is illustrated by inserting the National Commission’s definition of research into its definition of therapeutic research, as Levine does here:
There is, of course, no such thing as a ‘systematic collection of data or observations…designed to improve the health condition of a research subject…that departs from standard medical practice.’ Thus, the Commission developed recommendations for the conduct of a nonexistent set of activities….20
A further problem exists with this approach. The inclusion of one or more therapeutic procedures in a study will lead it to being identified as therapeutic research. Once this categorization has taken place, there is no limit to procedures without therapeutic intent that might be administered to research subjects. Thus, this approach not only leads to confusion, it leaves research subjects without adequate protection.
Levine correctly observes that “all ethical codes, regulations, and commentaries relying on the distinction between therapeutic and non-therapeutic research contain serious errors.”20 The Declaration of Helsinki, perhaps the source of the National Commission’s approach to risk in Research on the Fetus, relies on the distinction. Article III.2 requires of nontherapeutic biomedical research that “[t]he subjects should be volunteers—either healthy persons or patients for whom the experimental design is not related to the patient’s illness.” This would seem to require that research into the pathophysiology of disease be conducted (absurdly) on those who are either healthy or who have a disease other than that of interest. This sort of thinking continues to pervade the work even of well-known thinkers in research ethics. Baruch Brody, for instance, in his recent book The Ethics of Biomedical Research concludes that phase I chemotherapy studies are nontherapeutic research.21 Despite endorsing the Declaration of Helsinki, he fails to recognize the entailment that such toxic studies, posing a risk of death to participants, must be done on healthy volunteers or persons with some disease other than cancer.22 Despite its shortcomings, this approach to the ethical analysis of risk is found in current DHHS regulations on the protection of fetuses in research. The regulations divide research on the fetus into two categories: research “to meet the health needs of the particular fetus,” i.e., therapeutic research; and research for “the development of important of biomedical knowledge,” i.e., nontherapeutic research (45 CFR 208(a)). As this approach to the ethical analysis of risk is not found elsewhere in the federal Common Rule or DHHS regulations, one interpretation is that it is a historical artifact of Research on the Fetus in current regulation.
The ethical analysis of whole protocols with particular components.
Recognizing these problems with the distinction between therapeutic and nontherapeutic research, the National Commission largely abandoned the use of these terms in subsequent reports. In the preface to Research Involving Prisoners they state: “The Commission recognizes problems with employing the terms ‘therapeutic’ and ‘nontherapeutic’ research, notwithstanding their common usage, because they convey a misleading impression” (p. x).16 In Research Involving Prisoners the therapeutic research category is replaced with “research on practices which have the intent and reasonable probability of improving the health and well being of the subject” (p. xi).16 While cumbersome, this manner of speaking at least avoids the conceptual confusion pointed to by Levine supra. The National Commission recognizes that:
Additional interventions over and above those necessary for therapy may need to be done, e.g., randomization, blood drawing, catheterization; these interventions may not be ‘therapeutic’ for the individual. Some of these interventions may themselves present risk to the individual—risk unrelated to the therapy of the subject (p. xi).16
P-9
Despite this, it remains unclear in the report just how one is to determine whether such nontherapeutic risks are at an acceptable level. Indeed, Recommendation 4 merely states, in part, that “[a]ll research involving prisoners should be reviewed by at least one human subjects review committee or Institutional Review Board….[T]he committee or board [IRB] should consider at least the following: the risks involved…” (p. 20).16 Clearly, IRBs require more detailed guidance on the ethical analysis of risks and potential benefits in research than is provided in Research Involving Prisoners.
It has been suggested that this and other failings of the report may be due to the fact that members of the National Commission confused the need for prison reform with the need for protection of prisoners in research.23 Be this as it may, the report does contain early ruminations about the notion of “minimal risk.” Minimal risk is referred to in Research on the Fetus, but only in Research Involving Prisoners does one see recognizable beginnings of what would become a central concept in the moral analysis of risk. A standard similar to that of minimal risk is articulated for research without therapeutic procedures:
Research designed to determine the effects on general health of institutional diets and restricted activity, and similar studies that do not manipulate bodily conditions (except innocuously, e.g., obtaining blood samples) but merely monitor or analyze such conditions, also present little physical risk and are necessary to gain some knowledge of the effects of imprisonment (p.15).16
Furthermore, there is an explicit recognition that in determining which risks should be acceptable, comparison is to be made between risks of research and those of daily life, in this case, the daily lives of persons who are not incarcerated:
The risks involved in research involving prisoners should be commensurate with risks that would be accepted by non-prisoner volunteers. If it is questionable whether a particular project is offered to prisoners because of the risk involved, the review committee might require that non-prisoners be included in the same project (p. 20).16
Both of these standards find expression in current DHHS regulations (45 CFR 306(a)(2)(A); 45 CFR 46.303(d)).
The concept of minimal risk is first fully expressed in the National Commission’s report Research Involving Children.17 It is perhaps natural that the most detailed recommendations regarding the analysis of risks and potential benefits are found in this report. Levine explains that:
Because infants and very young children have no autonomy, there is no obligation to respond to it through the usual devices of informed consent. Rather, respect for infants and very small children requires that we protect them from harm. No discernable risk seemed to the commission to be virtually impossible; therefore, they stipulated a definition of ‘minimal risk’ as the amount that would be acceptable without unusual standards for justification.24
The National Commission defines minimal risk as “the probability and magnitude of physical or psychological harm that is normally encountered in the daily lives, or in the routine medical or psychological examination, of healthy children” (p. xx).17 This definition differs from that found in the DHHS regulations in its stipulation of healthy children; DHHS does not so limit minimal risk (45 CFR 46.120(i)). The National Commission provides a number of prima facie examples of procedures that pose no more than minimal risk, including “routine immunization, modest changes in diet or schedule, physical examination, obtaining blood and urine specimens, and developmental assessments [emphasis added]” (p. 20).17 Again, this differs from DHHS regulation in its inclusion of a procedure, routine immunization, administered with therapeutic intent.
P-10
The concept of minimal risk is central to the schema for risk analysis presented in Research Involving Children. Recommendation 2 requires that the IRB ensure that “[r]isks are minimized by using the safest procedures consistent with sound research design and by using procedures performed for diagnostic or treatment purposes whenever feasible” (p. 2).17 Thus, if a blood sample is needed from a child, one should, where possible, use blood left over from a venipuncture done for therapeutic purposes. If the research does not involve therapeutic or nontherapeutic procedures that present more than minimal risk, it may be approved provided the above condition is fulfilled. Recommendation 3 states:
Research that does not involve greater than minimal risk to children may be conducted or supported provided that an Institutional Review Board has determined that: (A) the conditions of Recommendation (2) are met; and (B) adequate provisions are made for assent of the children and permission of their parents or guardians, as set forth in Recommendations (7) and (8) (p. 5).17
Separate recommendations, as follows, apply to research involving therapeutic or nontherapeutic interventions that exceed the minimal risk threshold.
If research involving a therapeutic intervention poses more than minimal risk, the IRB must ensure that the balance of potential benefits and risks of the intervention is at least as favorable as alternatives.
Recommendation 4 follows:
Research in which more than minimal risk to children is presented by an intervention that holds out the prospect of direct benefit for the individual subjects, or by a monitoring procedure required for the well-being of the subjects, may be conducted or supported provided that an Institutional Review Board has determined that:
| (A) |
such risk is justified by the anticipated benefit to the subjects; |
| (B) |
the relation of anticipated benefit to such risk is at least as favorable to the subjects as that presented by available alternative approaches; |
| (C) |
the conditions of recommendation (2) are met; and |
| (D) |
adequate provisions are made for assent of the children and permission of their parents or guardians, as set forth in Recommendations (7) and (8) (pp. 5–6).17 |
In short, the IRB should evaluate such interventions in the same way as they are evaluated in clinical practice:
It should compare the risk and anticipated benefit of the intervention under investigation (including the monitoring procedures necessary for the care of the child) with those of available alternative methods for achieving the same goal, and should also consider the risk and possible benefit of attempting no intervention whatsoever (p. 7).17
If, on the other hand, the research involves a nontherapeutic intervention that poses more than minimal risk, the provisions of Recommendation 5 apply:
Research in which more than minimal risk to children is presented by an intervention that does not hold out the prospect of direct benefit for the individual subjects, or by a monitoring procedure not required for the well-being of the subjects, may be conducted or supported provided an Institutional Review Board has determined that:
(A) such risk represents a minor increase over minimal risk;
P-11
| (B) |
such intervention or procedure presented experiences to subjects that are reasonably commensurate with those inherent in their actual or expected medical, psychological or social situations, and is likely to yield generalizable knowledge about the subject’s disorder or condition; |
| (C) |
the anticipated knowledge is of vital importance for understanding or amelioration of the subject’s disorder or condition; |
| (D) |
the conditions of Recommendation (2) are met; and |
| (E) |
adequate provisions are made for assent of the children and permission of their parents or guardians, as set forth in Recommendations (7) and (8) (pp. 7–8).17 |
Risks presented by nontherapeutic procedures are justified, therefore, in part by the importance of the knowledge to be gained from the research study as a whole. However important the knowledge, risks associated with the nontherapeutic interventions are effectively limited to “a minor increase over minimal risk.” (Risks exceeding this threshold require the approval of a National Ethics Advisory Board and the Secretary of the responsible federal agency [Recommendation 6].) The majority of the members of the National Commission defend this threshold for permissible risk as posing no significant threat to the child’s health. The added requirement that such risks be commensurate to the child’s experience ensures that such risks will be familiar. “Such activities, then, would be considered normal for these children” (p. 139).17 Importantly, if the research involves both a therapeutic intervention and a nontherapeutic intervention that exceed minimal risk, then both Recommendations 4 and 5 are to be applied by the IRB.
This provision (Recommendation 5) was the subject of the most enduring disagreement among members of the National Commission. Turtle dissented from the provision arguing that it should be impermissible to expose children to nontherapeutic procedures that pose more than minimal risk. He objected strenuously to the suggestion that sick children might be exposed to greater nontherapeutic research risk than healthy children:
Children, who through no fault or choice of their own, are subjected to greater risks incident to their condition or treatment, cannot ethically be assumed to qualify for additional increments of risk. To do so, is to add to the potential burdens that result, directly or indirectly, from the child’s illness (p. 148).17
It scarcely needs to be observed that these provisions for the moral analysis of risk are complex. The recognition that a study may involve therapeutic procedures, nontherapeutic procedures, or both is a substantial leap forward over the schema for risk analysis found in Research on the Fetus. The members of the National Commission have solved both of the shortcomings associated with the attempt to classify research as therapeutic or nontherapeutic discussed supra. The solution nonetheless suffers from a number of problems of its own:
1) The concept of minimal risk is applied to both therapeutic and nontherapeutic procedures in both the examples provided and in Recommendation 3. It is unclear, moreover, in what meaningful way minimal risk can apply to therapeutic procedures. According to Recommendation 4, therapeutic procedures that are more than minimal risk are justified as they are in clinical practice. In other words, there is no limit to the risk that may be posed by such procedures so long as they are reasonable in relation to potential benefits. Only nontherapeutic procedures should be subject to a threshold for permissible risk, such as “a minor increase over minimal risk.”
2) The National Commission’s use of the concept of minimal risk in the recommendations seems at odds with its definition. Recall that the National Commission defines minimal risk as risks commensurate to those of daily life of healthy children. Fixing the standard to the daily lives of healthy children seems designed to protect sick children from being exposed to more nontherapeutic research risks than healthy children. This
P-12
presumed intention is contradicted by Recommendation 5 which allows nontherapeutic risks that are a “minor increase over minimal risk” so long as “such intervention of procedure presents experiences to subjects that are reasonably commensurate” with their experience (p. 7).17 Thus, a spinal tap done purely for research purposes may be permissible in a child with a neurological disorder in which such procedures are common, but not in a healthy child. The definition of minimal risk would be consistent with its use in this section if it omitted reference to healthy children, as is the case in current DHHS regulation (45 CFR 46.102(i)).
3) Little guidance is provided for the analysis of risks and potential benefits for procedures that pose no more than minimal risk (Recommendation 3). Recommendation 2 requires that “[r]isks are minimized by using the safest procedures consistent with sound research design” (p. 2).17 This cannot, however, sensibly apply to risks posed by therapeutic procedures, as considerations of research design are largely irrelevant to them. One might reasonably ask the following question: What ethical test should the IRB apply to research involving a therapeutic procedure posing no more than minimal risk? No answer is forthcoming in this report.
4) Research may involve both therapeutic and nontherapeutic procedures. Indeed, I think it is fair to say that this is often or always the case in clinical research. If a study involves a therapeutic intervention and a nontherapeutic intervention, then multiple recommendations may apply. The various possibilities are summarized in Table 1. If both procedures present only minimal risk, then only Recommendation 3 applies. If the therapeutic procedure is more than minimal risk but the nontherapeutic procedure is minimal risk, then Recommendations 3 and 4 apply. If the reverse, then Recommendations 3 and 5 apply. Finally, if both procedures present more than minimal risk, then Recommendations 4 and 5 apply. Since each of the recommendations refer to a research study as a whole involving a particular type of intervention, it is unclear how multiple recommendations are to be applied to a particular study. Without doubt, it is a cumbersome approach and, worse, it may easily lead to confusion or conflict.
Table 1. Applicability of Differing Recommendations from Research Involving Children in a Mixed Clinical Study
| Therapeutic procedure | ||
| Nontherapeutic procedure | No more than minimal risk | More than minimal risk |
| Recommendation 3 only | Recommendation 3 and | |
| No more than minimal risk | Recommendation 4 | |
| Recommendation 3 and | Recommendation 4 and | |
| More than minimal risk | Recommendation 5 | Recommendation 5 |
Despite these difficulties, the model for risk assessment found in Research Involving Children is clearly reflected in current DHHS regulations for the protection of children in research. Indeed, there is a one-to-one correspondence between certain regulations and recommendations made by the National Commission.
45 CFR 46.404, “Research not involving greater than minimal risk,” corresponds to Recommendation 3; 46.405, “Research involving greater than minimal risk but presenting the prospect of direct benefit to the individual subjects,” corresponds to Recommendation 4; 46.606, “Research involving greater than minimal risk and no prospect of direct benefit to individual subjects, but likely to yield generalizable knowledge about the subject’s disorder or condition,” corresponds to Recommendation 5; and 46.407, “Research not otherwise approvable which presents an opportunity to understand, prevent, or alleviate a serious problem affecting the
P-13
health or welfare of children,” corresponds to Recommendation 6. Note that the conceptual model for risk analysis underlying 45 CFR 46.404-407 differs from that underlying protections for the fetus, 45 CFR 46.208(a), noted supra.
The schema for the analysis of risks and potential benefits of research found in Research Involving Those Institutionalized as Mentally Infirm is essentially identical to that found within Research Involving Children.18
Accordingly, only a few comments need to be added at this point. The report refers primarily to persons who are both incapable of providing informed consent and institutionalized. It addressed problems of including such persons in research by incorporating elements of Research Involving Prisoners and Research Involving Children. The definition of minimal risk refers to the “risk…normally encountered in the daily lives…of normal persons” (p. 8).18 Thus, the risks associated with institutionalization may not be used to justify exposing subjects to greater research risks. Recommendations 1 through 5 map onto Recommendations 2 through 6 found in Research Involving Children, and they will not be further elaborated here.
Perhaps the most remarkable fact about the report is its failure to be translated into regulation. Like children, adults incapable of providing informed consent are a vulnerable population in need of protection. Some have suggested that this failure is in part the result of the report’s exclusive focus on institutionalized incapable persons. On this point, the National Commission merely responded to the charge provided to it by the Congress. The President’s Commission (1980–1983) repeatedly called for the entrenchment of protections for incapable adults in regulation.25,26 According to Levine:
The Secretary of the Department of Health and Human Services (DHHS) responded that, ‘while continuing to consider specific issues regarding protections for institutionalized mental patients, the Department is not intending to issue additional regulations in the near future.’ He provided two justifications: ‘first, that the rules proposed by the Department in November 1978 had produced a lack of consensus and, second, that the basic regulations on human subjects research adequately respond to the recommendations made by the National Commission to protect persons institutionalized as mentally disabled….’27
This is a remarkable assertion considering the fact that DHHS regulations contain no special protections for incapable adults in research.
The ethical analysis of risks and potential benefits of components of a research study.
The final works of the National Commission are typified by a move towards a model of the analysis of risks and potential benefits of components of studies, be they therapeutic interventions or nontherapeutic interventions. The move is, however, incomplete. Previous work of the National Commission has focused on risk analysis for particular vulnerable populations. In Institutional Review Boards, members of the National Commission articulate for the first time ethical standards to apply to the review of all human subjects research. The report acknowledges explicitly that a protocol may contain therapeutic procedures, nontherapeutic procedures, or both:
A research project is described in a protocol that sets forth explicit objectives and formal procedures designed to reach those objectives. The protocol may include therapeutic and other activities intended to benefit the subjects, as well as procedures to evaluate such activities (p. xx).19
Risks must be analyzed systematically and should involve a procedure-by-procedure review of risks, benefits, and alternatives. In the words of the National Commission, “[t]his evaluation should include an arrayal of alternatives to the procedures under review and the possible harms and benefits associated with each alternative”
P-14
(p. 23).19 The risks associated with particular procedures are acceptable only if “risks to subjects are minimized by using the safest procedures consistent with sound research design and, wherever appropriate, by using procedures being performed for diagnostic or treatment purposes; [and] risks to subjects are reasonable in relation to anticipated benefits to subjects and importance of knowledge to be gained….” (Recommendation 4; pp.
19 –20).19
The Belmont Report surprisingly provides little additional detail with regard to this model for the ethical analysis of risk. It famously articulated three ethical principles guiding the conduct of clinical research: respect for persons, beneficence, and justice. Beneficence demands that one 1) do no harm and 2) maximize possible benefits while minimizing harms.19 The translation of this principle into practice requires that the IRB ensure that research participation presents subjects with a favorable balance of possible benefits and risks. The Belmont Report once again emphasizes that this is to be done in a systematic and rigorous manner:
…the idea of systematic, nonarbitrary analysis of risks and benefits should be emulated insofar as possible. This ideal requires those making decisions about the justifiability of research to be thorough in the accumulation and assessment of information about all aspects of the research, and to consider alternatives systematically. This procedure renders the assessment of research more rigorous and precise, while making communication between review board members and investigators less subject to misinterpretation, misinformation and conflicting judgments.19
While it encourages the IRB to be explicit about precisely how risks and potential benefits are analyzed, it is itself not more explicit than the previous report, Institutional Review Boards.
Levine, a staff member and consultant to the National Commission, renders the thinking of the National Commission somewhat clearer in two papers contained within the appendix to the Belmont Report. In “The Boundaries Between Biomedical or Behavioral Research and the Accepted and Routine Practice of Medicine,” the existence of “complex activities” in research is recognized.4 Such activities involve procedures administered with different intent in the research setting. Some interventions may be administered for therapeutic purposes, while other procedures are done solely to answer a scientific question. It is this difference in intent that drives the ensuing moral analysis of components of research.
Levine illustrates this point in a lengthy but instructive example of just such a complex research study. In it he weighs the risks and potential benefits of each component of the research separately:
…the benefit [to the subject] will ordinarily derive from those aspects of the complex activity that may be considered practice rather than research. For example, if one wishes to study the effects of chlorothiazide (a diuretic) on sodium balance in patients with congestive heart failure and if one selects subjects in whom chlorothiazide is indicated and administers the drug in appropriate doses, the subject may receive direct therapeutic benefit. This study might be accomplished in a metabolic research ward. It might involve a period of two or three weeks of eating a constant diet with precise control of sodium content. It might involve repeated sampling of venous blood and collection of all urine excreted during those two or three weeks for purposes of sodium assay. The patient may be expected to receive direct therapeutic benefit through administration of the drug; however, this is only technically a research intervention. The subject will not benefit ordinarily from repeated blood sampling and urine collection. The subject may or may not benefit from the constant diet; it might be more or less nutritious and/or palatable that the diet to which he [sic] is ordinarily accustomed. The subject might also benefit from a two or three week period of relative rest on a metabolic research ward. If the subject required hospitalization for that long a period of time anyhow it is likely that he will find the accommodations better on the metabolic ward than on the usual hospital ward;
P-15
however, this is not true in all hospitals. If the subject did not require hospitalization for therapeutic purposes, the period of incarceration might be viewed as more an inconvenience than as a benefit. Further, owing to the customary practices of very careful scrutiny of all activities on metabolic research wards, the subject might derive additional benefits as follows: Any adverse effects of the chlorothiazide are likely to be discovered earlier than they would in the course of the ordinary practice of medicine. Thus, the risks of taking the drug would be reduced accordingly. Further, any complications of the subject’s basic disease are likely to be found and tended to quite promptly. Additional ramifications may be provided if desired….”5
The view is further elucidated in comments by Levine in his book Ethics and Regulation of Clinical Research on the work of the National Commission. He states:
…the Commission calls for an analysis of the various components of the research protocol. Procedures that are designed solely to benefit society or the class of children of which the particular child-subject is representative are to be considered as the research component. Judgements about the justification of the risks imposed by such procedures are to be made in accord with other recommendations. For example, if the risk is minimal, the research may be conducted as described in Recommendations 3 and 7 [of Research Involving Children], no matter what the risks are of the therapeutic components. The components of the protocol ‘that hold out the prospect of direct benefit for the individual subjects’ are to be considered precisely as they are in the practice of medicine.24
Levine’s description is clearly at variance with the actual text of Research Involving Children. The passage is significant, I believe, as an account of Levine’s own views on the ethical analysis of risk, as developed for the National Commission. It may also be an accurate description of the view of the National Commission itself as reflected in Institutional Review Boards and the Belmont Report. A formal articulation of what we have called a component analysis of the potential benefits and risks of research would not, however, come until long after the close of the National Commission’s work.
It is this last model of risk assessment, “component analysis,” that serves as the conceptual framework for the analysis of risk found within the Common Rule. Risks associated with nontherapeutic procedures must be minimized and “reasonable in relation to…the importance of the knowledge that may reasonable be expected to result” (45 CFR 46.111(a)). Risks associated with therapeutic procedures must be “reasonable in relation to anticipated benefits…to subjects” (45 CFR 46.111(a)). Thus, reflecting their historical origins, DHHS regulations protecting fetuses, children, and research subjects in general are based on three different approaches to the ethical analysis of the risks and potential benefits in research.
Toward a Comprehensive Approach for the Ethical Analysis of Potential Benefits and Risks in Research
What conceptual framework should guide the ethical analysis of risk? In this paper’s introduction we noted that:
The moral analysis of risk is neither obvious nor intuitive. Rules, including those of the Common Rule, are not self-interpreting. They must be situated within a conceptual framework which facilitates their interpretation by the IRB. The articulation of a conceptual framework for the ethical analysis of risk might therefore be a project assisting IRBs in fulfilling their mandate—the protection of research subjects.
P-16
Our historical analysis reveals that differing aspects of current DHHS regulations are supported by differing and mutually incompatible conceptual frameworks for the moral analysis of risk:
The proliferation of conceptual frameworks underlying current regulation is obviously problematic. It has surely lead to ambiguity in regulation and confusion among IRBs attempting to implement the regulations in a consistent manner. One conceptual framework should guide the moral analysis of risks and potential benefits in research.
Of the three historical approaches to risk analysis it is clear that an approach based on “component analysis” is preferred. A “whole protocol” approach suffers from two problems: 1) Therapeutic research is a contradiction in terms and describes the null set and 2) research subjects are inadequately protected as any number of procedures not for the benefit of subjects may be added to a therapeutic study. An analysis of “protocols with particular components” also suffers from shortcomings:
1) The concept of minimal risk is applied to both therapeutic and nontherapeutic procedures, but sets a threshold for allowable risk only to nontherapeutic procedures.
2) The anchoring the concept of minimal to the risks of daily life for healthy, persons seems to run counter to the use of the notion of commensurability.
3) Little guidance is given for the analysis of research that presents less than minimal risk.
4) Since clinical research often contains a mixture of procedures, differing rules for whole protocols may simultaneously apply leading to confusion and conflict.
The ethical analysis of the various “components” in a research study presents a number of advantages:
1) It acknowledges that clinical research often contains a mixture of procedures, some administered with therapeutic intent and others that answer the research question.
2) Therapeutic procedures and nontherapeutic procedures are, by definition, administered with differing intent. This difference is morally relevant.
3) Therapeutic procedures are justified by their potential to benefit the subject, while nontherapeutic procedures are justified by their potential to generate knowledge. These two benefits are largely incommensurable.
4) Rigorous separate moral calculi for therapeutic and nontherapeutic procedures protect research subjects better than other approaches. It prevents the justification of risky nontherapeutic procedures by the benefits that may flow from therapeutic procedures.
P-17
5) It is a more parsimonious model for analysis than other alternatives, and therefore avoids confusion and conflict.
A comprehensive approach to the ethical analysis of research risk was first formalized by Freedman and colleagues.1,28 This approach is summarized in Figure 1. Three main topics will be discussed here: the moral analysis of potential benefits and risks presented by therapeutic procedures; the moral analysis of potential benefits and risks of nontherapeutic procedures; and the role of the concept of minimal risk in the protection of vulnerable research subjects.
Figure 1. The Ethical Review of the Potential Benefits and Risks in Research
Protocol
![]()
Therapeutic Procedures
| n |
Procedures administered with therapeutic warrant |
Must pass test of
Clinical Equipoise
| n |
At the start of the study there must exist a state of genuine uncertainty in the community of expert practitioners as to the preferred treatment |
IRB review:
Nontherapeutic Procedures
| n |
Procedures administered without therapeutic warrant |
Risks Must Be Minimized
Risks Reasonable in Relation to Knowledge to Be Gained
![]()
ACCEPTABLE
| n |
Only if ethical tests for both therapeutic and nontherapeutic procedures are passed |
P-18
Therapeutic procedures.
Therapeutic procedures are those interventions in research—drug, surgical procedure, device, or psychological procedure—administered with therapeutic intent (Figure 1). This category also encompasses monitoring procedures that optimally guide the administration of treatment, even if these procedures are not routinely administered in clinical practice. Let us consider what procedures might be considered therapeutic in the four example studies from the beginning of this paper.
Having determined which procedures are administered with therapeutic warrant, how do we determine whether they are morally acceptable?
Therapeutic procedures must pass the test of clinical equipoise (Figure 1).29 A major competing notion, the uncertainty principle, has recently been shown inferior to clinical equipoise.30–32 Clinical equipoise is normally developed in response to the following question: When may the ethical physician offer trial participation to her patient? It begins from the recognition that competent medical practice is defined as that falling within the bounds of standard of care—that is, practice accepted by at least a respectable minority of expert practitioners. The innovation of clinical equipoise is the recognition that study treatments—be they experimental or control treatments—may be consistent with this standard of care. Thus, a physician, in keeping with his or her duty of care to the patient, may offer trial enrollment when “[t]here exists…an honest, professional disagreement among expert clinicians about the preferred treatment.”29 A state of clinical equipoise may arise in a number of ways. Evidence may emerge from early clinical studies that a new treatment offers advantages over standard treatment. Alternatively, there may be a split within the clinical community, with some physicians preferring one treatment and other physicians preferring another. This latter scenario is well documented in the literature and calls for a randomized controlled trial (RCT) to settle which is the better treatment.33 Clinical equipoise permits these important RCTs. It would have physicians respect the fact that “their less favored treatment is preferred by colleagues whom they consider to be responsible and competent.”29 When evaluating a study containing one or more therapeutic procedures, the IRB must take reasonable steps to assure itself that a state of clinical equipoise exists. This will involve a critical evaluation of the study’s justification. In selected cases, it may also require searches of the medical literature or consultation with relevant experts who have no connection with the study or its sponsor. A variety of treatment-related factors are also likely to contribute to this determination: the efficacy of the treatment; side effects, both reversible and
P-19
irreversible; ease of administration; patient compliance; and perhaps even cost. It is important to recognize that clinical equipoise does not require numeric equality of treatment risks (or benefits, for that matter). It is more accurate to say that equipoise requires approximate equality in treatments’ therapeutic index—a compendious measure of potential benefits, risks, and uncertainty. Thus, a novel treatment may pose considerably more risk to subjects, so long as it also offers the prospect of considerably greater benefit. With novel interventions, the uncertainty associated with their effects will almost always be greater than treatments currently used in practice.
Study A is the only one of our four examples that involves the use of therapeutic procedures. The question the IRB must ask itself is as follows: Does a state of clinical equipoise exist among the new antipsychotic, placebo, and alternatives available in clinical practice? It follows from clinical equipoise that placebo controls will generally only be permissible for first generation treatments, when no standard treatment is available. Once effective treatment exists, new interventions must be tested against best available standard treatment. Freedman describes five circumstances in which placebo controls may be employed legitimately: 1) when there is no standard treatment; 2) when standard treatment is no better than placebo; 3) when standard treatment is placebo; 4) when the net therapeutic advantage of standard treatment has been called into question by new evidence; and 5) when effective treatment exists but is not available due to cost or short supply (although caveats apply to this criterion).34 Effective treatment exists for the treatment of schizophrenia, and, hence, the use of placebo in this case is impermissible.35 The IRB must not approve the study unless either an active control is used or the patient population is restricted to those who have no response to standard therapy, including any routinely used second- or third-line agents. A detailed rebuttal of scientific arguments made in favor of the routine use of placebo controls can be found elsewhere.8,36,37
Nontherapeutic procedures.
The remaining procedures administered in a clinical study are, by definition, not administered with therapeutic warrant and are properly referred to as “nontherapeutic procedures” (Figure 1). Such procedures are administered solely for scientific purposes, to answer the research question at hand. As all research is a “systematic investigation…designed to develop or contribute to generalizable knowledge” (45 CFR 46.102(d)), it is difficult to imagine a study that does not include a nontherapeutic procedure. A nontherapeutic procedure may be as simple—and innocuous—as randomization, chart review, a questionnaire, an interview, or data that is recorded in some other manner; it may, however, be invasive or otherwise fraught with risk, as with genetic testing, organ biopsy, or the collection of information related to illegal practices. All four of the examples discussed at the beginning of this paper include nontherapeutic procedures:
P-20
| n |
Study D (breast cancer genes) also involves only nontherapeutic procedures. The epidemiological survey and genetic tests may generate information that is anxiety provoking or that indeed may lead to workplace or insurance discrimination. Beyond risks to the individual study participants, the Jewish community as a whole may be wrongly labeled as “cancer prone” and subjected to discrimination and stigmatization. |
By definition, risks associated with nontherapeutic procedures cannot be justified by the prospect of benefits to individual research subjects and, hence, a risk-benefit calculus is inappropriate to assessing their acceptability. The IRB must first ensure that the risks associated with nontherapeutic procedures are minimized “by using procedures which are consistent with sound research design and which do not unnecessarily expose subjects to risk, and whenever appropriate, by using procedures already being performed on the subjects for diagnostic or treatment purposes” (Figure 1) (45 CFR 46.111(a)(1)). Second, the IRB must ascertain that the risks of such procedures are reasonable in relation to the knowledge to be gained (Figure 1) (45 CFR 46.111(a)(2)). Thus, the ethical analysis of risks associated with nontherapeutic procedures involves a risk-knowledge calculus. The knowledge that may result from a study is essentially its scientific value. Freedman has argued that the proper assessment of the scientific value of a study requires not only the opinion of experts from relevant disciplines, but also of representatives of the community-at-large.38 In study A, the IRB will wish to ensure that all of the tests administered are required and consider whether psychometric tests administered routinely might provide equivalent information. In study B, hypnosis and hypnotic suggestion present worrisome risks. Can the information be gained in another way, for example, by studying those who are already deaf? Can the risks associated with hypnosis be minimized? Study C also presents nontrivial risk, in part because the questionnaire is administered in a high school setting. Paying careful attention to the protection of anonymity, allowing students to opt out of the questionnaire (or certain questions) unobtrusively, and seating students so they cannot see one the answers of others will minimize risk. In study D, risks to subjects of genetic information are considerably alleviated by destroying identifiers and by not informing participants of the results of genetic testing. In all cases, the risks of these procedures must be reasonable in relation to the knowledge to be gained.
Study D poses one category of risk that is not dealt with by this model—risks to the community. The Ashkenazi community has expressed the concern that such studies may lead to discrimination:
Such findings, which have already lead to Jewish groups being targeted as a potential market for commercial genetic tests, could create the perception that Jewish people are unusually susceptible to disease….As a result…anyone with a Jewish sounding name could face discrimination in insurance and employment as companies struggle to keep down healthcare costs.39
The protection of communities in research is a novel area of inquiry in research ethics. Another paper commissioned by NBAC argues for a new ethical principle of respect for communities.40 Subsequent work has detailed possible protections for communities in research.41 Most recently, a rational schema for mapping appropriate protections onto specific communities, such as Ashkenazic Jews, has been reported.42 More work will be required to determine how the ethical analysis of risk for communities in research should proceed.
Minimal risk.
Minimal risk is a widely used concept in the regulation of research internationally. It can be found in contemporary guidelines from Australia,43 Canada,44 CIOMS,45 the Council of Europe,46 the United Kingdom [Physicians, 1996 #220], and the United States (45 CFR 46). That a research study poses minimal risk “means that the risks of harm anticipated in the proposed research are not greater, considering probability and magnitude, than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests” (45 CFR 46.102(i)).
P-21
Minimal risk has been the subject of considerable debate and confusion in the literature. As we have seen, the concept of minimal risk was applied to both research with a therapeutic procedure and research with a nontherapeutic procedure in Research Involving Children. In the context of our schema for the ethical analysis of risk, this makes little sense. If a state of clinical equipoise exists, it follows that the therapeutic indices of the various study treatments (and alternatives available in clinical practice) are roughly equivalent. Thus, when considering the limits of risk to which research subjects may be exposed, we must focus on nontherapeutic risks. The risks of nontherapeutic procedures are the incremental risks associated with study participation.
Freedman and colleagues have argued that the definition of minimal risk found in the Common Rule is best understood as a core definition with examples.47 Minimal risk refers to risks “ordinarily encountered in daily life”—or, shorter, risks of daily life (45 CFR 46.102(i)). The second part of the definition provides two examples of minimal risk: procedures encountered “during the performance of routine physical or psychological examinations or tests” (45 CFR 46.102(i)). The concept has been criticized on the grounds that it is difficult to know what counts as a risk of daily life and that the quantification of such risks is elusive.48 Freedman and colleagues conclude that the first claim is untrue and the second irrelevant.47 The risks of daily life are familiar to us all. Minimal risk does not refer to any risk encountered by any person, as some individuals engage in hazardous professions and pastimes. Rather it refers to the risks that are common to us all—driving to work, crossing the street, exchanging information over the internet, or getting a blood test at the doctor’s office. While it may be difficult to quantify the precise probability of given outcomes associated with each of these behaviors, we can nonetheless easily identify them as risks of daily life. As Freedman and colleagues observe: “We are, by definition, each of us acquainted with them; and, almost by definition, if we are unsure whether they belong within the set of common tasks then they don’t.”47 The assessment of whether a procedure is minimal risk is not primarily a quantitative determination; rather, it is a qualitative or categorical judgment made by the IRB. Research interventions may be determined to be of minimal risk because either the procedure is in fact encountered in daily life or it is sufficiently similar to those routinely encountered.
The threshold of “a minor increase over minimal risk” corresponds to the custodial duty that parents have for their children. Responsible parents makes decisions regarding new activities for their child based on the daily life of the child (“minimal risk”) and make allowances for the importance of new experiences (“a minor increase over”). While the majority of researchers and parents are scrupulous, some are not. The IRB acts in loco parentis by evaluating nontherapeutic risks as a responsible parent would, thereby ensuring that parents, scrupulous or not, will have the opportunity to enroll a child only in a study that would have passed such a test.
The concept of minimal risk serves two basic functions in regulation. First, it may be used as a “sorting mechanism,” directing the attention of the IRB to studies posing greater risk. Second, it serves as a threshold limiting the amount of nontherapeutic risk to which vulnerable research subjects may be exposed. The provision in the Common Rule allowing for expedited review is an example of the use of minimal risk as a sorting mechanism. If a study is found to pose only minimal risk, it may, with certain other caveats, receive approval by the IRB chair without full IRB review. The regulations state:
An IRB may use the expedited review procedure to review either or both of the following:
| (1) |
some or all of the research appearing on the list and found by the reviewer(s) to involve no more than minimal risk, |
| (2) |
minor changes in previously approved research during the period (of one year or less) for which approval is authorized. |
Under an expedited review procedure, the review may be carried out by the IRB chairperson or by one or more experienced reviewers designated by the chairperson from among members of the IRB. In reviewing the research, the reviewers may exercise all of the authorities of the
P-22
IRB except that the reviewers may not disapprove the research. A research activity may be disapproved only after review in accordance with the non-expedited procedure set forth in
§ 46.108(b) (45 CFR 46.110(b)).
Several problems are apparent with this provision. First, the requirement that nontherapeutic risks be both minimal risk and included in the list of “research activities which may be reviewed through expedited review procedures” (45 CFR 46) is curious. The list is obviously designed to include procedures that pose minimal risk to healthy adult subjects. For example, “moderate exercise by healthy volunteers” and “collection of blood samples by venipuncture…from subjects 18 years of age or older” are permitted procedures. This effectively eliminates any study involving venipuncture in children or exercise testing of adults with illness from expedited review. This seems inconsistent with minimal risk as defined, which does not limit the standard to healthy persons or adults (45 CFR 46.102(i)).
Second, the expedited review provision as stated must surely be an incomplete set of criteria. A given study might pose only minimal risk to subjects and yet raise serious ethical concerns that should make it ineligible for expedited review. One such case is a study that involves a vulnerable population. Studies involving vulnerable populations require special scrutiny by IRBs and should not be eligible for expedited review. It may be that the current regulation attempts to so restrict the use of expedited review by limiting approvable “activities” to those administered to healthy adults. A more direct (and effective) regulatory stance on this issue would be preferable. Another such case is a study that has serious methodological flaws. Freedman observes that the ethical requirement that a study have a sound research design (validity) is absolute.38 Thus, a study should be eligible for expedited review only if three conditions are fulfilled: 1) the study poses no more than minimal risk to participants; 2) it does not involve a vulnerable population; and 3) no serious methodological flaws are apparent.
Most important is minimal risk’s role as a threshold concept for allowable nontherapeutic risk in research on vulnerable populations (Figure 2). Vulnerable populations in the Common Rule include children, prisoners, pregnant women, mentally disabled persons, or economically or educationally disadvantaged persons (45 CFR 111(b)). Given the heterogeneity of these groups, vulnerability itself must be a complex notion. Indeed, it encompasses groups who have one or more of the following characteristics: undue susceptibility to harm; incapability of providing informed consent to study participation; or being so situated as to render the voluntariness of consent suspect.49 In light of these characteristics, the vulnerable are entitled to special protections in research. Three protections are often invoked. First, members of a vulnerable group may be included in research only when their participation is essential to the hypothesis being tested. Second, if persons are incapable of providing informed consent, the consent of a proxy decisionmaker is required. Third, the amount of nontherapeutic risk to which persons may be exposed is limited to either minimal or a minor increase over minimal.
The importance of the last protection can scarcely be over emphasized. Clinical equipoise ensures that therapeutic procedures in a study are comparable with each other and alternatives in clinical practice in terms of their therapeutic indices. Thus, the incremental risk posed by study participation is that posed by nonthera-peutic procedures. If vulnerable populations, such as children or incapable adults, are to be protected in any meaningful way, the risks of nontherapeutic procedures to which they may be exposed must be limited to a minor increase above minimal risk. As we have discussed, the standard has the advantage of mirroring the custodial duties of parents to children and caretakers to incapable adults.
NBAC proposes to eliminate this important protection.50 In its report Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity, no limit is placed on the nontherapeutic risk to which an incapable adult may be exposed, provided certain consent provisions obtain (Recommendation 12). This is shortsighted. When the limit of a minor increase above minimal risk is eliminated as a threshold for permissible
P-23
Figure 2. The Ethical Review of the Potential Benefits and Risks in Research Involving a Vulnerable Population
Protocol
Study Hypotheses Requires Inclusion of Vulnerable Pop’n
Therapeutic Procedures
| n |
Procedures administered with therapeutic warrant |
Nontherapeutic Procedures
| n |
Procedures administered without therapeutic warrant |
Must pass test of
Clinical Equipoise
| n |
At the start of the study there must exist a state of genuine uncertainty in the community of expert practitioners as to the preferred treatment |
IRB review:
Risks Must Be Minimized
Risks Reasonable in Relation to Knowledge to Be Gained
No More Than a Minor Increase Above Minimal Risk
ACCEPTABLE
n Only if ethical tests for both therapeutic and nontherapeutic procedures are passed
P-24
nontherapeutic risk, no amount of risk is ruled out for research involving incapable persons. So long as the research question is important enough (and informed consent provisions fulfilled), any amount of nonthera-peutic risk is permissible. This change, if translated into regulation, will effectively undermine protections for incapable persons in research. Incapable persons will then be exposed to exploitation legitimated by the very regulations that were to protect them.
Implications for U.S. Regulations Protecting Research Subjects
What changes to U.S. regulations would the implementation of such a framework require? As we remarked supra, rules are not self-interpreting, and a conceptual framework is required. What follows is a summary of the changes to aspects of the Common Rule and DHHS regulations pertaining to risk analysis required to achieve this end. Proposed text is highlighted in bold. Specific recommendations for changes to subparts B (pregnant women) and C (prisoners) are not included.
| 1. |
It is clear that the U.S. regulations protecting research subjects found in the Common Rule and the DHHS regulations were profoundly influenced by the works of the National Commission. The differing models of risk analysis with which the National Commission worked influenced different parts of the regulations. These inconsistencies must surely be corrected. IRBs require a single conceptual framework for the ethical analysis of the risks and benefits in research if they are to apply regulations consistently. The “component” approach described in the last section of this paper is the preferred conceptual model. |
| 2. |
The concepts of therapeutic and nontherapeutic procedures should be included and defined, as they are central to this approach to risk analysis. 46.102(k) Therapeutic procedures are study interventions administered with the intent of providing direct benefit to the research subject. 46.102(l) Nontherapeutic procedures are study interventions that are not administered with therapeutic intent, and are only intended to answer the scientific question of the study. |
| 3. |
The IRB’s general obligations regarding the ethical analysis of the potential benefits and risks of research should be stated more clearly. 46.111(a) In order to approve research covered by this policy the IRB shall determine that all of the following requirements are satisfied: 1) Therapeutic procedures fulfill the requirements of clinical equipoise. That is, at the start of the study there must exist a state of genuine uncertainty in the community of expert practitioners as to the preferred treatment. 2) The risks associated with nontherapeutic procedures must be minimized i) by using procedures which are consistent with sound research design and which do not unnecessarily expose subjects to risk and ii) whenever appropriate, by using procedures already being performed on the subjects for diagnostic or treatment purposes. Risks of nontherapeutic procedures must be reasonable in relation to the knowledge to be gained. Both 46.111(a)(1) and 46.111(a)(2) must be satisfied if a given study is to be approved. |
| 4. |
The definition of minimal risk has been a source of considerable controversy and confusion. The definition should be simplified and clarified. 46.102(i) Minimal risk means that the probability and magnitude of harm is no greater than that encountered in daily lives of all (or the great majority) of persons in the population from which research subjects are to be recruited. It refers only to the risks associated with nontherapeutic procedures. |
P-25
| 5. |
The role of the concept of minimal risk in expedited review needs to be clarified. The use of a list of procedures drawn up only for healthy adults is inconsistent with the concept’s definition and use. Furthermore, minimal risk is not a sufficient condition for a research protocol to receive expedited review. Generally speaking, the study protocol must also be methodologically sound and not involve a vulnerable population. 46.110(a) deleted 46.110(b) An IRB may use the expedited review procedure to review either an entire protocol or a protocol amendment provided the review(s) determine: 1) The study methods are valid; 2) The study does not involve a vulnerable population; and 3) The study poses no more than minimal risk. |
| 6. |
The ethical analysis of risk as pertains to children as research subjects can be simplified greatly with this conceptual approach. Simplifying these regulations will avoid confusion and help IRBs protect children who are research subjects. 46.404 delete 46.405 delete 46.406 delete 46.407 delete 46.404 (new) In order to approve research involving children covered by this policy the IRB shall determine that all of the following requirements are satisfied: a) the conditions of 46.111(a)(1), 46.111(a)(2), and 46.111(a)(3); b) answering the study’s scientific hypothesis requires the inclusion of children as research subjects; and c) risks associated with nontherapeutic procedures are no more than a minor increase over minimal risk. |
| 7. |
A new section must be added to the DHHS regulations detailing protections for adults incapable of providing informed consent. The protections for incapable adults will for the most part be similar to those for children. 46.500 In order to approve research involving incapable adults covered by this policy the IRB shall determine that all of the following requirements are satisfied: a) the conditions of 46.111(a)(1), 46.111(a)(2), and 46.111(a)(3); b) answering the study’s scientific hypothesis requires the inclusion of incapable adults as research subjects; and c) risks associated with nontherapeutic procedures are no more than a minor increase over minimal risk. |
Acknowledgments
I am grateful for the helpful comments on earlier drafts of this paper by Drs. Chris MacDonald, Eric Meslin, Paul Miller, and Marjorie Speers.
P-26
References
| 1. |
Weijer C. Thinking clearly about research risk: Implications of the work of Benjamin Freedman. IRB: A Review of Human Subjects Research 1999; 21:1–5. |
| 2. |
Freedman B, Shapiro S. Ethics and statistics in clinical research: Towards a more comprehensive examination. Journal of Statistical Planning and Inference 1994; 42:223–240. |
| 3. |
National Commission on the Protection of Humans Subjects of Biomedical and Behavioral Research. The Belmont Report: Ethical Principles for the Protection of Human Subjects of Biomedical and Behavioral Research. Washington, D.C.: DHEW Publication no. (OS) 78-0012, 1978. |
| 4. |
Levine R. The boundaries between biomedical or behavioral research and the accepted and routine practice of medicine. In: National Commission on the Protection of Humans Subjects of Biomedical and Behavioral Research. The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research, Appendix 1. Washington, D.C.: DHEW Publication no. (OS) 78-0013, 1978. |
| 5. |
Levine R. The role of assessment of risk benefit criteria in the determination of the appropriateness of research involving human subjects. In: National Commission on the Protection of Humans Subjects of Biomedical and Behavioral Research. The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research, Appendix 1. Washington, D.C.: DHEW Publication no. (OS) 78-0013, 1978. |
| 6. |
Levine R. Balance of Harms and Benefits. Ethics and the Regulation of Clinical Research (2nd ed.). New Haven: Yale University Press, 1988:37–65. |
| 7. |
Chouinard G. A placebo-controlled clinical trial of remoxipride and chlorpromazine in newly admitted schizophrenia patients with acute exacerbation. Acta Psychiatrica Scandinavica 1990; 358:111–119. |
| 8. |
Weijer C. Placebo-controlled trials in schizophrenia: Are they ethical? Are they necessary? Schizophr Res 1999; 35:211–218; discussion 227–236. |
| 9. |
Zimbardo P. Inducing hearing deficit generates experimental paranoia. Science 1981; 212:1529–1531. |
| 10. |
Lewis M, Zimbardo P. The ethics of inducing paranoia in an experimental setting [letter and response]. IRB: A Review of Human Subjects Research 1981; December:9–11. |
| 11. |
Phillips S. Asking the sensitive question: the ethics of survey research and teen sex. IRB: A Review of Human Subjects Research 1994; 16:1–7. |
| 12. |
Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, Timmerman MM, Brody LC, Tucker MA. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews [see comments]. N Engl J Med 1997; 336:1401–1408. |
| 13. |
Glass K, Weijer C, Lemmens T, Palmour R, Shapiro S. Structuring the review of human genetics protocols part II: Diagnostic and screening studies. IRB: A Review of Human Subjects Research 1997; 19:1–11, 13. |
| 14. |
Gray B. Bioethics commissions: What can we learn from past successes and failures? In: Bulger R, Bobby E, Fineberg H, eds. Society’s Choices: Social and Ethical Decision Making in Biomedicine. Washington, D.C.: National Academy Press, 1995:261–306. |
| 15. |
National Commission on the Protection of Humans Subjects of Biomedical and Behavioral Research. Research on the Fetus: Report and Recommendations. Washington, D.C: DHEW Publication no. (OS) 76-127, 1975. |
| 16. |
National Commission on the Protection of Humans Subjects of Biomedical and Behavioral Research. Research Involving Prisoners: Report and Recommendations. Washington, D.C.: DHEW Publication no. (OS) 76-131, 1976. |
| 17. |
National Commission on the Protection of Humans Subjects of Biomedical and Behavioral Research. Research Involving Children: Report and Recommendations. Washington, D.C.: DHEW Publication no. (OS) 77-0004, 1977. |
| 18. |
National Commission on the Protection of Humans Subjects of Biomedical and Behavioral Research. Research Involving Those Institutionalized as Mentally Infirm: Report and Recommendations. Washington, D.C.: DHEW Publication no. (OS) 78-0006, 1978. |
| 19. |
National Commission on the Protection of Humans Subjects of Biomedical and Behavioral Research. Institutional Review Boards: Report and Recommendations. Washington, D.C.: DHEW Publication no. (OS) 78-0008, 1978. |
| 20. |
Levine R. The Fetus and the Embryo. Ethics and Regulation of Clinical Research. New Haven: Yale University Press, 1988:297–320. |
P-27
| 21. |
Brody B. The Ethics of Biomedical Research: An International Perspective. New York: Oxford University Press, 1998. |
| 22. |
Weijer C. Trial by error. Hastings Center Report 2000; in press. |
| 23. |
Levine R. Prisoners. Ethics and Regulation of Clinical Research (2nd ed.). New Haven: Yale University Press, 1988:277–295. |
| 24. |
Levine R. Children. Ethics and Regulation of Clinical Research (2nd ed.). New Haven: Yale University Press, 1988:235–256. |
| 25. |
The President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research. Protecting Human Subjects: The Adequacy and Uniformity of Federal Rules and Their Implementation. Washington, D.C.: U.S. Government Printing Office, Stock no. 040-000-00452-1, 1981. |
| 26. |
The President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research. Implementing Human Research Regulations: The Adequacy and Uniformity of Federal Rules and of Their Implementation. Washington, D.C.: U.S. Printing Office, Stock no. 040-000-004781-8, 1983. |
| 27. |
Levine R. Those Institutionalized as Mentally Infirm. Ethics and Regulation of Clinical Research (2nd ed.). New Haven: Yale University Press, 1988:257–276. |
| 28. |
Freedman B, Fuks A, Weijer C. Demarcating research and treatment: a systematic approach for the analysis of the ethics of clinical research. Clin Res 1992; 40:65360. |
| 29. |
Freedman B. Equipoise and the ethics of clinical research. N Engl J Med 1987; 317:141–145. |
| 30. |
Peto R, Pike M, Armitage P, Breslow NE, Cox DR, Howard SV, Mantel N, McPherson K, Peto J, Smith PG. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. I. Introduction and design. British Journal of Cancer 1976; 34:585–612. |
| 31. |
Peto R, Baigent C. Trials: The next 50 years. British Medical Journal 1998; 317:1170–1171. |
| 32. |
Weijer C, Shapiro SH, Cranley Glass K. For and against: Clinical equipoise and not the uncertainty principle is the moral underpinning of the randomised controlled trial. British Medical Journal 2000; 7263:756–758. |
| 33. |
Taylor KM, Margolese RG, Soskolne CL. Physicians’ reasons for not entering eligible patients in a randomized clinical trial of surgery for breast cancer. New England Journal of Medicine 1984; 310:1363–1367. |
| 34. |
Freedman B. Placebo-controlled trials and the logic of clinical purpose. IRB: A Review of Human Subjects Research 1990; 12:1–6. |
| 35. |
Kane J. Schizophrenia. New England Journal of Medicine 1996; 334:34–41. |
| 36. |
Freedman B, Weijer C, Glass KC. Placebo orthodoxy in clinical research. I: Empirical and methodological myths. J Law Med Ethics 1996; 24:243–251. |
| 37. |
Freedman B, Glass KC, Weijer C. Placebo orthodoxy in clinical research. II: Ethical, legal, and regulatory myths. J Law Med Ethics 1996; 24:252–259. |
| 38. |
Freedman B. Scientific value and validity as ethical requirements for research: a proposed explication. IRB: A Review of Human Subjects Research 1987; 9:7–10. |
| 39. |
Lehrman S. Ashkenazi signals are timely. Nature 1997; 389:315–316. |
| 40. |
Weijer C. Protecting communities in research: philosophical and pragmatic challenges. Camb Q Healthc Ethics 1999; 8:501–513. |
| 41. |
Weijer C, Goldsand G, Emanuel EJ. Protecting communities in research: current guidelines and limits of extrapolation. Nat Genet 1999; 23:275–280. |
| 42. |
Weijer C, Emanuel EJ. Protecting communities in biomedical research. Science 2000; 289:1142–1144. |
| 43. |
Australia National Health and Medical Research Council. National Statement on Ethical Conduct in Research Involving Humans. Canberra: NHMRC, 1999. |
| 44. |
Medical Research Council of Canada, Natural Sciences and Engineering Research Council of Canada, Social Sciences and Humanities Research Council of Canada. Tri-Council Policy Statement: Ethical Conduct for Research Involving Humans. Ottawa: Public Works and Government Services Canada, 1998. |
| 45. |
Council for International Organizations of Medical Sciences (CIOMS). International Ethical Guidelines for Biomedical Research Involving Human Subjects. Geneva: CIOMS, 1993. |
P-28
| 46. |
Council of Europe. Convention for the Protection of Human Rights and Dignity of the Human Being with Regard to the Application of Biology and Medicine: Convention on Human Rights and Biomedicine. Oviedo: European Treaty Series, 1997. |
| 47. |
Freedman B, Fuks A, Weijer C. In loco parentis: Minimal risk as an ethical threshold for research upon children. Hastings Center Report 1993; 23:13–19. |
| 48. |
Kopelman L. Estimating risk in human research. Clinical Research 1981; 29:1–8. |
| 49. |
Weijer C. Research involving the vulnerable sick. Accountability in Research 1999; 7:21–36. |
| 50. |
National Bioethics Advisory Commission. Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity. Rockville, Maryland: NBAC, 1998. |
P-29
