
The National Bioethics Advisory Commission (NBAC) was established by Executive Order 12975, signed by President Clinton on October 3, 1995. NBAC’s functions are defined as follows:
a) NBAC shall provide advice and make recommendations to the National Science and Technology Council and to other appropriate government entities regarding the following matters:
1) the appropriateness of departmental, agency, or other governmental programs, policies, assignments, missions, guidelines, and regulations as they relate to bioethical issues arising from research on human biology and behavior; and 2) applications, including the clinical applications, of that research.
b) NBAC shall identify broad principles to govern the ethical conduct of research, citing specific projects only as illustrations for such principles.
c) NBAC shall not be responsible for the review and approval of specific projects.
d) In addition to responding to requests for advice and recommendations from the National Science and Technology Council, NBAC also may accept suggestions of issues for consideration from both the Congress and the public. NBAC also may identify other bioethical issues for the purpose of providing advice and recommendations, subject to the approval of the National Science and Technology Council.
National Bioethics Advisory Commission
6705 Rockledge Drive, Suite 700, Bethesda, Maryland 20892-7979 Telephone: 301-402-4242 • Fax: 301-480-6900 • Website: www.bioethics.gov
ISBN 1-931022-16-X

Ethical and Policy Issues in Research Involving Human Participants
Volume I
Report and Recommendations of the National Bioethics Advisory Commission
Bethesda, Maryland August 2001
Table of Contents
Letter of Transmittal to the President National Bioethics Advisory Commission
National Bioethics Advisory Commission Staff and Consultants
| Prologue | i |
| Summary of Recommendations | xi |
| Chapter 1: Introduction—The Need for Change | 1 |
| Introduction | 1 |
| The Value of Research | 1 |
| The Need for Oversight of Research Involving Human Participants | 2 |
| Current Factors Influencing the Research Enterprise | 4 |
| Major Challenges Facing the Current Oversight System | 8 |
| Consequences of a Flawed System | 16 |
| Notes | 16 |
| References | 18 |
| Chapter 2:A Proposal for Oversight | 21 |
| Introduction | 21 |
| Purpose of Oversight | 21 |
| Functions and Responsibilities of the Oversight System | 23 |
| Scope and Structure of a Revised Oversight System | 26 |
| The Need for a Unified, Comprehensive Federal Policy | 28 |
| Definition of Research Involving Human Participants | 32 |
| Summary | 42 |
| Notes | 42 |
| References | 43 |
| Chapter 3:Methods for Ensuring Protection: | |
| Education, Certification, and Accreditation | 45 |
| Introduction | 45 |
| Education and Competence in Conducting Research Involving | |
| Human Participants | 45 |
| Mechanisms to Ensure Institutional, Institutional Review Board, | |
| and Investigator Compliance | 50 |
| Conflicts of Interest | 58 |
| Summary | 64 |
| Notes | 64 |
| References | 65 |
| Chapter 4:Assessing Risks and Potential Benefits and | |
| Evaluating Vulnerability | 69 |
| Introduction | 69 |
| Institutional Review Board Approval of Research | 69 |
| Risks of Harms and Potential Benefits to Participants and to Society | 71 |
| Vulnerability | 85 |
| Summary | 93 |
| Notes | 93 |
| References | 95 |
| Chapter 5:Ensuring Voluntary Informed Consent and | |
| Protecting Privacy and Confidentiality | 97 |
| Introduction | 97 |
| Voluntary Informed Consent | 97 |
| Privacy and Confidentiality | 104 |
| Summary | 107 |
| Notes | 108 |
| References | 108 |
| Chapter 6:Conduct of Research—Selected Issues for | |
| Local Institutions | 111 |
| Introduction | 111 |
| Monitoring of Ongoing Research | 111 |
| Review of Cooperative Research Studies | 117 |
| Compensation for Research-Related Injuries | 123 |
| Summary | 126 |
| Notes | 127 |
| References | 128 |
| Chapter 7:Implications of This Report | 131 |
| Introduction | 131 |
| Resources | 131 |
| Future Research | 133 |
| The New System as a Dynamic and Responsive Structure | 134 |
| General Themes in the Recommendations | 135 |
| What This Report Means to Those Who Use the System | 136 |
| How This Report Will Affect Institutional Review Boards, | |
| Investigators, and Institutions | 136 |
| How This Report Will Affect Research Participants | 137 |
| Summary | 137 |
| Notes | 137 |
| References | 138 |
| Appendices | |
| A. Acknowledgments | 139 |
| B. May 1999 Letter from Harold T. Shapiro to | |
| President William J. Clinton and President Clinton’s | |
| Response | 141 |
| C. The Current Oversight System: History and Description | 151 |
| D.The Belmont Report | 161 |
| E. Code of Federal Regulations, Title 45 Part 46 | 169 |
| F. Code of Federal Regulations, Title 21 Parts 50 and 56 | 187 |
| G.Statutory Authority for Agency Human Subjects Protection | |
| Regulations (Common Rule) | 205 |
| H.Food and Drug Administration Regulations Requiring | |
| Reporting of Adverse Events | 213 |
| I. Public Comments on NBAC’s December 19, 2000, Draft | 217 |
| J. Public and Expert Testimony | 221 |
| K. Commissioned Papers and Staff Analysis | 225 |
| Index | 227 |


National Bioethics Advisory Commission
Harold T. Shapiro, Ph.D., Chair
President Emeritus and Professor of Economics and Public Affairs The Woodrow Wilson School of Public and International Affairs Princeton University Princeton, New Jersey
Patricia Backlar
Research Associate Professor of Bioethics Department of Philosophy Portland State University Assistant Director Center for Ethics in Health Care Oregon Health Sciences University Portland, Oregon
Arturo Brito, M.D.
Assistant Professor of Clinical Pediatrics University of Miami School of Medicine Miami, Florida
Alexander Morgan Capron, LL.B.
Henry W. Bruce Professor of Law
University Professor of Law and Medicine
Co-Director, Pacific Center for Health Policy and Ethics University of Southern California Los Angeles, California
Eric J. Cassell, M.D., M.A.C.P.
Clinical Professor of Public Health
Weill Medical College of Cornell University New York, New York
R. Alta Charo, J.D.
Professor of Law and Bioethics Law School and Medical School University of Wisconsin Madison, Wisconsin
James F. Childress, Ph.D.
Kyle Professor of Religious Studies Professor of Medical Education Director, Institute for Practical Ethics Department of Religious Studies University of Virginia Charlottesville, Virginia
David R. Cox, M.D., Ph.D.
Scientific Director Perlegen Sciences Santa Clara, California
Rhetaugh Graves Dumas, Ph.D., R.N.
Vice Provost Emerita, Dean Emerita, and Lucille Cole Professor of Nursing University of Michigan Ann Arbor, Michigan
Laurie M. Flynn*
Senior Research and Policy Associate
Department of Child and Adolescent Psychiatry Columbia University New York, New York
Carol W. Greider, Ph.D.
Professor of Molecular Biology and Genetics Department of Molecular Biology and Genetics Johns Hopkins University School of Medicine Baltimore, Maryland
Steven H. Holtzman
Chief Business Officer
Millennium Pharmaceuticals, Inc. Cambridge, Massachusetts
Bette O. Kramer
Founding President
Richmond Bioethics Consortium Richmond, Virginia
Bernard Lo, M.D.
Director
Program in Medical Ethics Professor of Medicine
University of California, San Francisco San Francisco, California
Lawrence H. Miike, M.D., J.D.
Kaneohe, Hawaii
Thomas H. Murray, Ph.D.
President
The Hastings Center Garrison, New York
William C. Oldaker, LL.B.
Senior Partner
Oldaker & Harris, L.L.P. Washington, D.C.
Co-Founder and General Counsel NeuralStem Biopharmaceuticals Ltd. College Park, Maryland
Diane Scott-Jones, Ph.D.
Professor
Psychology Department Boston College Chestnut Hill, Massachusetts
*Resigned on May 10, 2001
National Bioethics Advisory Commission Staff and Consultants
Executive Director
Eric M. Meslin, Ph.D.*
Project Director
Marjorie A. Speers, Ph.D.
Research Staff
Douglas Berger, M.Litt., Program Analyst**
Glen Drew, M.S., J.D., Senior Research Policy Analyst Elisa Eiseman, Ph.D., Senior Research Analyst
Ellen L. Gadbois, Ph.D., Senior Policy Analyst Stu Kim, J.D., Program Analyst*** Kerry Jo Lee, Program Analyst* Ayodeji Marquis, Intern (June–August 2000) Debra McCurry, M.S., Information Specialist
Alice K. Page, J.D., M.P.H., Senior Policy Analyst*
Consultants
Burness Communications, Communications Consultant
Sara Davidson, M.A., Editor
Liza Dawson, Ph.D., Research Consultant (December 2000–March 2001) Debra DeBruin, Ph.D., Bioethics Consultant (July–December 2000) Kathi E. Hanna, M.S., Ph.D., Editorial Consultant
The Hill Group, Meeting Consultant Tamara Lee, Graphic Designer
Administrative Staff
Nicole Baker, Administrative Technician Jody Crank, Assistant to the Executive Director Evadne Hammett, Administrative Officer
Margaret C. Quinlan, Office Manager Sherrie Senior, Secretary
| * |
Until July 2001 |
| ** |
Until June 2001 |
| *** |
Until November 2000 |
Prologue
Protecting Research Participants—A Time for Change
Introduction
Protecting the rights and welfare of those who volunteer to participate in research is a fundamental tenet of ethical research. A great deal of progress has been made in recent decades in changing the culture of research to incorporate more fully this ethical responsibility into protocol design and implementation. In the 1960s and 1970s, a series of scandals concerning social science research and medical research conducted with the sick and the illiterate underlined the need to systematically and rigorously protect individuals in research (Beecher 1966; Faden and Beauchamp 1986; Jones 1981; Katz 1972; Tuskegee Syphilis Study Ad Hoc Advisory Panel 1973). However, the resulting system of protections that evolved out of these rising concerns—although an improvement over past practices—is no longer sufficient. It is a patchwork arrangement associated with the receipt of federal research funding or the regulatory review and approval of new drugs and devices. In addition, it depends on the voluntary cooperation of investigators, research institutions, and professional societies across a wide array of research disciplines. Increasingly, the current system is being viewed as uneven in its ability to simultaneously protect the rights and welfare of research participants and promote ethically responsible research.
Research involving human participants has become a vast academic and commercial activity, but this country’s system for the protection of human participants has not kept pace with that growth. On the one hand, the system is too narrow in scope to protect all participants, while on the other hand, it is often so unnecessarily bureaucratic that it stifles responsible research. Although some reforms by particular federal agencies and professional societies
are under way,1 it will take the efforts of both the executive and legislative branches of government to put in place a streamlined, effective, responsive, and comprehensive system that achieves the protection of all human participants and encourages ethically responsible research.
Clearly, scientific investigation has extended and enhanced the quality of life and increased our understanding of ourselves, our relationships with others, and the natural world. It is one of the foundations of our society’s material, intellectual, and social progress. For many citizens, scientific discoveries have alleviated the suffering caused by disease or disability. Nonetheless, the prospect of gaining such valuable scientific knowledge need not and should not be pursued at the expense of human rights or human dignity. In the words of philosopher Hans Jonas, “progress is an optional goal, not an unconditional commitment, and...its tempo... compulsive as it may become, has nothing sacred about it” (Jonas 1969, 245).
Since the 1974 formation of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research and the activities in the early 1980s of the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research, American leaders have consistently tried to enhance the protections for human research participants. The research community has, in large part, supported the two essential protections for human participants: independent review of research to assess risks and potential benefits and an opportunity for people to voluntarily and knowledgeably decide whether to participate in a particular research protocol.
i
Prologue
The charter of the National Bioethics Advisory Commission (NBAC), a presidential commission created in 1995, makes clear the Commission’s focus: “As a first priority, NBAC shall direct its attention to consideration of protection of the rights and welfare of human research subjects.” In our first five years, we focused on several issues concerning research involving human participants, issuing five reports and numerous recommendations that, when viewed as a whole, reflect our evolving appreciation of the numerous and complex challenges facing the implementation and oversight of any system of protections.2 The concerns and recommendations addressed in these reports reflect our dual commitment to ensuring the protection of those who volunteer for research while supporting the continued advance of science and understanding of the human condition. This report views the oversight system as a whole, provides a rationale for change, and offers an interrelated set of recommendations to improve the protection of human participants and enable the oversight system to operate more efficiently.
Respecting Research Participants
Whether testing a new medical treatment, interviewing people about their personal habits, studying how people think and feel, or observing how they live within groups, research seeks to learn something new about the human condition. Unfortunately, history has also demonstrated that researchers sometimes treat participants not as persons but as mere objects of study. As Jonas observed: “Experimentation was originally sanctioned by natural science. There it is performed on inanimate objects, and this raises no moral questions. But as soon as animate, feeling beings become the subject of experiment...this innocence of the search for knowledge is lost and questions of conscience arise” (Jonas 1969, 219).
How, then, should people be studied? For over half a century, since the revelations of medical torture under the guise of medical experimentation were described at the Nuremberg Trials,3 it has been agreed that people should participate in research only when the study addresses important questions, its risks are justifiable, and an individual’s participation is voluntary and informed.
The principles underlying the Belmont Report: Ethical Principles and Guidelines for the Protections of Human Subjects of Research (Belmont Report) (National Commission 1979) have served for over 20 years as a leading source of guidance regarding the ethical standards that should govern research with human participants in the United States. The Belmont Report emphasized that research must respect the autonomy of participants, must be fair in both conception and implementation, and must maximize potential benefits while minimizing potential harms. The report’s recommendations provided a coherent rationale for the federal policies and rules that created the current U.S. system of decentralized, independent research review coupled with some degree of federal oversight. But although the Belmont Report is rightly hailed as a key source of guidance on informed consent, assessment of risk, and the injustice of placing individuals (and groups) in situations of vulnerability, the principles the report espouses and the regulations adopted as federal policy 20 years ago have often fallen short in achieving their overarching goal of protecting human research participants. Moreover, since the Belmont Report was published, additional concerns have arisen that require much-needed attention today.
Ensuring Independent Review of Risks and Potential Benefits
A central protection for research participants is the guarantee that someone other than the investigator will assess the risks of the proposed research. No one should participate in research unless independent review concludes that the risks are reasonable in relation to the potential benefits. In the United States, the Institutional Review Board, or IRB, has been the principal structure responsible for conducting such reviews.
Independent review of research is essential because it improves the likelihood that decisions are made free from inappropriate influences that could distort the central task of evaluating risks and potential benefits. Certainly, reviewers should not have a financial interest in the work, but social factors may be just as crucial. Reviewers may feel constrained because they are examining the work of their colleagues or their supervisors, and they should not participate in protocol review unless they are able to separate these concerns from their task. All
ii
reviewers who themselves are members of the research community should recognize that their familiarity with research and (perhaps) their predilection to support research are factors that could distort their judgment. Truly independent and sensitive review requires more involvement of individuals drawn from the ranks of potential research participants or those who can adequately represent the interests of potential research participants.
A critical purpose of independent review is to ensure that risks are reasonable in relation to potential personal and societal benefits. This is a precondition to offering people the opportunity to volunteer, since informed consent alone cannot justify enrollment. When reviewed for risks and potential benefits, research studies must be evaluated in their entirety. Studies often include different components, however, and the risks and potential benefits of each should also be examined separately, lest the possibility of great benefit or monetary enticement in one component cause potential participants or IRBs to minimize or overlook risk in another. No matter what potential benefit is offered to individual participants or society at large, the possibility of benefit from one element of a study should not be used to justify otherwise unacceptable elements.
In our view, IRBs should appreciate that for some components of a study, participants might incur risks with no personal potential benefit—for example, when a nondiagnostic survey is included among the components of a psychotherapy protocol or when placebos are given to some participants in a drug trial. For these elements, there should be some limitation on the amount of social and physical risk that can be imposed, regardless of the participants’ willingness to participate or the monetary (or other) enticement being offered. Further, the possibility of some benefit from one element of a study should not be used to justify otherwise unacceptable elements of research whose potential benefits, if any, accrue, solely to society at large. If aspects of a study present unacceptable risks, protocols should not be approved until these elements are eliminated. If removing the risky component would impair the study as a whole, then the entire study should be redesigned so that each of its elements presents risks that are reasonable in relation to potential benefits.
National Bioethics Advisory Commission
Other parts of studies can obscure risks, such as when standard medical interventions are compared in a patient population, leading some participants and researchers to discount the risks because they are associated with known therapies. It is essential that participants and investigators not be led to believe that participating in research is tantamount to being in a traditional therapeutic relationship. Regardless of whether there is the possibility or even the likelihood of direct benefit from participation in research, such participation still alters the relationship between a professional and the participant by introducing another loyalty beyond that to the participant, to wit, loyalty to doing good science. It is too often forgotten that even though the researchers may consider participants’ interests to be important, they also have a serious, and perhaps conflicting, obligation to science.
Years of experience with the current system of independent review have demonstrated that there are enduring questions about how to arrive at such impartial judgments and how to go about deciding when potential benefits justify risks that are incurred solely by participants or the community from which they come. In recent years, increasing strains on the system have undermined the practice of independent review. IRBs are overburdened by the volume of research coming before them, a strain that is compounded by concerns about training of IRB members and possible conflicts of interest. In addition, the constantly changing nature of research challenges existing notions about what constitutes risks and potential benefits.
Because IRBs are so central to the current oversight system, they need better guidance on how to review and monitor research, how to assess potential benefits to research participants and their communities, and how to distinguish among levels of risk. This report provides such guidance in the following areas: determining the type of review necessary for minimal risk research; ensuring that research participants are able to make voluntary decisions and are appropriately informed prior to giving consent; providing adequate protections for privacy and confidentiality; identifying appropriate measures needed when participants are susceptible to coercion or are otherwise placed in vulnerable situations; and monitoring ongoing research. In addition, the report recommends
iii
Prologue
that IRB members and staff complete educational and certification programs on research ethics before being permitted to review research studies.
Obtaining Voluntary Informed Consent
Even when risks are reasonable, however, no one should participate in research without giving voluntary informed consent (except in the case of an appropriate authorized representative or a waiver). Investigators must make appropriate disclosures and ensure that participants have a good understanding of the information and their choices, not only at the time of enrollment, but throughout the research. Engaging in this process is one of the best ways researchers can demonstrate their concern and respect for those they aim to enroll in a study. It also serves as the best means for those who do not wish to participate to protect themselves.4 Recommendations from our previous reports are reinforced in this report, which emphasizes the process of providing information and ensuring comprehension rather than the form of documentation of the decision to give consent. Both the information and the way it is conveyed—while meeting full disclosure requirements—must be tailored to meet the needs of the participants in the particular research context. In addition, documentation requirements must be adapted for varying research settings, and the criteria for deciding when informed consent is not necessary must be clarified so that participants’ rights and welfare are not endangered.
The decision to participate in research must not only be informed, it must be voluntary. Even when risks are reasonable and informed consent is obtained, it may nonetheless be wrong to solicit certain people as participants. Those who are not fully capable of resisting the request to become participants—such as prisoners and other institutionalized or otherwise vulnerable persons—should not be enrolled in studies merely because they are easily accessible or convenient. This historic emphasis on protecting people from being exploited as research participants, however, has failed to anticipate a time when, at least for some areas of medical research, people would be demanding to be included in certain studies because they might provide the only opportunity for receiving medical care for life-threatening diseases.
Making Research Inclusive While Protecting Individuals and Categorized as Vulnerable
Vulnerable individuals need additional protection in research. Although certain individuals and populations are more vulnerable as human participants than others, people whose circumstances render them vulnerable should not be arbitrarily excluded from research for this reason alone. This includes those viewed as more open to harm (e.g., children), more subject to coercion (e.g., institutionalized persons), more “complicated” (e.g., women, who are considered more biologically complicated than men), or more inconvenient (e.g., women with small children, who are viewed as less reliable research participants due to conflicting demands on time). Calling competent people intrinsically “vulnerable” can be both insulting and misleading. It is not their gender or other group designation that exposes them to injury or coercion, but rather their situation that can be exploited by ethically unacceptable research. That is, it is their circumstances, which are situational, that create the vulnerability. At other times it is the intrinsic characteristics of the person—for example, children or those with certain mental or developmental disorders—that make them generally vulnerable in the research setting.
The response, whenever possible, should not be to exclude people from research, but instead to change the research design so that it does not create situations in which people are unnecessarily harmed. To do otherwise is to risk developing knowledge that helps only a subset of the population. To the extent that the results are not generalizable, the potential societal benefits that justify doing the research are attenuated. Research participants must be treated equally and with respect. Whenever possible, research should be designed to encourage the participation of all groups while protecting their rights and welfare.
To accomplish this, we recommend that rather than focusing primarily on categorizing groups as vulnerable, investigators and IRBs should also recognize and avoid situations that create susceptibility to harm or coercion. Such situations may be as varied as patients being recruited by their own physicians; sick and desperate patients seeking enrollment in clinical trials; participants
iv
being recruited by those who teach or employ them; or studies involving participants with any characteristic that may make them less likely to receive care and respect from others (e.g., convicted criminals or intravenous drug users). In these circumstances, rather than excluding whole groups of people, researchers should design studies that reduce the risk of exploitation, whether by using a different method of recruitment, by using a recruiter who shares the participants’ characteristics, or by some other technique. This is not always easy. It requires researchers to consider carefully their research design and the potential pool of participants. At times, it will mean anticipating that otherwise seemingly benign situations may become more complex because a particular participant or group of participants will be unusually susceptible to harm or manipulation. At other times, the nature of the vulnerability may require using a different research design. Ethical research does not avoid complexity. Rather, it acknowledges the full range and realities of the human condition.
Compensating for Harms
Despite all these precautions, however, some research participants might be harmed. Participants who are harmed as a direct result of research should be cared for and compensated. This is simple justice. The fact that they offered to participate in no way alters the view that mere decency calls for us to take care of these volunteers. Unfortunately, this is a greater challenge than it might appear. For those who endure harm while participating in research, it is often very difficult to separate injuries traceable to the research from those that stem from the underlying disease or social condition being studied. For others, appropriate care and compensation would be far beyond the means of the researchers, their sponsors, and their institutions. Two decades ago, the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research called for pilot studies of compensation programs—a recommendation that was not pursued. It is time to reconsider the need for some type of compensation program and to explore the possible mechanisms that could be used were one to be adopted. Regardless of individual
National Bioethics Advisory Commission
motives, research participants are providing a service for society, and justice requires that they be treated with great respect and receive appropriate care for any related injuries. It should always be remembered that it is a privilege for any researcher to involve human participants in his or her research.
Establishing a Comprehensive, Effective, and Streamlined System
In the United States, government regulations, professional guidelines, and the general principles highlighted in the Belmont Report (1979) form the basis of the current system of protections. In the earliest stages of adoption, the federal regulations were fragmented and confusing. Even today, they apply to most—but not all—research funded or conducted by the federal government, but have inconsistent and sometimes no direct application to research funded or conducted by state governments, foundations, or industry. They apply to medical drugs and devices and vaccines approved for interstate sale, but not to some medical innovations that would remain wholly within state borders. And they apply to other research only when the investigators and their institutions volunteer to abide by the rules.
A comprehensive and effective oversight system is essential to uniformly protect the rights and welfare of participants while permitting ethically and scientifically responsible research to proceed without undue delay. A fundamental flaw in the current oversight system is the ethically indefensible difference in the protection afforded participants in federally sponsored research and those in privately sponsored research that falls outside the jurisdiction of the Food and Drug Administration. As a result, people have been subjected to experimentation without their knowledge or informed consent in fields as diverse as plastic surgery, psychology, and infertility treatment. This is wrong. Participants should be protected from avoidable harm, whether the research is publicly or privately financed. We have repeated this assertion throughout our deliberations, and recommendations in this regard appear in four previous reports (NBAC 1997; NBAC 1999a; NBAC 1999b; NBAC 2001).
v
Prologue
In this report, we recommend that the protections of an oversight system extend to the entire private sector for both domestic and international research. A credible, effective oversight system must apply to all research, and all people are entitled to the dignity that comes with freely and knowingly choosing whether to participate in research, as well as to protection from undue research risks. This is consistent with our 1997 resolution that no one should be enrolled in research absent the twin protections of independent review and voluntary informed consent.
Even when current protections apply, the interpretation of the federal regulations can vary unpredictably, depending on which federal agency oversees the research. Even the most basic, common elements of the federal rules took a decade to develop into regulations, because there was no single authority within the government to facilitate and demand cooperation and consistency. There still is no such single authority.5 This has slowed the diffusion of basic protections and made it almost impossible to develop consistent interpretations of the basic protections or those relevant to especially problematic research, such as studies involving children or the decisionally impaired. Nor has there been a unified response to emerging areas of research, such as large-scale work on medical records and social science databases or on stored human biological materials.
Today’s research protection system cannot react quickly to new developments. Efforts to develop rules for special situations, such as research on those who can no longer make decisions for themselves, have languished for decades in the face of bureaucratic hurdles, and there is no reason to believe that efforts to oversee other emerging research areas will be any more efficient. In addition, the current system leaves people vulnerable to new, virtually uncontrolled experimentation in emerging fields, such as some aspects of reproductive medicine and genetic research.
Indeed, some areas of research are not only uncontrolled, they are almost invisible. In an information age, poor management of research using medical records, human tissue, or personal interview data could lead to employment and insurance discrimination, social stigmatization, or even criminal prosecution.6 The privacy and
confidentiality concerns raised by this research are real, but the federal response has often been illusory. There is almost no guidance and certainly no coordination on these topics. The time has come to have a single source of guidance for these emerging areas, one that would be better positioned to effect change across all divisions of the government and private sector, as well as to facilitate development of specialized review bodies, as needed.
In this report we propose a new independent oversight office that would have clear authority over all other segments of the federal government and extend protections to the entire private sector for both domestic and international research. A single office would decide how to introduce consistency or reforms, and only that office would develop mechanisms to provide specialized review when needed. We recognize the challenges to such a proposal. For example, an independent office might lack the political support accorded an existing cabinet-level department. Although assigning one department, such as the Department of Health and Human Services, the role of “first among equals” would allow it to advocate forcefully for uniform rules across the government, without special provisions it would not have the authority to require other departments to comply, nor is it certain to escape the temptation to develop rules premised on a traditional, biomedical model rather than the wider range of research to be covered.
Federal research protections should be uniform across all government agencies, academe, and the private sector, but they should be flexible enough to be applied in widely different research settings or to emerging areas of research. Furthermore, any central coordinating body should be open to public input, have significant political or legal authority over research involving human participants—whether in the public or private sector—and have the support of the executive and legislative branches of government.
Education as the Key to Promoting Local Responsibility
Currently, federal protections depend on a decentralized oversight system involving IRBs, institutions, investigators, sponsors, and participants. We endorse the spirit
vi
and intent of this approach, specifically its contention that the ethical obligation to protect participants lies first with researchers, their sponsors, and the IRBs that review their research. Protecting research participants is a duty that researchers, research institutions, and sponsors cannot delegate completely to others or to the government. In addition, merely adhering to a set of rules and regulations does not fulfill this duty. Rather, it is accomplished by acting within a culture of concern and respect for research participants.
It is unrealistic to think that ethical obligations can be fully met without guidance and resources. To help researchers and IRBs fulfill their responsibilities, the federal government should promote the development of education, certification, and accreditation systems that apply to all researchers, all IRB members and staff, and all institutions. These tools should help researchers craft and IRBs review studies that pose few problems and to know when their work requires special oversight. Today, investigators and IRBs are rightly confused over issues as basic as which areas of inquiry should be reviewed and who constitutes a human participant.
Education is the foundation of the oversight system and is essential to protecting research participants. In all of our reports, we have highlighted the need to educate all those involved in research with human participants, including the public, investigators, IRB members, institutions, and federal agencies. In Cloning Human Beings (1997), we recommended federal support of public education in biomedical sciences that increasingly affect our cultural values. In Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity (1998), we called for practice guidelines and ethics education on special concerns regarding this population. In Ethical and Policy Issues in International Research: Clinical Trials in Developing Countries (2001), we recommended measures to help developing countries build their capacity for designing and conducting clinical trials, for reviewing the ethics and science of proposed research, and for using research results after a trial is completed.
In this report, we again acknowledge the inadequacy of educational programs on research ethics in the United States. This deficiency begins at the highest level within the federal oversight system and extends to the local level
National Bioethics Advisory Commission
at individual institutions. We recommend that investigators and IRB members and staff successfully complete educational programs on research ethics and become certified before they perform or review research, that research ethics be taught to the next generation of scientists, and that research ethics be included in continuing education programs.
Clarifying the Scope of Oversight
Many areas of scientific inquiry are “research,” and many of these involve human participants, but only some need federal oversight, while others might be better regulated through professional ethics, social custom, or other state and federal law. For example, certain types of surveys and interviews are considered research, but they can be well managed to avoid harms without federal oversight, as the risks are few and participants are well situated to decide for themselves whether to participate. On the other hand, certain studies of medical records, databases, and discarded surgical tissue are often perceived as something other than human research, even when the information retrieved is traceable to an identifiable person. Such research does need oversight to avoid putting people at risk of identity disclosure or discrimination without their knowledge. Federal policies should clearly identify the kinds of research that are subject to review and the types of research participants to whom protections should apply. When research poses significant risks or when its risks are imposed on participants without their knowledge, it clearly requires oversight. However, meaningless or overly rigid oversight engenders disdain on the part of researchers, creates an impossible and pointless workload for IRBs, and deters ethically sound research from going forward.
Ensuring That the Level of Review Corresponds to the Level of Risk
Even within areas of research that need oversight, many individual studies will involve little or no risk to participants. Although current federal policies allow for some distinction between research involving minimal risk and research involving more than minimal risk, the distinction operates mostly in terms of how the research will be
vii
Prologue
reviewed—that is, how procedures are to be followed. But the distinction should be based on how the research is pursued, how the participants are treated, and how the work is monitored over time. Overall, the emphasis should be on knowing how to protect participants rather than on knowing how to navigate research regulations. Instead of focusing so much on the period during which a research design is reviewed, oversight should also include an ongoing system of education and certification that helps researchers to anticipate and minimize research risks. Oversight should also make it easier for researchers to collaborate with their colleagues here and abroad without the burden of redundant reviews.
Research review and monitoring should be intensified as the risk and complexity of the research increase and at all times should emphasize protecting participants rather than following rigid rules. In addition, the review process should facilitate rather than hinder collaborative research among institutions and across national boundaries, provided that participants are protected.
Providing Resources for the Oversight System
Creating a system that protects the rights and welfare of participants and facilitates responsible research demands political and financial support from the federal government as well as the presence of a central coordinating body to provide guidance and oversee education and accreditation efforts. The oversight system should be adequately funded at all levels to ensure that research continues in a manner that demonstrates respect and concern for the interests of research participants.
Conclusions
The current system for protecting human participants in research is in need of reform. It does not protect all research participants, and where protection is offered, it is often burdened by excessive bureaucracy, confusing or conflicting interpretations of rules, and an inability to respond to emerging areas of research. We recommend that a new oversight system be adopted that is led by a responsive and authoritative federal office, that
emphasizes researcher education and research design as the primary means to protect participants, and that encourages responsible research while protecting all research participants.
Notes
1 For example, the Office for Human Research Protections is implementing a new process by which institutions assure future compliance with human participant protections. The Institute of Medicine has recently issued a report on accreditation standards for IRBs (IOM 2001). Public Responsibility in Medicine and Research has established training programs and has co-founded a new organization, the Association for the Accreditation of Human Research Protection Programs.
2 To date, NBAC has issued five reports: Cloning Human Beings
(NBAC 1997), Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity (NBAC 1998), Ethical Issues in Human Stem Cell Research (NBAC 1999a), Research Involving Human Biological Materials: Ethical Issues and Policy Guidance (NBAC 1999b), and Ethical and Policy Issues in International Research: Clinical Trials in Developing Countries (NBAC 2001).
3 United States v. Karl Brandt et al., Trials of War Criminals Before the Nuremberg Military Tribunals Under Control Council Law 10. Nuremberg, October 1946–April 1949. Volumes I–II. Washington, D.C.: U.S. Government Printing Office.
4 There are, of course, some circumstances in which consent cannot be obtained and in which an overly rigid adherence to this principle would preclude research that is either benign or potentially needed by the participant him- or herself. Thus, NBAC endorses the current exceptions for research that is of minimal risk to participants and for potentially beneficial research in emergency settings where no better alternative for the participants exists. NBAC also urges attention to emerging areas of record, database, and tissue bank research in which consent serves only as a sign of respect and in which alternative ways to respect participants do exist (NBAC 1999b; 21 CFR 50.24). In a previous report, the Commission made recommendations regarding persons who lack decisionmaking capacity and from whom informed consent cannot be obtained (NBAC 1998).
5 Porter, J., Testimony before NBAC. November 23, 1997. Bethesda, Maryland. See McCarthy, C.R., “Reflections on the Organizational Locus of the Office for Protection from Research Risks.” This background paper was prepared for NBAC and is available in Volume II of this report.
6 See Goldman, J., and A. Choy, “Privacy and Confidentiality in Health Research” and Sieber, J., “Privacy and Confidentiality: As Related to Human Research in Social and Behavioral Science.” These background papers were prepared for NBAC and are available in Volume II of this report. See also Ferguson v. City of Charleston 121 S. Ct. 1281. (2001).
viii
References
Beecher, H.K. 1966. “Ethics and Clinical Research.” New England Journal of Medicine 274(24):1354–1360.
Faden, R.R., and T.L. Beauchamp. 1986. A History and Theory of Informed Consent. New York: Oxford University Press.
Institute of Medicine (IOM). 2001. Preserving Public Trust: Accreditation and Human Research Participant Protection Programs.
Washington, D.C.: National Academy Press.
Jonas, H. 1969. “Philosophical Reflections on Experimenting with Human Subjects.” Daedalus 98:219–247.
Jones, J.H. 1981. Bad Blood: The Tuskegee Syphilis Experiment.
New York: The Free Press.
Katz, J. 1972. Experimentation with Human Beings. New York: Russell Sage Foundation.
National Bioethics Advisory Commission (NBAC). 1997. Cloning Human Beings. 2 vols. Rockville, MD: U.S. Government Printing Office.
National Bioethics Advisory Commission
———. 1998. Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity. 2 vols. Rockville, MD: U.S. Government Printing Office.
———. 1999a. Ethical Issues in Human Stem Cell Research. 3 vols. Rockville, MD: U.S. Government Printing Office.
———. 1999b. Research Involving Human Biological Materials: Ethical Issues and Policy Guidance. 2 vols. Rockville, MD: U.S. Government Printing Office.
———. 2001. Ethical and Policy Issues in International Research: Clinical Trials in Developing Countries. 2 vols. Bethesda, MD: U.S. Government Printing Office.
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission). 1979.
Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research. Washington, D.C.: U.S. Government Printing Office.
Tuskegee Syphilis Study Ad Hoc Advisory Panel. 1973. Final Report. Washington, D.C.: U.S. Department of Health, Education, and Welfare.
ix
Summary of Recommendations
The charter of the National Bioethics Advisory Commission (NBAC), a presidential commission created in 1995, states that “[a]s a first priority, NBAC shall direct its attention to consideration of protection of the rights and welfare of human research subjects.” During NBAC’s first five years, the Commission focused on several special issues surrounding research involving human participants, issuing five reports and numerous recommendations that, when viewed as a whole, reflect an evolving appreciation of the various and complex challenges facing the implementation and oversight of the system that protects those who volunteer to participate in research.
In May 1997, NBAC unanimously resolved that “[n]o person in the United States should be enrolled in research without the twin protections of informed consent by an authorized person and independent review of the risks and benefits of the research.” In 1999, NBAC indicated to the White House the following areas of concern regarding the oversight of human research in the United States: 1) not all research participants are protected by the federal oversight system; 2) a number of federal departments and agencies that sponsor primarily nonbiomedical research or modest amounts of research have failed to implement fully the federal protections available; 3) the federal protections do not always include specific provisions for those individuals who are especially vulnerable; and 4) the federal protections are difficult to enforce and improve effectively across the government, in part, because no single authority or office oversees research protections across all agencies and departments.
Based on these findings and in response to a special request from the White House Office of Science and Technology Policy to further develop recommendations
for improving the system for protecting research participants, NBAC undertook a comprehensive examination of the various aspects of the oversight system, including its purpose; its structure, including its local configuration—composed of investigators, institutions, and Institutional Review Boards (IRBs); and the ethical issues relevant to review of research. The recommendations contained in this report reflect a dual commitment to ensuring the protection of those who volunteer for research while supporting the continued advancement of science. The recommendations are based on a view of the oversight system as a whole and provide both a rationale for change and an interrelated set of proposals to improve the protection of human participants and enable the oversight system to operate more efficiently.
Readers should note that three general themes are found in the recommendations. First, they suggest that there should be fewer federal regulations and more guidance. Second, they generally focus attention on research for which participants need the most protection and strive to make the level of protection commensurate with the level of risk involved in the research. This theme is strongest in recommendations regarding continuing review, verification of compliance, adverse event reporting, informed consent, and the review of minimal risk research. Third, the recommendations both increase the scope of regulated research and streamline the process of regulatory compliance.
The most significant proposed change is that of consolidation. In place of the current multiple sets of overlapping regulations, a unified comprehensive policy is proposed in the form of a single set of regulations with a single source of guidance and interpretation. Six distinct functions or responsibilities of a consolidated oversight
xi
Summary of Recommendations
system are recommended: policy development through regulations and guidance; educational programs aimed at the responsibilities of all parties; ethical review of proposed research; monitoring of ongoing research; enforcement of the requirements; and accountability for all those involved in the review and conduct of research. By focusing attention and resources on these functions, the system will provide the necessary infrastructure for protecting research participants and promoting ethical research. To ignore any one of them is to have a system that functions less than optimally.
Scope and Structure of the Oversight System
The entitlements due to all research participants of a prior independent review of risks and potential benefits and the opportunity to exercise voluntary informed consent are the most basic and essential protections for all research participants. However, not all research participants receive these entitlements and not all are protected by the existing oversight system. The commitment to protect participants should not be voluntary, nor should requirements be in place for only some human research. Extending current protections to all research, whether publicly or privately funded, and making uniform all federal regulations and guidance cannot be accomplished within the current oversight system, in which no entity has the authority to act on behalf of all research participants. Thus, to facilitate the extension of the same protections to all humans participating in research, a unified, comprehensive federal policy promulgated and interpreted by a single office is needed.
Recommendation 2.1: The federal oversight system should protect the rights and welfare of human research participants by requiring 1) independent review of risks and potential benefits and 2) voluntary informed consent. Protection should be available to participants in both publicly and privately sponsored research. Federal legislation should be enacted to provide such protection.
Recommendation 2.2: To ensure the protection of the rights and welfare of all research participants, federal legislation should be enacted to create a single, independent federal office, the National
Office for Human Research Oversight (NOHRO), to lead and coordinate the oversight system. This office should be responsible for policy development, regulatory reform (see Recommendation 2.3), research review and monitoring, research ethics education, and enforcement.
Recommendation 2.3: A unified, comprehensive federal policy embodied in a single set of regulations and guidance should be created that would apply to all types of research involving human participants (see Recommendation 2.2).
Determining whether particular research activities involving human participants should be subject to a federal oversight system has been a source of confusion for some time. No regulatory definition of covered research can be provided that has the sensitivity and specificity required to ensure that all research activities that include human participants that should be subject to oversight are always included and all activities that should be excluded from oversight protections are always excluded. Clarification and interpretation of the definition of what constitutes research involving human participants will invariably be required if the oversight system is to work effectively and efficiently. Moreover, there will always be cases over which experts disagree about the research status of a particular activity. One of the important leadership roles the proposed oversight office should fulfill is that of providing guidance on determining whether an activity is research involving human participants and is therefore subject to oversight.
Recommendation 2.4: Federal policy should cover research involving human participants that entails systematic collection or analysis of data with the intent to generate new knowledge. Research should be considered to involve human participants when individuals 1) are exposed to manipulations, interventions, observations, or other types of interactions with investigators or 2) are identifiable through research using biological materials, medical and other records, or databases. Federal policy also should identify those research activities that are not subject to federal oversight and outline a procedure for determining whether a particular study is or is not covered by the oversight system.
xii
The proposed federal office should initiate a process in which representatives from various disciplines and professions (e.g., social science, humanities, business, public health, and health services) contribute to the development of the definition and the list of research activities subject to the oversight system.
Level of Review
Although the definition of research involving human participants should be applied to all disciplines, the risks differ both qualitatively and quantitatively across the spectrum of research. Therefore, the oversight system should ensure that all covered research is subject to basic protections—such as a process of informed consent—with the exceptions of the specified conditions for which these protections can be waived, including protection of privacy and confidentiality and minimization of risks. Because the proposed oversight system may include more research activities, it is more critical than ever that review mechanisms and criteria for various types of research are suited to the nature of the research and the likely risks involved. More specific guidance is needed for review of different types of research, including appropriate review criteria and IRB composition. For example, procedures other than full board review could be used for minimal risk research, and national level reviews could supplement local IRB review of research involving novel or controversial ethical issues.
Recommendation 2.5: Federal policy should require research ethics review that is commensurate with the nature and level of risk involved. Standards and procedures for review should distinguish between research that poses minimal risk and research that poses more than minimal risk. Minimal risk should be defined as the probability and magnitude of harms that are normally encountered in the daily lives of the general population (see Recommendation 4.2). In addition, the federal government should facilitate the creation of special, supplementary review bodies for research that involves novel or controversial ethical issues.
National Bioethics Advisory Commission
Education, Certification, and Accreditation
Protecting the rights and welfare of research participants is the major ethical obligation of all parties involved in the oversight system, and to provide these protections, all parties must be able to demonstrate competence in research ethics—that is, conducting, reviewing, or overseeing research involving human participants in an ethically sound manner. Such competence entails not only being knowledgeable about relevant research ethics issues and federal policies, but also being able to identify, disclose, and manage conflicting interests for institutions, investigators, or IRBs. Finally, the oversight system must include a sufficiently robust monitoring process to provide remedies for lapses by institutions, IRBs, and investigators.
Recommendation 3.1: All institutions and sponsors engaged in research involving human participants should provide educational programs in research ethics to appropriate institutional officials, investigators, Institutional Review Board members, and Institutional Review Board staff. Among other issues, these programs should emphasize the obligations of institutions, sponsors, Institutional Review Boards, and investigators to protect the rights and welfare of participants. Colleges and universities should include research ethics in curricula related to research methods, and professional societies should include research ethics in their continuing education programs.
Recommendation 3.2: The federal government, in partnership with academic and professional societies, should enhance research ethics education related to protecting human research participants and stimulate the development of innovative educational programs. Professional societies should be consulted so that educational programs are designed to meet the needs of all who conduct and review research.
Educating all parties in research ethics and human participant protections is effective only when it results in the necessary competence for designing and conducting ethically sound research, including analyzing, interpreting, and disseminating results in an ethically sound
xiii
Summary of Recommendations
manner. Such competence, however, cannot be assumed to follow from exposure to an educational course or program. As the complexion of research continues to change and as technology advances, new and challenging ethical dilemmas will emerge. And, as more people become involved in research as investigators or in roles that are specifically related to oversight, it becomes increasingly important for all parties to be able to demonstrate competence in the ethics of research involving human participants.
Although accreditation and certification do not always guarantee the desired outcomes, these programs, which generally involve experts and peers developing a set of standards that represents a consensus of best practices, can be helpful in improving performance. Therefore, the choice of standards for these programs and the criteria for evaluating whether an institution has met them are critically important. Accreditation and certification programs should emphasize providing education and assuring that appropriate protections are in place, while avoiding excessively bureaucratic procedures.
Recommendation 3.3: All investigators, Institutional Review Board members, and Institutional Review Board staff should be certified prior to conducting or reviewing research involving human participants. Certification requirements should be appropriate to their roles and to the area of research. The federal government should encourage organizations, sponsors, and institutions to develop certification programs and mechanisms to evaluate their effectiveness. Federal policy should set standards for determining whether institutions and sponsors have an effective process of certification in place.
Recommendation 3.4: Sponsors, institutions, and independent Institutional Review Boards should be accredited in order to conduct or review research involving human participants. Accreditation should be premised upon demonstrated competency in core areas through accreditation programs that are approved by the federal government.
Assessing and Monitoring Compliance
Assessing institutional, IRB, and investigator compliance can help to ensure that standards are being followed
consistently. Current mechanisms for assessment include assurances of compliance issued by the Department of Health and Human Services and several other federal departments, site inspections of IRBs conducted by the Food and Drug Administration, other types of site inspections for participant protection, and institutional audits. In addition, some institutions have established ongoing mechanisms for assessing investigator compliance with regulations. However, institutions vary considerably in their efforts and abilities to monitor investigator compliance, from those that have no monitoring programs to those that conduct random audits. Assessing the behavior of investigators is an important part of protecting research participants and should be taken seriously as a responsibility of each institution. Investigators, IRBs, and institutions should discuss the many practical issues involved in monitoring investigators as they conduct their research studies and provide input into the regulatory process.
Recommendation 3.5: The process for assuring compliance with federal policy should be modified to reduce any unnecessary burden on institutions conducting research and to register institutions and Institutional Review Boards with the federal government. The assurance process should not be duplicative of accreditation programs for institutions (see Recommendation 3.4).
Recommendation 3.6: Institutions should develop internal mechanisms to ensure Institutional Review Board compliance and investigator compliance with regulations, guidance, and institutional procedures. Mechanisms should be put in place for reporting noncompliance to all relevant parties.
Managing Conflicts of Interest
A research setting that involves human participants necessarily creates a conflict of interest for investigators who seek to develop or revise knowledge by enrolling individuals in research protocols to obtain that knowledge. Overzealous pursuit of scientific results could lead to harm if, for example, investigators design research studies that pose unacceptable risks to participants,
xiv
enroll participants who should not be enrolled, or continue studies even when results suggest they should have been modified or halted. Conflicts of interest can also exist for IRB members or the institutions in which the research will be conducted. Thus, it is important to address prospectively the potentially harmful effects on participants that conflicts of interest might cause.
Organizations, particularly academic institutions, should become more actively involved in managing investigators’ and IRB members’ conflicts of interest and increase their efforts for self-regulation in this arena. IRB review of research studies is one method for identifying and dealing with conflicts of interest that might face investigators. By having IRBs review research studies prospectively and follow an IRB-approved protocol, investigators and IRBs together can manage conflict between the investigators’ desire to advance scientific knowledge and to protect the rights and welfare of research participants. Financial and other obvious conflicts for IRB members, such as collaboration in a research study, are often less difficult to identify and manage than some of the more subtle and pervasive conflicts. Guidance should be developed to assist IRBs in identifying various types of conflict.
Recommendation 3.7: Federal policy should define institutional, Institutional Review Board, and investigator conflicts of interest, and guidance should be issued to ensure that the rights and welfare of research participants are protected.
Recommendation 3.8: Sponsors and institutions should develop policies and mechanisms to identify and manage all types of institutional, Institutional Review Board, and investigator conflicts of interest. In particular, all relevant conflicts of interest should be disclosed to participants. Policies also should describe specific types of prohibited relationships.
IRB Membership
Appropriate composition of IRB membership ensures that research studies are reviewed with the utmost regard for protecting the rights and welfare of research participants. Current federal regulations require that each IRB have “at
National Bioethics Advisory Commission
least one member who is not otherwise affiliated with the institution and who is not part of the immediate family of a person who is affiliated with the institution” (45 CFR 46.107(d); 21 CFR 56.107(d)). The regulations also require that each IRB include “at least one member whose primary concerns are in scientific areas and at least one member whose primary concerns are in non-scientific areas.” Some have raised the concern of whether only 1 unaffiliated member on an IRB is sufficient to avoid institutional influence, especially when IRBs have 15 to 21 members on average. In addition, unaffiliated members do not have to be present for an IRB to conduct review and approve research studies. Thus, IRBs can approve research with only institutional representation present as long as a nonscientist and a quorum are also present. IRBs should strive to complement their membership by having clearly recognizable members who are unaffiliated with the institutions, members who are nonscientists, and members who represent the perspectives of participants. However, it is difficult to require that IRBs increase the presence and participation of more unaffiliated members to reduce the influence of institutional interests on IRB decisionmaking, because finding them can be difficult. Currently, there are no rules or guidance that describe criteria for meeting the definition of an unaffiliated member, that specify how long such members should serve, or that provide guidance regarding under what circumstances they may be removed or what payment should be provided. Institutions should be careful to select unaffiliated members who are truly separated from the institution, except for their role on the IRB. Procedures for the selection and removal of unaffiliated members should be established in a way that empowers the independent voices of those members. In addition, providing reasonable payment to IRB members who are otherwise unaffiliated with the institution can be a valuable way to strengthen these members’ role.
Recommendation 3.9: Federal policy should establish standards and criteria for the selection of Institutional Review Board members. The distribution of Institutional Review Board members with relevant expertise and experience should be commensurate with the types of research reviewed by the Institutional Review Board (see Recommendation 3.10).
xv
Summary of Recommendations
Recommendation 3.10: Institutional Review Boards should include members who represent the perspectives of participants, members who are unaffiliated with the institution, and members whose primary concerns are in nonscientific areas. An individual can fulfill one, two, or all three of these categories. For the purposes of both overall membership and quorum determinations 1) these persons should collectively represent at least 25 percent of the Institutional Review Board membership and 2) members from all of these categories should be represented each time an Institutional Review Board meets (see Recommendation 3.9).
Guidance for Assessing Risks and Potential Benefits
In addition to protecting the rights and welfare of research participants, it is equally important to protect them from avoidable harm. Thus, an IRB’s assessment of the risks and potential benefits of research is central to determining whether a research study is ethically acceptable. Yet, this assessment can be a difficult one to make, as there are no clear criteria for IRBs to use in judging whether the risks of research are reasonable in terms of what might be gained by the individual or society.
IRBs should be able to identify whether a clear and direct benefit to society or the research participants might result from participating in the study. However, IRBs should be cautious in classifying procedures as offering the prospect of direct benefit. In fact, if it is not clear that a procedure also offers the prospect of direct benefit, IRBs should treat the procedure as one solely designed to answer the research question(s). A major advantage of this approach is that it avoids justifying the risks of procedures that are designed solely to answer the research question(s) based on the likelihood that another procedure in the protocol would provide a benefit.
Recommendation 4.1: An analysis of the risks and potential benefits of study components should be applied to all types of covered research (see Recommendation 2.4). In general, each component of a study should be evaluated separately,
and its risks should be both reasonable in themselves as well as justified by the potential benefits to society or the participants. Potential benefits from one component of a study should not be used to justify risks posed by a separate component of a study.
Minimal Risk
Determining whether a study poses more than minimal risk is a central ethical and procedural function of the IRB. The definition of minimal risk in federal regulations (45 CFR 46.102(i); 21 CFR 56.102(i)) provides an ambiguous standard by which risks involved in a research study are compared to those encountered in daily life. However, it is unclear whether this applies to those risks found in the daily lives of healthy individuals or those of individuals who belong to the group targeted by the research. If it refers to the individuals to be involved in the research, then the same intervention could be classified as minimal risk or greater than minimal risk, depending on the health status of those participants and their particular experiences. According to this understanding, the standard for minimal risk is a relative one.
This report recommends that IRBs use a standard related to the risks of daily life that are familiar to the general population for determining whether the level of risk is minimal or more than minimal, rather than using a standard that refers to the risks encountered by particular persons or groups. These common risks would include, for example, driving to work, crossing the street, getting a blood test, or answering questions over the telephone. Thus, research would involve no more than minimal risk when it is judged that the level of risk is no greater than that encountered in the daily lives of the general population.
Recommendation 4.2: Federal policy should distinguish between research studies that pose minimal risk and those that pose more than minimal risk (see Recommendation 2.5). Minimal risk should be defined as the probability and magnitude of harms that are normally encountered in the daily lives of the general population. If a study that would normally be considered
xvi
minimal risk for the general population nonetheless poses higher risk for any prospective participants, then the Institutional Review Board should approve the study only if it has determined that appropriate protections are in place for all prospective participants.
Evaluating Vulnerability
All segments of society should have the opportunity to participate in research, if they wish to do so and if they are considered to be appropriate participants for a given protocol. However, some individuals may need additional protections before they can fully participate in the research study; otherwise they might be more susceptible to coercion or exploitation. Individuals might be considered vulnerable within the research context because of intrinsic characteristics (e.g., they are children or have mental illness or retardation) or because of the situation in which they find themselves (e.g., they are impoverished, unemployed, or incarcerated). Recognizing various types of vulnerability and providing adequate safeguards can prove challenging for IRBs.
Appropriate and specific safeguards should be established to protect persons who are categorized as vulnerable. Once safeguards are established, investigators should not exclude persons categorized as vulnerable from research involving greater than minimal risk because this would deprive them of whatever potential direct benefits they might receive from the research and deprive their communities and society from the benefit of the knowledge such research might generate.
Recommendation 4.3: Federal policy should promote the inclusion of all segments of society in research. Guidance should be developed on how to identify and avoid situations that render some participants or groups vulnerable to harm or coercion. Sponsors and investigators should design research that incorporates appropriate safeguards to protect all prospective participants.
National Bioethics Advisory Commission
Emphasizing the Informed Consent Process
Rather than focusing on the ethical standard of informed consent and what is entailed in the process of obtaining informed consent, IRBs and investigators have followed the lead of the federal regulations and have tended to focus on the disclosures found in the consent form. However, from an ethics perspective, the informed consent process, not the form of its documentation, is the critical communication link between the prospective participant and the investigator throughout a study, beginning when the investigator initially approaches the participant. Informed consent should be an active process through which both parties share information and during which the participant at any time can freely decide whether to withdraw from or continue to participate in the research. It is time to place the emphasis on the process of informed consent to ensure that information is fully disclosed, that competent participants fully understand the research in order to make informed choices, and that decisions to participate or not are always made voluntarily.
Recommendation 5.1: Federal policy should emphasize the process of informed consent rather than the form of its documentation and should ensure that competent participants have given their voluntary informed consent. Guidance should be issued about how to provide appropriate information to prospective research participants, how to promote prospective participants’ comprehension of such information, and how to ensure that participants continue to make informed and voluntary decisions throughout their involvement in the research.
Waiver of Informed Consent
Obtaining voluntary informed consent should not be a requirement for every research study. In fact, waiving the informed consent process is justifiable in research studies that include no interaction between investigators and participants, such as in studies using existing identifiable data (e.g., studies of records) and in studies in which risks generally are not physical. In these kinds of research,
xvii
Summary of Recommendations
risks are likely to arise from the acquisition, use, or dissemination of information resulting from the study and are likely to involve threats to privacy and breaches in confidentiality. The criteria for waiving informed consent in such instances should be revised, so that if such studies have protections in place for both privacy and confidentiality, IRBs may waive the requirement for informed consent.
Recommendation 5.2: Federal policy should permit Institutional Review Boards in certain, limited situations (e.g., some studies using existing identifiable data or some observational studies) to waive informed consent requirements if all of the following criteria are met:
a) all components of the study involve minimal risk or any component involving more than minimal risk must also offer the prospect of direct benefit to participants; b) the waiver is not otherwise prohibited by state, federal, or international law; c) there is an adequate plan to protect the confidentiality of the data; d) there is an adequate plan for contacting participants with information derived from the research, should the need arise; and e) in analyzing risks and potential benefits, the Institutional Review Board specifically determines that the benefits from the knowledge to be gained from the research study outweigh any dignitary harm associated with not seeking informed consent.
Documentation of Informed Consent
Although the federal regulations may have been intended to reflect a legal standard for documentation of informed consent, NBAC is aware of no case law in which a signed, written consent form is required. To fulfill the substantive ethical standard of informed consent, depending on the type of research proposed, it may be more appropriate to use other forms of documentation, such as audiotape, videotape, witnesses, or telephone calls to participants verifying informed consent and participation in the research study.
Recommendation 5.3: Federal policy should require investigators to document that they have obtained voluntary informed consent, but should be flexible with respect to the form of such documentation. Especially when individuals can easily refuse or discontinue participation, or when signed forms might threaten confidentiality, Institutional Review Boards should permit investigators to use other means of verifying that informed consent has been obtained.
Protecting Privacy and Confidentiality
Privacy and confidentiality are complex and poorly understood concepts in the context of some research. Privacy refers to the ways and circumstances under which investigators access information from participants. Because privacy concerns vary by type and context of research and the culture and individual circumstances of participants, investigators should be well informed and mindful of the cultural norms of the participants. In addition, investigators should be aware of the various research procedures and methods that can be used to respect privacy. Needed is a clear, comprehensive regulatory definition of privacy along with guidance for protecting privacy in various types of research.
Like privacy concerns, concerns about confidentiality vary by the type and context of the research. No one set of procedures can be developed to protect confidentiality in all research contexts. Thus, IRBs and investigators must tailor confidentiality protections to the specific circumstances and methods used in each specific research study. Further, IRBs and investigators are encouraged to consider the use of strong confidentiality protections, which can also reduce some of the violations associated with privacy. A clear, comprehensive definition of confidentiality is needed, along with guidance for protecting confidentiality in various types of research.
Recommendation 5.4: Federal policy should be developed and mechanisms should be provided to enable investigators and institutions to reduce threats to privacy and breaches of confidentiality. The feasibility of additional mechanisms should be examined to strengthen confidentiality protections in research studies.
xviii
Monitoring of Ongoing Research
Continual review and monitoring of research that is in progress is a critical element of the oversight system. Such review is necessary to ensure that emerging data or evidence have not altered the risks/potential benefits assessment so that risks are no longer reasonable. In addition, mechanisms are needed to monitor adverse events, unanticipated problems, and changes to the protocol. IRBs can do a better job in this area with the appropriate guidance and some restructuring of the review and monitoring process.
Currently, the requirement of continuing review is overly broad. The frequency and need for continuing review vary depending on the nature of research, with some protocols not requiring continuing review. In research involving high or unknown risks, the first few trials of a new intervention may substantially affect what is known about the risks and potential benefits of that intervention. Even if the knowledge does not warrant changes in study design, it may warrant changes in the information presented to prospective and enrolled participants.
On the other hand, the ethics issues and participant protections necessary in minimal risk research are unlikely to be affected by developments from within or outside the research—for example, research involving the use of existing data or research that will no longer involve contact with participants because it is in the data analysis phase. Continuing review of such research should not be required because it is unlikely to provide any additional protection to research participants and merely increases the burden of IRBs. However, because minimal risk research does involve some risk, IRBs may choose to require continuing review. In these cases, other types of monitoring may be more appropriate, such as assessing investigator compliance with the approved protocol or reporting of protocol changes and unanticipated problems. Clarifying the nature of the continuing review requirements would allow IRBs to better focus their efforts on reviewing riskier research and would increase protections for participants where they are most needed.
National Bioethics Advisory Commission
Recommendation 6.1: Federal policy should describe how sponsors, institutions, and investigators should monitor ongoing research.
Recommendation 6.2: Federal policy should describe clearly the requirements for continuing Institutional Review Board review of ongoing research. Continuing review should not be required for research studies involving minimal risk, research involving the use of existing data, or research that is in the data analysis phase when there is no additional contact with participants. When continuing review is not required, other mechanisms should be in place for ensuring compliance of investigators and for reporting protocol changes or unanticipated problems encountered in the research.
Recommendation 6.3: Federal policy should clarify when changes in research design or context require review and new approval by an Institutional Review Board.
Adverse Event Reporting
Assessing adverse events reports can be a major burden for IRBs and investigators because of the high volume and ambiguous nature of such events and the complexity of the pertinent regulatory requirements. Investigators have reported frustration in attempting to understand what constitutes an adverse event, the required reporting times, and to whom adverse events should be reported. The regulations need to be simplified, and one set of regulations should be available for safety monitoring. Regulations and guidance should be written so that investigators and sponsors understand what constitutes an adverse event, what type of event must be reported within what time period, and to whom it should be reported. In addition, regulations and guidance should be clear regarding whose responsibility it is to analyze and evaluate adverse event reports and should describe the required communication and coordination channels for these reports among IRBs and safety monitoring entities, such as Data Safety Monitoring Boards, investigators, sponsors, and federal agencies.
xix
Summary of Recommendations
Recommendation 6.4: The federal government should create a uniform system for reporting and evaluating adverse events occurring in research, especially in multi-site research. The reporting and evaluation responsibilities of investigators, sponsors, Institutional Review Boards, Data Safety Monitoring Boards, and federal agencies should be clear and efficient. The primary concern of the reporting system should be to protect current and prospective research participants.
Review of Cooperative or Multi-Site Research Studies
One of the greatest burdens on IRBs and investigators is the review of multi-site studies. Requiring multiple institutions to review the same protocol is unnecessarily taxing and provides no additional protection to participants. In addition, such review poses problems in the initial stages of review as well as in the continual review and monitoring stages and is especially problematic in the evaluation of adverse events in clinical research.
Innovative and creative alternative mechanisms and processes for reviewing protocols in multi-site research are needed. To allow for such projects and to support a change in the current system toward a more flexible review system, federal policy should be clear about the functions that must be performed, but be less restrictive about who performs each function.
Recommendation 6.5: For multi-site research, federal policy should permit central or lead Institutional Review Board review, provided that participants’ rights and welfare are rigorously protected.
Compensation for Research-Related Injuries
Participants who volunteer to be in a research study and are harmed as a direct result of that study should be cared for and compensated. However, no adequate database exists that describes the number of injuries or illnesses that are suffered by research participants, the proportion of these illnesses or injuries that are caused by the research, and the medical treatment and rehabilitation
expenses that are subsequently borne by the participants. It may be argued that regardless of the magnitude of the problem, the costs of research injuries should never be borne by participants. If individuals are injured by research participation, those who benefit from the research (e.g., institutions and sponsors) bear some obligation to compensate those who risked and suffered injury on their behalf. At this time, injured research participants alone bear both the cost of lost health and the expense of medical care, unless they have adequate health insurance or successfully pursue legal action to gain compensation from the specific individuals or organizations that were involved in conducting the research.
A comprehensive system of oversight of human research should include a mechanism to compensate participants for medical and rehabilitative costs resulting from research-related injuries.
Recommendation 6.6: The federal government should study the issue of research-related injuries to determine if there is a need for a compensation program. If needed, the federal government should implement the recommendation of the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (1982) to conduct a pilot study to evaluate possible program mechanisms.
The Need for Resources
Adopting the recommendations made in this report will generate additional costs for institutions, sponsors, and the federal government (through the establishment of a new federal oversight office). Sponsors of research, whether public or private, should work together with institutions carrying out the research to make the necessary funds available.
Recommendation 7.1: The proposed oversight system should have adequate resources to ensure its effectiveness and ultimate success in protecting research participants and promoting research:
a) Funds should be appropriated to carry out the functions of the proposed federal oversight office as outlined in this report.
xx
b) Federal appropriations for research programs should include a separate allocation for oversight activities related to the protection of human participants. c) Institutions should be permitted to request funding for Institutional Review Boards and other oversight activities. d) Federal agencies, other sponsors, and institutions should make additional funds available for oversight activities.
Future Research
This report raises many questions about ethical issues that cannot be answered because of insufficient or nonexistent empirical evidence. Current thinking about ethical issues in research—such as analysis of risks and potential benefits, informed consent, privacy and confidentiality, and vulnerability—would greatly benefit from additional research. Deserving of more study, for example, are questions regarding the development of effective approaches for assessing cognitive capacity, for evaluating what participants want to know about research, and for determining how to ascertain best practices for seeking informed consent. Clearer and more effective guidance could be developed from a stronger knowledge base. In general, understanding the ethical conduct of research
National Bioethics Advisory Commission
would be advanced by increased interdisciplinary discussion that would include biomedical and social scientists, lawyers, and historians.
Recommendation 7.2: The federal government, in partnership with academic institutions and professional societies, should facilitate discussion about emerging human research protection issues and develop a research agenda that addresses issues related to research ethics.
Conclusions
This report proposes 30 recommendations for changing the oversight system at the national and local levels to ensure that all research participants receive the appropriate protections. The adoption of these recommendations, which are directed at all who are involved in the research enterprise, will not only lead to better protection for the participants of research, but will also serve to promote ethically sound research while reducing unnecessary bureaucratic burdens. Achieving these goals will, in turn, restore the respect of investigators for the system used to oversee research, support the public’s trust in the research enterprise, and enhance public enthusiasm for all research involving human beings.
xxi
Chapter One 1
Introduction—The Need for Change
Introduction
The charter of the National Bioethics Advisory Commission (NBAC), a presidential commission created in 1995, states that “[a]s a first priority, NBAC shall direct its attention to consideration of protection of the rights and welfare of human research subjects.” During NBAC’s first five years, the Commission focused on several issues concerning research involving human participants,1 issuing five reports and numerous recommendations that, when viewed as a whole, reflect an evolving appreciation of the various and complex challenges facing the implementation and oversight of the system that protects those who volunteer to participate in research.2 In May 1997, NBAC unanimously resolved that “[n]o person in the United States should be enrolled in research without the twin protections of informed consent by an authorized person and independent review of the risks and benefits of the research.”3 In 1999, NBAC indicated to the White House several areas of concern regarding the oversight of human research in the United States and provided preliminary findings. (See Appendix B.) The key concerns identified were as follows:
1) not all research participants are protected by the federal oversight system; 2) a number of federal departments and agencies that sponsor primarily nonbiomedical research or modest amounts of research have failed to implement fully the federal protections available; 3) the federal protections do not always include specific provisions for those individuals who are especially vulnerable; and
4) the federal protections are difficult to enforce and improve effectively across the government, in part, because no single authority or office oversees research protections across all agencies and departments.
Based on these findings, and in response to a special request from the White House Office of Science and Technology Policy to further develop recommendations for improving the system for protecting research participants, NBAC undertook a comprehensive examination of the various aspects of the oversight system, including its purpose; its structure, including its local configuration—composed of investigators, institutions, and Institutional Review Boards (IRBs); and the ethical issues relevant to review of research. The recommendations contained in this report reflect a dual commitment to ensuring the protection of those who volunteer for research while supporting the continued advancement of science. They are based on a view of the oversight system as a whole and provide both a rationale for change and an interrelated set of proposals to improve the protection of human participants and enable the oversight system to operate more efficiently.
The Value of Research
Throughout history, the pursuit of knowledge has been a highly valued human endeavor, and research through systematic, empirical investigation has become an essential method of attaining this goal. Like other forms of learning, research is worthwhile because it helps to make sense of and give meaning to the world and contributes to a growing knowledge base that also gives rise to a wide variety of practical benefits. Indeed, the contributions of
1
Chapter 1: Introduction—The Need for Change
science and technology to our daily lives are so ubiquitous that they are easily taken for granted. Knowledge developed from a constant and broad-based national investment in research has resulted in improvements in health, created valuable new products for everyday living, provided the capacity to sustain cleaner environments in a rapidly industrializing world, and facilitated better personal and family relationships.
This investment in basic science and clinical and public health research also has yielded a steady decline in mortality since the 1950s (National Center for Health Statistics 2000). Significant advances in treatment and prevention have reduced the impact of deadly diseases, such as some cancers (National Center for Health Statistics 2000; Ries et al. 2000) and cardiovascular diseases (National Center for Health Statistics 2000), as well as diseases causing morbidity, such as lead poisoning (CDC 1999a), vaccine-preventable diseases (CDC 1999a), depression (Frank et al. 1993), and sexually transmitted diseases (CDC 1999b).
The humanities and social sciences are also central to society’s capacity to understand human nature and biology by informing public and private decisionmaking and by clarifying the effects of human behavior on well-being. For example, long-term studies have increased our understanding of poverty and the effects of family stability on economic well-being, leading to changes in welfare policy and the tax code (Hurst et al. 1998), and numerous studies from developmental psychology and cognitive science have articulated the processes by which people learn, with important implications for education (National Research Council 2000). By illuminating the practices of others, anthropology research has also contributed to better understanding of certain societal groups, such as the homeless (Baxter and Hopper 1981).
Many important issues involving health and well-being can be studied by looking at how they interface at the intersection of the humanities, the social sciences, and the biological sciences. That is, prevention and amelioration of many diseases require attention to the interfaces that exist at the molecular, organismal, psychosocial, and environmental levels. For example, emotional states and the availability of social resources can influence disease survival rates and recovery and even the
likelihood of developing certain illnesses, indicating that one’s position in the social hierarchy can be related to morbidity and mortality. Even gene expression at the fundamental level may depend on the general environmental conditions experienced by an organism. Thus, meaningful studies that will enhance our understanding of human health and disease will include the study of biological, psychological, environmental, and societal factors and will involve the participation of a wide range of individuals—including the healthy and the sick and the affluent and the less fortunate—all of whom deserve to have their rights and welfare protected.
The Need for Oversight of Research Involving Human Participants
Although the rewards of research for society can be great, in some cases research can seriously harm participants. However noble an investigator’s intentions may be, the uncertainties that are inherent in any research study raise the prospect of harms that may be difficult to fully anticipate.4 Thus, a system of protections is needed to minimize harms that might occur. In the United States, the core aspect of Federal Policy for the Protection of Human Subjects, known as the Common Rule (Code of Federal Regulations, Title 45 Part 46 Subpart A), has been the regulatory policy followed by 17 federal departments and agencies for protecting human research participants (see Appendix C for a history of the Common Rule’s development and Appendix E for the regulations [45 CFR 46]). Each codification of the Common Rule by a department or agency is equivalent to 45 CFR 46.101–46.124 (Subpart A), the Department of Health and Human Services (DHHS) codification. Some agencies have promulgated additional regulations concerning the protection of human participants in research, for example, those related to privacy. The Common Rule applies to all research involving human participants “conducted, supported or otherwise subject to regulation by any federal department or agency which takes appropriate administrative action to make this policy applicable to such research.” The Food and Drug Administration (FDA) also has its own regulatory authority over research involving food and color additives, investigational drugs for human use, medical devices
2
for human use, biological products for human use being developed for marketing, and electronic products that emit radiation (21 CFR 50, 56; see Appendix F). To this research, FDA applies its own set of regulations, which is generally but not entirely the same as the Common Rule. Even though the federal regulations cover a large portion of human research conducted domestically, and in some cases overseas, they are limited in their reach. In fact, if federal funds are not involved or if regulatory approval is not required, research activities involving human participants might not be subject to any form of oversight.
In general, the current research oversight system, when applicable, adequately protects the rights and welfare of research participants. However, the consequences of it not working can be tragic. Several recent cases point to the need for improvements in the current oversight system.
For example, in California, a research study of schizophrenic disorders raised concerns about the quality and completeness of informed consent and about the risks of research when one of the participants committed suicide (Appelbaum 1996; Katz 1993; OPRR 1994). The informed consent process for the study did not adequately explain the risks associated with receiving fixed rather than individually tailored doses of medication, receiving no medication at all, or the alternatives for treatment that were available outside of research (OPRR 1994).
In 1994, a healthy 19-year-old student at the University of Rochester died from complications related to a research study in which she underwent a bron-choscopy, during which investigators took more samples and used more anesthetic than were called for in the research protocol as approved by the IRB (New York State Department of Health 1996; Rosenthal 1996). Her death illustrates the need for independent review of protocols accompanied by the assurance that investigators will adhere to the approved protocol.
In 1999, the death of a young man, Jesse Gelsinger, in a gene transfer trial highlighted a number of concerns—including the role of federal oversight—that arise when researchers began human trials of new and experimental approaches to treatment (Marshall 2000; Wolf and Lo 2000). Gelsinger, who had ornithine transcarbamylase
National Bioethics Advisory Commission
deficiency, a rare genetic disorder that affects the body’s ability to eliminate ammonia, participated in a gene transfer trial conducted at the University of Pennsylvania. The Phase I study was designed to test the safety of a gene transfer vector that, if successful, would have been used to treat infants with the fatal form of the disorder. Gelsinger was in a group receiving the highest dose. Although he was aware that he was in a research study, the research may not have been fully or adequately explained to him. During this study, participants were not informed about serious adverse events that had been previously reported, such as significant elevations in liver enzymes experienced by other participants.5 In addition, FDA was not notified of results from preclinical animal studies as required,6 and some participants, including Gelsinger, did not fit the revised inclusion criteria.7 Moreover, the lead investigator had financial interests in the company that developed the gene transfer techniques being studied (Wolf and Lo 2000). Finally, the death of Jesse Gelsinger raised questions about federal and local IRB monitoring of previous gene transfer studies when it was discovered that adverse events from other trials had not been reported to the National Institutes of Health (NIH) in a timely manner. As this incident was investigated, it became clear that a mechanism for federal agencies to adequately share information was lacking and that NIH was unaware of the exact nature of adverse event reports provided to FDA. In addition, an amendment broadening the inclusion criteria for the trial was implemented without specific FDA approval.8 A number of other cases highlight the limits of the current oversight system. In the early 1990s, for example, plastic surgeons at a New York City hospital compared two common surgical procedures for facelifts by performing both procedures on each individual participant, one procedure on each side of the face. The study was not reviewed by an IRB, and the participants were not told that they were participating in a research study (Hilts 1998). The Office for Protection from Research Risks (OPRR) halted its investigation of the case when it learned that the research, which involved no federal funds, was not subject to the federal oversight system.9 In another case, in 1996, according to one news account, an eye surgeon at the University of South
3
Chapter 1: Introduction—The Need for Change
Florida performed an experiment on at least 60 people using a cutting tool he developed to accelerate the healing process after corneal transplantation. However, the surgeon did not have IRB approval to use the experimental tool on human participants,10 and participants did not give informed consent.11 Moreover, the press raised concerns about conflicts of interest because the university held a patent on the cutting tool and listed the surgeon as a co-inventor. Both stood to benefit financially from the marketing of the tool (Klein 2000).
Although the more dramatic examples have occurred in clinical research, problematic cases are not limited to this area of investigation. The real and potential harms involved in these and other well-publicized examples of the failure to protect the rights and welfare of research participants erode public trust in the research enterprise and make it clear that a viable and credible oversight system should aim first to protect participants from undue harm, with the additional goal of creating an environment in which ethically sound and meritorious research can be conducted with society’s support and trust. Ideally, the oversight system should avoid needless complexities and regulation, enhance the quality of research, and protect participants, a difficult but achievable balance.
Current Factors Influencing the Research Enterprise
The conduct of research has been transformed by many factors over the past 25 years, resulting in a much larger and more complex enterprise. Changes include shifts in patterns of research investment; growing stresses on academic medical centers and research universities; the emergence of independent IRBs; changing public perceptions and expectations about research participation; new technologies that affect risks and potential benefits in research; and growing consideration of the roles of groups and communities in research design and implementation.
Changes in Trends in Research Investment
In the past two decades, phenomenal growth has occurred in federally and industry-sponsored biomedical research. Federal expenditures for medical and health
research conducted in the United States and in foreign countries almost doubled from $6.9 billion to $13.4 billion between 1986 and 1995. Roughly half of that funding went to university-based research programs, largely to academic medical centers.12 The federal investment in research involving human participants extends well beyond biomedical research and is extremely diverse (see Exhibit 1.1).
Industry expenditures for medical and health-related research conducted in the United States and in foreign countries have been rising even faster than those of the public sector, tripling from $6.2 billion to $18.6 billion during that same period.13 Research conducted in the United States sponsored by one segment of industry, pharmaceutical companies, has experienced particularly rapid growth, rising 14-fold from $1.5 billion to $22.4 billion between 1980 and 2000 (PhRMA 2000). As a result, industry funding is playing an increasingly important role in the support and conduct of medical and health-related research.
Not surprisingly, the rapid rise in industry investment in research funding has been matched by an accompanying rise in the number of clinical investigators connected with this activity. For example, the number of investigators participating in FDA-regulated research increased from 5,500 in 1990 to 25,000 in 1996 (Valigra 1997), and the total number of U.S. clinical investigators is now estimated to be between 45,000 and 50,000 (CenterWatch 2000). Thus, the sheer volume and diversity of research have placed new strains on the system designed to oversee the protection of research participants.
Stresses on Academic Medical Centers and Research Universities
Academic medical centers, traditionally the principal sites of clinical research, have experienced certain stresses that offset, in part, the effects of this growth in research funding. In particular, managed care, price competition in health care, and cost containment efforts (e.g., the Balanced Budget Act of 1997)14 have resulted in reductions in net clinical income to academic institutions. This trend negatively influences their capacity for research and education, because excess clinical revenue traditionally has been the means by which academic medical centers subsidize these activities (Crowley and Thier 1996;
4
National Bioethics Advisory Commission
Exhibit 1.1: The Size and Scope of Federally Funded Research Involving Human Participants
Sixteen federal departments and agencies reported to NBAC that they conduct or support research involving human participants, although some components within departments reported that they do not sponsor or conduct such research (e.g., the DHHS Administration on Aging). Each agency’s research program involving human participants is distinctive in terms of its size, scope, organization, and focus, all of which reflect its primary mission. The following examples illustrate the diverse types of research conducted or supported by federal agencies:
At least 69 federal departments and agencies are not covered by the Common Rule. NBAC was unable to determine which of these departments and agencies might sponsor or conduct research with human participants; however, at least some of them are involved in such activities.
5
Chapter 1: Introduction—The Need for Change
Mechanic and Dobson 1996). In addition, clinical income has been affected by managed care’s scrutiny of patient-related costs, whereby much routine patient care is deemed unreimbursable when associated with a clinical trial or an “experimental” therapy.
One set of responses has been the establishment of revenue-generating centers for clinical research and the development of new relations with industry (Gallin and Smits 1997). Another is the creation of research partnerships with health maintenance organizations (Donahue et al. 1996). In addition, the burgeoning number of academic investigators competing for funding has stimulated many institutions to seek financial support from industry (Henderson 1999). In some cases, this shift to more private funding has changed the nature of regulatory oversight.
At the same time, industry-sponsored research is spreading between and beyond academic medical centers. In 1998, only 40 percent of industry funding for clinical trials went to academic medical centers, down from 80 percent in 1991 (Henderson 1999). Large amounts of research are now managed by private Contract Research Organizations (CROs), rather than academic investigators, and there has been significant growth in Site Management Organizations (SMOs), which conduct research in dedicated facilities and through various types of physician networks (Association of Clinical Research Professionals 1997). Research also continues to be performed in private medical and diagnostic practices unaffiliated with an SMO (CenterWatch 1998). Thus, increasingly some avenues of clinical research fall outside the strongest and most experienced part of the current system of oversight.
To find the large numbers of participants needed to enroll in clinical studies, sponsors and CROs often conduct a single research study at dozens or even hundreds of sites. A study may involve numerous academic centers, as well as community hospitals and private practice physicians. In order to compete, some academic medical centers are forming research networks and attempting to provide services similar to those of for-profit companies (Bodenheimer 2000). Consequently, the traditional biomedical research model of one research study led by one
investigator at one academic institution now occurs much less frequently than in the past, a situation that complicates and often prolongs the review and approval of research studies (OIG 1998a, 4–5).
The Emergence of Independent IRBs
As clinical research has spread beyond academic institutions, the locus of ethics review also has shifted. In the United States, the committees that review research with the mandate to protect the rights and welfare of human participants—IRBs—have traditionally been located in the institution in which the research is conducted. However, IRBs also now exist as separate entities that are not part of the organizational structure of an institution that conducts or funds research. Although many labels are used to describe these groups, this report will use the term independent IRBs.16 Independent IRBs, which have existed for more than 30 years and are growing both in size and in the number of protocols they review, usually are for-profit entities that operate on a fee-for-service basis (OIG 1998b). Traditionally, independent IRBs primarily have reviewed industry-funded clinical research, but since 1995 they also have been permitted to review federally funded research.17 It should be noted that some institutionally affiliated IRBs have begun to charge for review of certain types of protocols (e.g., industry-sponsored research), and may even conduct reviews for other, unaffiliated or loosely affiliated groups, thus acting much like independent IRBs.
Changing Public Perspectives About Research Participation
The growth and spread of clinical research also reflects a growing demand by patients for access to clinical trials. People with difficult-to-treat, life-threatening diseases often see clinical trials as offering the benefits of cutting-edge medicine. In this context, trial participation is viewed as a benefit to be sought rather than a burden to be avoided (Kahn et al. 1998). This sentiment was expressed forcefully by HIV/AIDS activists (Rothman and Edgar 1991), some of whom adopted the slogan, “A Drug Trial Is Health Care Too” (Annas 1990, 35). Disease-oriented patient activists have also emphasized the collective benefits of research for all individuals with
6
a specific condition. Advocacy groups commonly lobby Congress and NIH for more research funding (in particular for clinical trials) for specific diseases and conditions, not only to benefit individual research participants, but also to improve treatment for all who are affected by a given disease or condition.
These calls for access to trials have spurred a “recon-ceptualization of the concept of justice” in clinical research (Brody 1998). That is, although the application of the principle of justice has traditionally focused on fairly distributing the risks of research—selecting participants equitably meant not targeting individuals considered vulnerable for participation in risky research from which they were unlikely to receive any direct benefit—applying the principle of justice now focuses also on fairly distributing the potential benefits of research. Selecting participants equitably means not unfairly excluding certain subgroups of the population from research and working to ensure that the knowledge gained in research applies as appropriate across all groups in society. Routine exclusion of groups—such as women of childbearing age—once seen as appropriate and protective, is now seen as arbitrary and paternalistic. Therefore, several federal agencies have developed policies to promote, for example, inclusion of women and/or minorities in research, as well as data analysis relevant to these groups.18 Other policies that reflect a growing emphasis on access to participation in research include FDA’s regulations granting an exception from informed consent requirements for some emergency research (21 CFR 50.24), the promotion of the inclusion of children in research,19 and the provision of Medicare payment for the routine costs of clinical trials and items and services that are otherwise generally available to beneficiaries.20
New Technologies Posing New Challenges to Risk/Potential Benefit Analysis
Research both produces and is affected by advances in technology. However, although advances in genetics, the rise of the Internet, and the growth of informatics are all providing important new capabilities for research, these advances also can raise new ethical challenges. For example, although genetic research may pose no physical risk beyond that of drawing blood, it can pose significant
National Bioethics Advisory Commission
psychological and economic risks if participants—or their insurers or employers—learn that they are predisposed to an untreatable condition. NBAC has addressed some of these issues in a previous report (NBAC 1999b).
New information technologies can provide opportunities for medical, health-related, and social science research while also raising ethical challenges regarding the protection of confidentiality of the resulting data. The computerization of medical records, which greatly facilitates retrospective analysis of patients’ medical records, has also prompted discussion about the legitimate access to and use of medical records in the new electronic environment (Etzioni 1999; National Research Council 1997). Such new technologies might, in this case, increase threats to privacy by making it easier to identify patients from combinations of seemingly unidentifiable data, such as age and date of hospital admission (Sweeney 1997; Woodward 1999). As with medical records, computerization has prompted discussion about the ability to restrict access to and use of employment or school records, financial information, and large survey data sets (Garfinkel 2000; White 2000).
The Internet has also given rise to new research opportunities and risks by allowing investigators to reach a wide pool of participants, although participants’ assumptions about the anonymity, security, and privacy of Internet connections might not be justified. The ease with which investigators can misrepresent themselves online raises new questions about the propriety of deception research carried out in this context. The possibility for online misrepresentation by participants is also of concern; for example, investigators may have no way of knowing whether children are participants in online research and are therefore in need of special protections (Frankel and Siang 1999).
Growing Consideration of Groups and Communities
Social science research also is undergoing a number of important changes that affect the protection of research participants. Beginning with the cardiovascular disease primary prevention trials conducted in various communities and sponsored by the National Heart, Lung, and Blood Institute in the 1980s (Carleton et al.
7
Chapter 1: Introduction—The Need for Change
1995; Farquhar et al. 1985; Jacobs et al. 1986), there has been an increase in the number of research studies conducted in community settings (Mittelmark et al. 1993). As the behavioral and social determinants of more diseases are known (e.g., HIV/AIDS, lung cancer, heart attack, stroke), the focus of intervention strategies has broadened from the individual to the population, and the research setting has in some cases moved into the community (Schneiderman and Speers 2000). For example, research on cigarette smoking once focused on cessation efforts, and interventions were targeted at individuals (DiClemente et al. 1991). Now, with the emphasis on prevention of smoking behavior, research interventions are often targeted at particular populations and carried out at the community level (Cummings 1999). With such community-oriented research interventions, defining the research participants and identifying the appropriate participant protections can be difficult.
Increasingly, research is conducted with communities, not on communities (Bracht 1991; George et al. 1996). Local community groups and organizations often act as collaborative investigators by sharing responsibility with academic investigators in designing and implementing a research study (Hatch et al. 1993). However, this new collaborative role of the community raises many issues related to research infrastructure and oversight. For example, it is unclear when community groups must have an IRB and how to build capacity within the community to carry out these regulatory responsibilities. Issues related to just what individual or which group speaks for the community as a whole and how to obtain community input or consent are continuing challenges to conducting such research.
Major Challenges Facing the Current Oversight System
Huge financial investments, expansion of the research enterprise, and new technology have all stressed a system of oversight that is less than optimal. A major overarching challenge that faces the entire system is a lack of adequate resources, both financial and human. Scarce resources limit the functioning of the oversight system at every level and often prevent federal offices, institutions, and
IRBs from implementing initiatives that would improve the system.
A 1996 General Accounting Office report, conducted at the request of the Senate Committee on Governmental Affairs, found the current system for protecting participants in scientific research to be deficient because of heavy workloads and competing demands on IRBs, a lack of preparedness of IRBs to review complex research, limited funds for federal inspections, and over-reliance on investigators’ willingness to comply with regulatory requirements (GAO 1996).
In June 1998, the DHHS Office of Inspector General (OIG) sounded a “warning signal” that the system had not adapted sufficiently to the changing research environment (OIG 1998a). This warning accompanied and was followed by a series of reports on specific aspects of the oversight of research, particularly the role of IRBs (OIG 1998b–e; OIG 2000a–d). The OIG reports found that many IRBs are simply overwhelmed by the volume and complexity of the research they review, by a lack of financial, administrative, and educational resources, and by a regulatory system that often distracts from rather than focuses on key ethical issues. These pressures make the system inefficient and strain its capacity to protect participants.
A related report sponsored by the NIH Office of Extramural Research provided quantitative information about IRBs’ workloads (Bell et al. 1998). Based on a survey of 491 institutions holding Multiple Project Assurances (MPAs),21 this report provided a sense of the scale of the human research enterprise and noted that some IRBs review a striking number of protocols. Indeed, the highest volume IRBs, about 10 percent, were found to account for 37 percent of the total reviews (Bell et al. 1998, 8).
Recent actions of the federal Office for Human Research Protections (OHRP) (formerly OPRR) within DHHS highlight the existence of systemic problems of the oversight system at the institutional level. For example, OHRP has restricted or suspended MPAs and required corrective actions at nearly a dozen academic institutions. These sanctions were imposed by OHRP when it found “numerous deficiencies and concerns regarding systemic protections for human subjects” (OHRP 2000). As previous reports have suggested, deficiencies occurred in areas
8
such as IRB membership, education of IRB members and investigators, institutional commitment, IRB initial and continuing review of protocols, review of protocols involving vulnerable persons, and procedures for obtaining voluntary informed consent.
The recent academic literature regarding the current oversight system for the protection of human research supports many of the findings from these reports (Edgar and Rothman 1995; Moreno et al. 1998; Phillips 1996; Snyderman and Holmes 2000; Woodward 1999). There is general recognition that because the nature and context of research have changed, the nature and structure of the oversight of research also must change. For example, Moreno et al. argue that the federal regulations should be revised to reflect changes that have affected the nature and context of research, such as the increased importance of multi-site studies (1998). They also argue that the federal regulations should be responsive to certain needed protections that have been identified, but were not enacted when the Common Rule was issued (e.g., protections for individuals categorized as vulnerable). Others, including Edgar and Rothman, argue that the expansion of the scientific frontier requires that ethics review mechanisms other than local IRBs should be considered, such as national, topic-specific advisory panels (1995). Edgar and Rothman also characterize the local IRB as a “paper tiger,” buried in paperwork and often unable to deal effectively with ethical issues (1995).
While more protection may be needed in some areas, another concern is the overwhelming burden that is placed on IRBs and investigators and the extent to which unnecessary paperwork requirements are displacing a focus on important ethical issues. For example, Phillips points to the growing frustration among investigators and IRBs that has resulted from the increase in administrative and regulatory requirements without a commensurate increase in protection (1996). Some support the need for oversight while still perceiving regulatory and compliance mechanisms, such as reporting requirements, as difficult to interpret, redundant, and inefficient (Snyderman and Holmes 2000).
Others perceive the problems with the current oversight system as failures to address such issues as inadequate funding, lack of adequate education for IRB
National Bioethics Advisory Commission
members and investigators, and insufficient focus on conflicts of interest in research (Amdur 2000; Shamoo 1994; Snyderman and Holmes 2000; Sugarman 2000). Overall, there is broad agreement in the academic literature that the current oversight system is in need of improvement.
Inconsistent Interpretation and Implementation of the Regulations by Federal Agencies
The creation of the Common Rule (see Appendix C) provided significant unification in the language of federal regulations for the protection of human research participants. However, the Common Rule did not create a shared mechanism for interpreting and implementing the regulations at the federal level. In the absence of a formal mechanism, OHRP sometimes acts as a de facto reference point and consensus builder among federal agencies, even though it has no congressional or executive authority to do so. Moreover, some other departments have not established offices comparable to OHRP for interpreting and implementing the regulations; in some cases, a single individual is responsible for oversight activities. Thus, the ability to coordinate oversight among the departments is weak, leading departments and agencies bound to the Common Rule potentially to interpret regulatory requirements differently (see Exhibit 1.2). In addition to varying substantive interpretations of the regulations, departments and agencies use different procedures to ensure compliance. This issue is further discussed in Chapter 2.
Incomplete Protection of Individuals Considered Vulnerable
Some federal departments have supplemented the Common Rule with additional regulations and policies. For example, DHHS provides additional protections for pregnant women and fetuses, prisoners, and children (45 CFR 46 Subparts B, C, and D).22 The Central Intelligence Agency and the Social Security Administration (SSA) also follow these regulations for groups that are considered vulnerable. The Department of Education has adopted protections for children (34 CFR 97 Subpart D), and the Department of Justice has adopted protections for research conducted within the Bureau of Prisons (28 CFR 512). In addition, although FDA’s regulations do not include
9
Chapter 1: Introduction—The Need for Change
Exhibit 1.2: Examples of Inconsistencies Among Agencies Bound to the Common Rule
There is wide variation among federal departments and agencies regarding their policies and procedures for determining whether a research activity is exempt from the federal regulations (45 CFR 46.101). Differences could be due to the variability in the types of research sponsored; however, they also could be due in part to inconsistent interpretation of the regulations. As shown in Table 1.1, many agencies report that all, or nearly all, of the research that they conduct or sponsor is exempt from the federal regulations.
The procedures used to make these determinations vary across agencies. In general, agencies use their IRB chair to determine whether research conducted by the agency is exempt, and a combination of technical and legal staff determine exemptions for human participant research sponsored through grants and contracts. Some agencies have customized administrative mechanisms for making these determinations. For example, the Census Bureau considers all of its research to be exempt under Federal Policy 15 CFR 27.101 (b)(3)(ii), which exempts survey procedures if “federal statute(s) require(s) without exception that the confidentiality of the personally identifiable information will be maintained throughout the research and hereafter.” However, privacy and confidentiality issues that relate to human participants are brought to the Census Bureau’s Policy Office. The Census Bureau’s Disclosure Review Board has primary responsibility for ensuring confidentiality in published reports and data products.
SSA does not have an IRB, because it claims that all of its research is exempt. This exemption took effect on April 4, 1983,23 as a result of a final DHHS rule published on March 4, 1983. Research carried out under section 1110(b) of the Social Security Act, however, remains subject to the Common Rule’s informed consent requirements. The 1983 notice states that “[i]n order to insure the continued protection of human subjects participating in such [otherwise exempt] research activity, the Department is adding a specific requirement of written, informed consent in any instance, not reviewed by an IRB, in which the Secretary determines that the research activity presents a danger to the physical, mental, or emotional well-being of a participant.”24 In the case of biomedical and behavioral research, in the 1983 Federal Register notice, DHHS makes clear the need for IRB review, but states that such review would be “unnecessary and burdensome in the context of research under the Social Security Act and otherwise.”25 DHHS discusses, but rejects, several proposals for IRB review of research and demonstrations to support public benefit or service programs and concluded that “ethical and other problems raised by research in benefit programs will be addressed by the officials who are familiar with the programs and responsible for their successful operations under state and federal law.”26 SSA reviewed the 1983 regulation with OHRP/OPRR and concluded that it continues to apply to SSA research and demonstrations. In 1999, SSA did not conduct any extramural human participant research or demonstrations under section 1110(b).
The Health Resources and Services Administration (HRSA, DHHS) reported that nearly all of its research activity comprises program evaluation or evaluation of demonstration projects, which are considered to be exempt from the federal regulations under the public “benefit and service” criterion. However, HRSA requires such a claim of exemption to be approved by the HRSA Human Subjects Committee. Otherwise IRB oversight is required.27 Furthermore, even within DHHS, both substantive and procedural differences can be found, notably between FDA and DHHS regulations. These differences relate to informed consent, the definition of research, emergency research, assurances of compliance, inspections by the sponsoring agency, sanctions for noncompliance, and additional protections for vulnerable populations.
Whatever the source, inconsistency among departments and agencies can lead to confusion and frustration among some investigators and IRBs28 and can render the oversight system unnecessarily confusing and open to misinterpretation. Not only do different rules apply to different research studies, but a single study may be subject to more than one set of regulations if it is sponsored or conducted by institutions that are required to follow more than one set of rules. IRBs and investigators are often uncertain which rules apply or to whom they must report. For example, an NIH-funded study involving an FDA-regulated investigational drug conducted in a VA hospital would be subject to the regulations and oversight of three different departments or agencies (45 CFR 46; 21 CFR 50,56; 38 CFR 16).
10
Table 1.1: Percent of Human Participants Research Determined to Be Exempt by Federal Agencies
Agency Estimated Percent
| of Human | |
| Participants | |
| Research | |
| Deemed Exempt* | |
| Central Intelligence Agency | 0 |
| Department of Commerce | 60–100 |
| Department of Defense | 80 |
| Department of Education | 60 |
| Department of Energy | unknown |
| Department of Health and Human Services | |
| Administration for Children and Families | 0 |
| Administration on Aging | unknown |
| Agency for Healthcare Research and Quality | 20–90 |
| Centers for Disease Control and Prevention | 10 |
| Food and Drug Administration | <10 |
| Health Care Financing Administration | 100 |
| Health Resources and Services Administration | 100 |
| Indian Health Service | 20 |
| National Institutes of Health | <10 |
| Substance Abuse and Mental Health | |
| Services Administration | ~100 |
| Department of Housing and Urban Development | 40 |
| Department of Justice | 0–80 |
| Department of Transportation | 5 |
| Department of Veterans Affairs | unknown |
| National Aeronautics and Space Administration | 0 |
| National Science Foundation | 80–100 |
| Social Security Administration | 100 |
| Agency for International Development | 50 |
| Consumer Product Safety Commission | 50–100 |
| Environmental Protection Agency | 3–5 |
*Some departments reported data for several units. The range represents the differences in data reported.
Source: NBAC, “Federal Agency Survey of Policies and Procedures for the Protection of Human Subjects in Research.” This staff analysis is available in Volume II of this report.
11
National Bioethics Advisory Commission
protections for vulnerable individuals analogous to the DHHS subparts, a law passed in 2000 requires DHHS to apply Subpart D of 45 CFR 46 to all research conducted, supported, or regulated by the department, including research regulated by FDA.29 FDA subsequently issued an interim rule incorporating Subpart D into its regulations.30 It is noteworthy, however, that so few Common Rule signatories have adopted additional protections for individuals who are considered vulnerable, in effect providing incomplete protection for human research participants. (The protection of individuals and groups that are categorized as vulnerable is discussed in detail in Chapter 4.)
Lack of Adaptability
Some of the regulatory challenges are exacerbated by the fact that the Common Rule is difficult to amend. Amending the Common Rule requires that each signatory agency first agree to a revision before the 15 agencies with regulations go through the rulemaking process to revise the regulations.31 This weakness results in a set of regulations for which no system-wide change is possible. Obtaining concurrence of the departments and agencies on any regulatory change so far has proven impossible, although not because of lack of need or effort.32 The addition of regulations to the Common Rule specific to classified research, for example, has not been achieved despite three years of effort, a Presidential Memorandum directing the change, and a challenge in a U.S. District Court.33 Unable to change the regulations, some departments have attempted to make modifications by issuing regulatory guidance, a strategy used by the VA in issuing regulations for providing treatment for injuries resulting from participation in research (38 CFR 17.85). However, the power of such changes is limited, and because guidance is usually department specific, it promotes inconsistency and undermines the very unification the Common Rule is
Chapter 1: Introduction—The Need for Change
supposed to establish. (Further discussion of this issue and recommendations appear in Chapter 2.)
Limited Scope
The Common Rule was intended to both provide uniformity across federal departments and expand the scope of regulations to federal departments that previously had none. However, although it marked a significant expansion in scope, the Common Rule still does not apply to all federally sponsored research.
Existing regulations also do not apply to many areas of research funded and conducted by businesses, private nonprofit organizations, and state or local agencies, although such research may be subject to federal regulation if it involves the development of medical devices or drugs requiring approval by the FDA or if it is conducted at an institution that has voluntarily agreed to apply Common Rule requirements to all research it conducts. An unknown amount of nonfederally funded research is completely unregulated under the federal system. This research may include experimental surgical techniques, research on reproductive technologies, some uses of approved drugs and medical devices, and research use of private, identifiable data.34 In some cases, nonfederally funded research may be subject to state regulations, or investigators may voluntarily meet federal requirements to reduce research-related liability. (Recommendations regarding expanding the scope of the system to include such research appear in Chapter 2.)
Enforcement Weaknesses
Even for research that is subject to federal regulations, the mechanisms for enforcing them suffer from three potential weaknesses; the lines of enforcement authority are awkward and sometimes isolated; there is a limited repertoire of sanctions to match the range of possible violations; and the oversight and monitoring process is uneven.
First, there is no clear line of authority or system for the federal government as a whole to sanction serious or repeated noncompliance by investigators or institutions. This results from the dispersion of enforcement functions among various departments and agencies, which weakens the sanctions any one department can impose because
investigators could continue research overseen by a different authority. Each federal department that adheres to the Common Rule has the authority to enforce its own codification of the Common Rule for research it conducts or sponsors. However, federal agencies and institutions with assurances of compliance (formal commitments by institutions to the government stating that they comply with federal regulations) from OHRP are subject to enforcement from that office as well. In the case of DHHS grantees and contractors, the enforcement authority is clear because OHRP is part of DHHS. But, when the assurance holder is the grantee of another department, OHRP decisions come from outside the regular reporting line of authority. Additionally, departments that do use the OHRP assurance process may have their own separate systems for enforcement, and there is little coordination among the various offices responsible for ensuring compliance with the Common Rule.
Second, concerns have emerged that enforcement authorities do not have or use an adequate range of sanctions to respond to various forms of noncompliance. Federal regulations give department and agency heads the authority to terminate or suspend funding for research projects that are not in compliance with the regulations (45 CFR 46.123(a)). Common enforcement tools are the requirement of written responses or the enactment of specific changes to address the identified deficiencies; those who grant assurances can also restrict or suspend institutional assurances. Under its regulations, FDA can withhold approval of new studies, prohibit enrollment of new subjects, and terminate studies. FDA can also issue warning letters and can restrict or disqualify investigators, IRBs, or institutions from conducting or reviewing research with investigational products.35 However, a more complete range of sanctions should be considered for enforcement authorities.
Third, any system of sanctions can only be as good as the monitoring and investigating processes that are used to determine their need. The Common Rule does not set out agency responsibilities for monitoring IRBs or investigators. Some agencies, such as DOE, have a program of routine site reviews.36 Other agencies, such as DHHS, conduct only “for cause” investigations, generally because limited budgets do not permit more proactive
12
monitoring. Investigations often take a long time (in some instances over a year)37 and usually do not include on-site visits (OIG 1998a).
This lack of centralized enforcement authority, proportionate sanctions, and active research oversight serves to weaken severely the system for protecting human research participants. Any one of these weaknesses alone or in combination can lead to unnecessary bureaucracy and burden that could be reduced by a unified office for oversight, which could serve to simplify the bureaucratic complexity and lead to improved monitoring and enforcement.
Overemphasis on Procedural Requirements Rather Than Ethical Principles
In addition to the challenges described above, the current regulations suffer from other weaknesses. For example, they do not sufficiently embody and reflect the substantive ethical principles and standards that should govern behavior, but instead focus on the procedural aspects of IRB review. Thus, although IRBs may review research in accordance with an appropriate focus on ethical behavior, they are ultimately held responsible primarily for procedure and documentation. OHRP’s
Compliance Activities: Common Findings and Guidance
(OHRP 2000) reflects this emphasis on the regulations by focusing on the procedures by which protocols are reviewed—for example, inappropriate use of expedited review and exemptions, lack of a quorum, less than annual continuing review, and failure to document required findings or votes (OHRP 2000). The emphasis by regulators on procedure is frustrating to IRBs and investigators38 and also contributes to an atmosphere in which review of research becomes an exercise in avoiding sanctions and liability rather than in maintaining appropriate ethical standards and protecting human participants. (Chapters 4 and 5 offer recommendations regarding IRB review and the emphasis on procedural requirements.)
Regulations Not Adequately Addressing All Types of Research
Another weakness in the current regulations is that they fail to adequately address unique ethical issues that arise in different types of research. Although federal
National Bioethics Advisory Commission
regulations for human research have long applied to the social sciences and humanities as well as biomedical research, their articulation reflects a persistent emphasis on clinical or biomedical research. Indeed, with regard to nonclinical activities, it is sometimes difficult to determine which activities constitute human research and are therefore subject to the regulatory requirements. In addition, quality improvement studies in health care organizations, public health studies, program evaluation, and humanities research may require review by an IRB in some institutions, but not in others.
Applied to nonclinical research and particularly to the humanities and social science research, the regulatory requirements seem to be either irrelevant or insufficient to provide protection, depending on the type of research. For example, requirements for written documentation of consent may be inappropriate for some survey and anthropological research. Recently, the Association of American University Professors issued a report stating that IRBs often “mistakenly apply standards of clinical and biomedical research” to social science and historical research, which adversely affects not only the quality of the research but inadequately protects human research participants (AAUP 2000). In other areas, the regulations are insufficient—for example, with regard to protecting privacy and confidentiality. Although the regulations require “adequate provisions” to protect privacy and confidentiality, nonphysical harms, such as those resulting from breaches of confidentiality, are often difficult for IRBs to assess without more specific regulatory guidance (45 CFR 46.111(a)(7); 21 CFR 56.111(a)(7)). Much of the difficulty in applying the federal regulations is due to differences in the nature of the risks associated with non-clinical research. For example, physical harms are rarely a concern in nonclinical research, while psychological, social, economic, and legal harms are more likely to occur and should be the primary concern of IRB review.
IRB Burdens
The quality of IRB review is often compromised by the burden of excessive paperwork, because although IRBs are broadly charged with ethical review, in practice they also must fulfill many procedural requirements. While some of these requirements are designed to ensure compliance with ethical standards (e.g., documentation
13
Chapter 1: Introduction—The Need for Change
of waiver of informed consent), others appear to have little relevance to ethical standards or the protection of participants (e.g., requirements for documentation in meeting minutes). In all of their deliberations, IRBs must keep track of a range of detailed regulations and document the grounds on which they make decisions in accordance with them. In addition, IRBs must comply with numerous regulations regarding their operations. However, some of the regulatory and paperwork requirements governing IRBs are difficult to interpret (NBAC 1999b), unnecessarily burdensome, and often not commensurate with their contribution to protecting research participants.
One particularly time-consuming task for investigators, IRBs, and institutional officials is the preparation of assurances. Although many domestic research institutions have an MPA, which covers all research and generally needs to be renewed every five years, other institutions must obtain a separate assurance for each funded project (i.e., a Single Project Assurance, or SPA). For multi-site and international research, this process can be particularly time consuming. Even institutions with an MPA must often revise or amend that document to include changing institutional affiliates as well as affiliation agreements between specific investigators and individual physicians, practice plans, and health care institutions.39 In addition, many IRBs lack the basic resources of staff, space, and technology (Sugarman 2000). Without strong professional and clerical support, busy IRBs remain mired in paperwork and are often unable to focus on ethical considerations. One can get a sense of the unmet resource needs of IRBs from the institutional responses to OPRR shutdowns; large institutions are routinely creating several new IRBs to share the review workload and adding several additional full-time IRB staff (Desruisseaux 2000; Phillips 2000).40 Because of the large workload, serving as an IRB member or chair requires a significant time commitment, with many hours spent in and out of meetings reviewing protocols and writing reports. Few IRB members receive compensation or recognition for their efforts. Thus, with little financial or academic support for IRB membership, IRBs must rely heavily on the goodwill of individual members, which can make it difficult to attract and retain members. (Recommendations related to reducing burdens on IRBs are discussed in Chapters 4, 5, and 6.)
Multi-Site Research Studies
Multi-site research, discussed further in Chapter 6, poses its own set of problems for local IRBs. The growing importance of multi-site research has challenged fundamental assumptions about the importance of local review, for the more IRBs duplicate each other’s work by reviewing the same protocols, the more pressure there is to show why multiple local reviews of identical research protocols are needed. Although local review can provide insight about the social and cultural context of a study, the facilities in which it will be carried out, and any local laws or policies that might affect the study, IRBs may be squandering precious resources when dozens or hundreds of them must review all aspects of a single, multi-site protocol when the design and methods are unlikely to be changed.
IRBs are not the only groups frustrated by multi-site research. Investigators and sponsors are discouraged by having to submit protocols to multiple boards, particularly because changes requested by one board usually have to be approved by the others, a repetitive process that is labor intensive and that can significantly delay research, with little resulting benefit.
Another difficulty is that local IRBs are sometimes poorly situated to review multi-site research. Although IRBs and institutions have the authority to require changes for their site or to refuse to approve a multi-site study about which they hold serious reservations, in practice they are hesitant to use that authority. Thus, although local IRBs may modify recruitment procedures and consent forms, it may be that no single IRB has the power to require substantive changes to a study design, which must remain standardized across sites.
Multi-site research also poses problems with regard to continuing review (mandated, periodic review of research in progress) and review of adverse events. Many IRBs find themselves reviewing a staggering number of reports of adverse events that have occurred at other sites, often without any context, such as the total number of participants in a protocol or whether an adverse event occurred with the experimental or control intervention. Even when they have this information, IRBs sometimes lack the expertise to assess its significance in terms of the risks and potential benefits to trial participants.
14
Educational Needs
Some of the weaknesses in the implementation of the federal regulations are being overcome by knowledgeable and creative IRBs, investigators, and institutional officials. But more can be done (see Chapter 3). Knowledgeable IRBs can find in the regulations extensive discretion in the types of protocols that may be approved. Investigators attentive to regulatory requirements can design research with protections that will easily satisfy ethical and regulatory requirements. Institutions can prepare policies and procedures that clarify, extend, and apply regulations to fit the local and institutional research context. Unfortunately, this kind of expertise is not widespread, and at all levels, from investigators to IRBs to institutions, there is frequently a lack of understanding of research ethics. In some cases, for example, investigators and IRB members might assume that research ethics means their own personal ethics rather than a common set of established ethical principles, standards, and procedures. The current system simply fails to ensure adequate education or preparation of individuals and institutions that wish to conduct and review research.
Two key tasks of institutions are supporting and educating IRB members. Currently, however, many IRB members receive little or no formal training in the relevant ethical analysis or federal regulations. A 1995 survey of 186 IRBs at major universities found that almost half provided no training or less than an hour of training to board members (Hayes et al. 1995). Without trained members, IRBs may act with little knowledge of or attention to the regulations or ethical principles they are supposed to implement. On-the-job-training of new IRB members reinforces IRBs’ isolation from each other and encourages inconsistency between IRBs. Most important, lack of training for IRB members leads even the best intentioned IRBs to consistently miss or ignore important ethical issues. IRB review can only be as good as the IRB members’ judgment. Without standards for IRBs and IRB members, IRB review is likely to be of uneven quality.
Education is essential not only for IRB members, but also for investigators and research staff. If investigators do not know that a specific project is subject to regulations, the entire system of protections is undermined. Even when investigators know that a research study must be
National Bioethics Advisory Commission
reviewed by an IRB, they may not understand their continuing responsibilities as well as the IRB’s responsibility for the continuing oversight of the research. Investigators may be unfamiliar with their obligations to report certain adverse events and have protocol amendments approved by the IRB and must be able to recognize their responsibilities beyond securing and maintaining IRB approval, including explaining protocols to prospective participants and answering their questions.
Responses to Concerns
Public and private sector groups have taken steps to improve protections for those who participate in research. A number of professional organizations, such as Public Responsibility in Medicine and Research, the Applied Research Ethics National Association, the Association of American Medical Colleges, and the Association of American Universities, have contributed to this area by issuing policy statements, instituting workshops and training, or encouraging their member organizations to strengthen their protections procedures. In addition, industry,41 advocacy groups (Shamoo and Irving 1993; Sharav 1994),42 and members of the media (Sloat and Epstein 1996; Whitaker and Kong 1998) have been vocal in calling for stronger protections.
To date, two federal departments also have moved to strengthen and streamline the oversight system. DHHS elevated its oversight office from NIH to the Office of the Secretary, reorganizing OPRR into OHRP.43 (Chapter 2 discusses this transition in more detail.) OHRP now has more visible authority over the 11 agencies within DHHS. However, OHRP’s authority does not extend to other departments and their research programs, although it often attempts to perform a government-wide role without specific authority. FDA has centralized and elevated its coordination of participant protection activities into a new Office of Clinical Science. DHHS instituted, through NIH or FDA, several initiatives to require education and training of investigators, improve monitoring of safety for those participating in clinical trials, address financial conflicts of interest, and seek civil, monetary penalties for noncompliance (DHHS 2000). In December 2000, OHRP revised its assurance process and is planning to work with other departments to create a unified
15
Chapter 1: Introduction—The Need for Change
system of registering IRBs. VA has strengthened oversight of research conducted at its facilities by developing a system of independent accreditation for all of its IRBs (VA 2000).
Notwithstanding these new initiatives, some of the basic problems with the current system have not been addressed and continue to burden sponsors, institutions, IRBs, and investigators with unnecessary delays and costs. While overall improvement of the system is needed, reform is particularly needed to direct attention to those research studies that pose the greatest risk to participants.
Consequences of a Flawed System
Faced with all of these challenges, the oversight system for protecting research participants is losing credibility among some investigators, IRBs, institutions, and, perhaps most important, the public, causing more frustration and less willingness to commit time and resources to the system. This could result in IRBs providing inadequate reviews; investigators not following the IRB-approved protocol or even submitting the protocol for IRB review; institutions not supporting their IRBs; and assurances being restricted or suspended. These possibilities are real and serious and are made more pressing by the continuing and rapid growth of the research enterprise.
The challenges facing the current system for research participant protection are significant and call for major change. Although there will always be ambiguity and difficult ethical decisions to make in reviewing the risks and potential benefits of research, and competing principles might apply in challenging new situations, the need for the protection of human participants requires a unified and consistent commitment on the part of the federal government, research organizations, sponsors, the research community, and the public.
Unfortunately, the history of human research protection demonstrates that knowing how to design research procedures generally has not always been translated into developing practices that are sound and ethical. Although there have been important improvements in research design over the past 50 years that enhance the protection of research participants, it is worth noting that many of these advances were motivated by reactions to
various problematic situations. However, given the great progress in all areas of research and the rapid increase in the number of research protocols that involve human participants, the time is right to create a system of oversight that provides appropriate participant protection and encourages ethically sound research.
With this objective in mind, this report offers a number of recommendations aimed at modifying the current oversight system, although this may involve certain trade-offs. For example, enhancing consistency across federal departments raises concerns that oversight mechanisms will be tailored primarily to the clinical or biomedical model and ignore the ethical and research issues in other disciplines,44,45,46,47 and increased oversight intended to provide more complete protections could lead to more unnecessary bureaucratic requirements and delays. It is not NBAC’s intention to recommend changes that will add burdens without demonstrable increases in protections for human research participants. It is important to understand that because comprehensive oversight is necessarily complex, with interconnected components, changes in any one part of the structure will affect the entire system. Chapter 7 discusses some of these interconnections and how the proposed system would function as a whole.
Notes
1 Traditionally, and in current regulations, those studied in experimental research are referred to as research subjects. This term accurately captures an important aspect of the relationship between an investigator and the individual being studied. However, the term subject also connotes society’s worst fears about research participation, rather than its aspirations. There are methods, such as obtaining voluntary informed consent, by which investigators can show respect for the human dignity of those involved in research. By securing consent, individuals become volunteers, not mere subjects, who are active participants in the research process. Therefore, this report uses the term human participants to refer to those who are studied in research protocols. See Chapter 2 for a more extensive discussion of this topic.
2 To date, NBAC has issued five reports: Cloning Human Beings
(NBAC 1997), Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity (NBAC 1998), Ethical Issues in Human Stem Cell Research (NBAC 1999a), Research Involving Human Biological Materials: Ethical Issues and Policy Guidance (NBAC 1999b), and Ethical and Policy Issues in International Research: Clinical Trials in Developing Countries (NBAC 2001).
16
3 NBAC Meeting, 17 May, 1997. Arlington, Virginia.
4 Those who conduct research are usually referred to as either researchers or investigators. NBAC uses the terms interchangeably.
5 FDA “Warning Letter” from S.A. Masiello, FDA, to James M. Wilson, University of Pennsylvania. March 3, 2000. Available at http://www.fda.gov/foi/warning.htm. Last accessed November 17, 2000.
6 Ibid. 7 Ibid. 8 Ibid.
9 OPRR letter from S.L. Crandall to G. Sarkar, Manhattan Eye, Ear, and Throat Hospital. May 8, 1998.
10 OHRP letter from M.A. Carome to G.R. Newkome, University of South Florida. September 28, 2000. Available at http://ohrp.osophs.dhhs.gov/detrm_letrs/sep00f.pdf. Last accessed May 21, 2001.
11 Ibid.
12 NIH Extramural Data and Trends. Available at http://grants. nih.gov/grants/award/trends95/annotate.htm. Last accessed November 13, 2000.
13 Estimates of National Support for Health R&D by Source or Performer, 1986–1995. Available at http://grants.nih.gov/ grants/award/trends95/pdfdocs/fedtabl1.pdf. Last accessed November 13, 2000.
14 Pub. Law 33, 105th Congress.
15 See NBAC, “Federal Agency Survey on Policies and Procedures for the Protection of Human Subjects in Research.” This staff analysis is available in Volume II of this report.
16 See Heath, E., “The History, Function, and Future of
Independent Institutional Review Boards.” This background paper was prepared for NBAC and is available in Volume II of this report.
17 Ibid.
18 FDA, 1993, “Guideline for the Study and Evaluation of Gender Differences in the Clinical Evaluation.” 22 July. 58 Fed. Reg. 39406; NIH, 1994, Guidelines on the Inclusion of Women and Minorities as Subjects in Clinical Research. 28 March. 59 Fed. Reg. 14508; CDC, 1995, Policy on the Inclusion of Women and Racial and Ethnic Minorities in Externally Awarded Research. 15 September.
60 Fed. Reg. 47947.
19 Pub. Law 115, 105th Congress.
20 White House, 2000, “Taking New Action to Encourage Participation in Clinical Trials.” [Press Release.] 7 June. Available at http://www.whitehouse.gov/WH/html/briefroom.html. Last accessed 12/10/00. See also Medicare Coverage Policy. Available at http://www.hcfa.gov/coverage/default.htm. Last accessed December 10, 2000.
National Bioethics Advisory Commission
21 A Multiple Project Assurance is an agreement between an institution and the federal government in which the institution pledges to comply with regulations.
22 Subpart B of 45 CFR 46 pertains to research involving fetuses, pregnant women, and human in vitro fertilization. Subpart C pertains to research involving prisoners. Subpart D pertains to research involving children.
23 48 Fed. Reg. 9266.
24 Ibid.
25 Ibid.
26 Ibid.
27 HRSA Circular 96.05.
28 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
29 Pub. Law 310, 106th Congress.
30 66 Fed. Reg. 20589.
31 Porter, J., Testimony before NBAC. November 23, 1997. Bethesda, Maryland. See McCarthy, C.R., “Reflections on the Organizational Locus of the Office for Protection from Research Risks.” This background paper was prepared for NBAC and is available in Volume II of this report.
32 There has been a change in the categories of research that IRBs may review through an expedited procedure (63 Fed. Reg. 60364). However, this change did not require amendment of the Common Rule. The Common Rule does not contain the list of categories, but rather refers to such a list, to be periodically revised and published by the Secretary, DHHS. There are no similar provisions for modification elsewhere in the Common Rule.
33 Russell-Einhorn, M., Testimony before NBAC. March 1, 2000. Herndon, Virginia.
34 See Gunsalus, C.K., “An Examination of Issues Presented by Proposals to Unify and Expand Federal Oversight of Human Subject Research.” This background paper was prepared for NBAC and is available in Volume II of this report.
35 21 CFR 56.120–121; 21 CFR 312.70; 21 CFR 812.119; Lepay, D., Testimony before NBAC, September 13, 2000. Washington, D.C.
36 See NBAC, “Federal Agency Survey on Policies and Procedures for the Protection of Human Subjects in Research.” This staff analysis is available in Volume II of this report.
37 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
38 Ibid.
39 Stinson, R., Testimony before NBAC. July 11, 2000. Bethesda, Maryland.
17
Chapter 1: Introduction—The Need for Change
40 Letter from E. Gislason, University of Illinois at Chicago to M. Carome, OPRR, and C. Weil, OPRR. September 30, 1999. Available at http://www.uic.edu/depts/ovcr/oprr/nih09-30-99.html. Last accessed December 5, 2000.
41 Spilker, B., Testimony before NBAC. April 7, 2000. Washington, D.C.
42 For example, National Organization for Rare Disorders, Inc.,
Policy Position: Patient Protection in Clinical Trials. Available at http://www.rarediseases.org/new/patpro.htm. Last accessed August 22, 2000.
43 65 Fed. Reg. 37136–37137.
44 Consortium of Social Science Associations, Public comment submitted to NBAC. Received February 16, 2001.
45 American Sociological Association, Public comment submitted to NBAC. Received February 20, 2001.
46 Shopes, L., Public comment submitted to NBAC. Received February 17, 2001.
47 American Psychological Association, Public comment submitted to NBAC. Received February 16, 2001.
References
Amdur, R.J. 2000. “Improving the Protection of Human Research Subjects.” Academic Medicine 75(7):718–720.
American Association of University Professors (AAUP). 2000.
Protecting Human Beings: Institutional Review Boards and Social Science Research. Available at http://www.aaup.org/irbdoc.htm. Last accessed December 15, 2000.
Annas, G.J. 1990. “FDA’s Compassion for Desperate Drug Companies.” Hastings Center Report 20(1):35–37.
Appelbaum, P.S. 1996. “Drug-free Research in Schizophrenia: An Overview of the Controversy.” IRB: A Review of Human Subjects Research 18(1):1–5.
Association of Clinical Research Professionals. 1997. White Paper on Future Trends: Faster Time to Market. Washington, D.C.: Association of Clinical Research Professionals. Available at http://www.acrpnet.org/publications/index.html#1998wp. Last accessed November 20, 2000.
Baxter, E., and K. Hopper. 1981. Private Lives/Public Spaces. Homeless Adults on the Streets of New York City. New York: Community Service Society.
Bell, J., J. Whiton, and S. Connelly. 1998. Final Report: Evaluation of NIH Implementation of Section 491 of the Public Health Service Act, Mandating a Program of Protection for Research Subjects.
Contract N01-OD-2-2109. Arlington, VA: James Bell Associates.
Bodenheimer, T. 2000. “Uneasy Alliance—Clinical Investigators and the Pharmaceutical Industry.” New England Journal of Medicine 342(20):1539–1544.
Bracht, N. 1991. Citizen Participation in Community Health: Principles for Effective Partnerships. In Health Promotion Research: Towards a New Social Epidemiology, eds. B. Badura and I. Kickbusch, 477–496. Copenhagen: World Health Organization.
Brody, B.A. 1998. Research on the Vulnerable Sick. In Beyond Consent: Seeking Justice in Research, eds. J.P. Kahn, A.C. Mastroinni, and J. Sugarman, 32–46. New York: Oxford University Press.
Carleton, R.A., T.M. Lasater, A.R. Assaf, H.A. Feldman, and S. McKinlay. 1995. “The Pawtucket Heart Health Program: Community Changes in Cardiovascular Risk Factors and Projected Disease Risk.” American Journal of Public Health 85(6):777–785.
Centers for Disease Control and Prevention (CDC). 1999a. An Ounce of Prevention…What Are the Returns? 2d ed. rev. Atlanta, GA: Department of Health and Human Services.
———. 1999b. Sexually Transmitted Disease Surveillance, 1998.
Atlanta, GA: Department of Health and Human Services.
CenterWatch. 1998. “Practice-Based Sites Prosper.” [Editorial.]
CenterWatch 5(3):1, 4–9.
———. 2000. “Turbulence and Transition: The Top Stories of 1999.” [Editorial.] CenterWatch 7(1):1, 5–10.
Crowley, Jr. W.F., and S.O. Thier. 1996. “The Continuing Dilemma in Clinical Investigation and the Future of American Health Care: A System-Wide Problem Requiring Collaborative Solutions.”
Academic Medicine 71(11):1154–1163.
Cummings, K.M. 1999. “Community-Wide Interventions for Tobacco Control.” Nicotine and Tobacco Research 1(Sup 1):S113–S116.
Department of Health and Human Services (DHHS). 2000. “Secretary Shalala Bolsters Protections for Human Research Subjects.” [Press Release.] 23 May. Available at http://www.hhs.gov/news/. Last accessed November 6, 2000.
Department of Veterans Affairs (VA). 2000. “VA Raises Standard for Protecting Human Research Participants.” [Press Release.] 2 May. Available at http://www.va.gov/opa. Last accessed December 8, 2000.
Desruisseaux, P. 2000. “U.S. Lifts Suspension of Research on Human Subjects at U. of Alabama at Birmingham.” Chronicle of Higher Education, 27 March, A-38.
DiClemente, C.C., J.O. Prochaska, S.K. Fairhurst, W.F. Velicer, M.M. Velasquez, and J.S. Rossi. 1991. “The Process of Smoking Cessation: An Analysis of Precontemplation, Contemplation, and Preparation Stages of Change.” Journal of Consulting and Clinical Psychology 59(2):295–304.
Donahue, D.C., B.E. Lewis, I.S. Ockene, and G. Saperia. 1996. “Research Collaboration Between an HMO and an Academic Medical Center: Lessons Learned.” Academic Medicine 71(2):126–132.
Edgar, H., and D.J. Rothman. 1995. “The Institutional Review Board and Beyond: Future Challenges to the Ethics of Human Experimentation.” Milbank Quarterly 73(4):489–506.
18
Etzioni, A. 1999. “Medical Records: Enhancing Privacy, Preserving the Common Good.” Hastings Center Report 29(2):14–23.
Farquhar, J.W., S.P. Fortman, N. Maccoby, W.L. Haskell, P.T. Williams, J.A. Flora, C.B. Taylor, B.W. Brown, D.S. Solomon, and SB Hulley. 1985. “The Stanford Five City Project: Design and Methods.” American Journal of Epidemiology 122(2):323–334.
Frank, E., J.F. Karp, and A.J. Rush. 1993. “Efficacy of Treatments for Major Depression.” Psychopharmacology Bulletin 29(4):457–475.
Frankel, M.S., and S. Siang. 1999. Ethical and Legal Aspects of Human Subjects Research on the Internet. Report of a Workshop, June 10–11, 1999. Washington, D.C.: American Association for the Advancement of Science.
Gallin, J.I., and H.L. Smits. 1997. “Managing the Interface Between Medical Schools, Hospitals, and Clinical Research.” Journal of the American Medical Association 277(8):651–654.
Garfinkel, S. 2000. Database Nation, ed. D. Russell. Sebastopol, CA: O’Reilly & Associates.
General Accounting Office (GAO). 1996. Scientific Research: Continued Vigilance Critical to Protecting Human Subjects.
GAO/HEHS-96-72. Washington, D.C.: GAO.
George, M.A., L.W. Green, and M. Daniel. 1996. “Evolution and Implications of P.A.R. for Public Health.” Promotion and Education 3(4):6–10.
Hatch J., N. Moss, A. Saran, L. Presley-Cantrell, and C. Mallory. 1993. “Community Research: Partnership in Black Communities.”
American Journal of Preventive Medicine 9(6 Sup.):27–31.
Hayes, G.J., S.C. Hayes, and T. Dykstra. 1995. “A Survey of University Institutional Review Boards: Characteristics, Policies, Procedures.” IRB: A Review of Human Subjects Research 17(3):1–6.
Henderson, L. 1999. “Centralized AMCs Growing Steadily.”
CenterWatch 6(2):4–5.
Hilts, P.J. 1998. “Study or Human Experimentation? Face-Lift Project Stirs Ethical Concerns.” New York Times, 21 June, A-25.
Hurst, E., M.C. Luoh, and F. Stafford. 1998. “The Wealth Dynamics of American Families, 1984-94.” Brookings Papers on Economic Activity 1:267–337.
Jacobs, D., R. Luepker, M. Mittelmark, A. Folsom, P. Pirie, S. Mascioli, P. Hanan, T. Pechacek, N. Bracht, R. Carlaw, F. Kline, and H. Blackburn. 1986. “Community-Wide Prevention Strategies: Evaluation Design of the Minnesota Heart Health Program.”
Journal of Chronic Disease 39:775–788.
Kahn, J.P., A.C. Mastroianni, and J. Sugarman, eds. 1998. Beyond Consent: Seeking Justice in Research. New York: Oxford University Press.
Katz, J. 1993. “Human Experimentation and Human Rights.”
Saint Louis University Law Journal 38(1):7–54.
Klein, B. 2000. “USF Must Inform 60 Research Subjects.”
St. Petersburg Times, 17 October, 1-B.
National Bioethics Advisory Commission
Marshall, E. 2000. “Gene Therapy on Trial.” Science 288(5468):951–957.
Mechanic, R.E., and A. Dobson. 1996. “The Impact of Managed Care on Clinical Research: A Preliminary Investigation.” Health Affairs 15(3):72–89.
Mittelmark, M.B., M.K. Hunt, G.W. Heath, and T.L. Schmid. 1993. “Realistic Outcomes: Lessons from Community-Based Research and Demonstration Programs for the Prevention of Cardiovascular Diseases.” Journal of Public Health Policy 14(4):437–462.
Moreno, J.D., A.L. Caplan, P.R. Wolpe, and the Members of the Project on Informed Consent, Human Research Ethics Group. 1998. “Updating Protections for Human Subjects Involved in Research.”
Journal of the American Medical Association 280(22):1951–1958.
National Bioethics Advisory Commission (NBAC). 1997. Cloning Human Beings. 2 vols. Rockville, MD: U.S. Government Printing Office.
———. 1998. Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity. 2 vols. Rockville, MD: U.S. Government Printing Office.
———. 1999a. Ethical Issues in Human Stem Cell Research. 3 vols. Rockville, MD: U.S. Government Printing Office.
———. 1999b. Research Involving Human Biological Materials: Ethical Issues and Policy Guidance. 2 vols. Rockville, MD: U.S. Government Printing Office.
———. 2001. Ethical and Policy Issues in International Research: Clinical Trials in Developing Countries. 2 vols. Bethesda, MD: U.S. Government Printing Office.
National Center for Health Statistics. 2000. Health, United States, 2000 with Adolescent Health Chartbook. Hyattsville, MD: DHHS.
National Research Council. 1997. For the Record: Protecting
Electronic Health Information. Washington, D.C.: National Academy Press.
———. 2000. How People Learn: Brain, Mind, Experience, and School. Washington, D.C.: National Academy Press.
New York State Department of Health. 1996. “Case Report on Death of University of Rochester Student Issued.” [Press Release.] 26 September. Available at http://www.health.state.ny.us/nysdoh/ consumer/pressrell/96/wan.htm. Last accessed May 22, 2001.
Office for Human Research Protections (OHRP). 2000. OHRP Compliance Activities: Common Findings and Guidance–9/21/2000.
Washington, D.C.: DHHS. Available at http://ohrp.osophs.dhhs. gov./references/findings.pdf. Last accessed November 7, 2000.
Office of Inspector General (OIG), DHHS. 1998a. Institutional Review Boards: A Time for Reform. OEI-01-97-00193. Washington, D.C.: DHHS.
———. 1998b. Institutional Review Boards: The Emergence of Independent Boards. OEI-01-97-00192. Washington, D.C.: DHHS.
19
Chapter 1: Introduction—The Need for Change
———. 1998c. Institutional Review Boards: Promising Approaches.
OEI-01-97-00191. Washington, D.C.: DHHS.
———. 1998d. Institutional Review Boards: Their Role in Reviewing Approved Research. OEI-01-97-00190. Washington, D.C.: DHHS.
———. 1998e. Low-Volume Institutional Review Boards.
OEI-01-97-00194. Washington, D.C.: DHHS.
———. 2000a. FDA Oversight of Clinical Investigators.
OEI-05-99-00350. Washington, D.C.: DHHS.
———. 2000b. Protecting Human Research Subjects: Status of Recommendations. OEI-01-97-00197. Washington, D.C.: DHHS.
———. 2000c. Recruiting Human Subjects: Pressures in Industry-Sponsored Clinical Research. OEI-01-97-00195. Washington, D.C.: DHHS.
———. 2000d. Recruiting Human Subjects: Sample Guidelines for Practice. OEI-01-97-00196. Washington, D.C.: DHHS.
Office for Protection from Research Risks (OPRR). 1994. Evaluation of Human Subject Protections in Schizophrenia Research Conducted by the University of California Los Angeles. Rockville, MD: OPRR.
Pharmaceutical Research and Manufacturers of America (PhRMA). 2000. Pharmaceutical Industry Profile 2000. Washington, D.C.: PhRMA.
Phillips, D.F. 1996. “Institutional Review Boards Under Stress: Will They Explode or Change?” Journal of the American Medical Association 276(20):1623–1626.
———. 2000. “IRBs Search for Answers and Support During a Time of Institutional Change.” Journal of the American Medical Association 283(6):729–730.
Ries, L.A.G., P.A. Wingo, D.S. Miller, H.L. Howe, H.K. Weir, H.M. Rosenberg, S.W. Vernon, K. Cronin, and B.K. Edwards. 2000. “The Annual Report to the Nation on the Status of Cancer, 1973–1997, with a Special Section on Colorectal Cancer.” Cancer 88(10):2398–2424.
Rosenthal, E. 1996. “New York Seeks to Tighten Rules on Medical Research.” New York Times, 27 September, B-4.
Rothman, D.J., and H. Edgar. 1991. “AIDS, Activism, and Ethics.”
Hospital Practice 26(7):135–142.
Schneiderman, N., and M.A. Speers. 2000. Behavioral Science, Social Science, and Public Health in the 21st Century. In Integrating Behavioral and Social Sciences with Public Health. Washington, D.C.: American Psychological Association.
Shamoo, A.E. 1994. “Our Responsibilities Toward Persons with Mental Illness as Human Subjects in Research.” Journal of the California Alliance for the Mentally Ill 5(1):14–16.
Shamoo, A.E., and D.N. Irving. 1993. “Accountability in Research: Using Persons with Mental Illness.” Accountability in Research 3:1–17.
Sharav, V.H. 1994. “What Is Ethical? What Is Not? Where Do You Draw the Line?” Journal of the California Alliance for the Mentally Ill
5(1):4–5.
Sloat, B., and K.C. Epstein. 1996. “Using Our Kids as Guinea Pigs, An Investigation of Medical Research Records Shows the U.S. Government Is Still in the Business of Conducting and Paying for Tests on Unsuspecting Americans.” Plain Dealer, 16 December, 1-A.
Snyderman, R., and E.W. Holmes. 2000. “Oversight Mechanisms for Clinical Research.” Science 287(5453):595–597.
Sugarman, J. 2000. “The Role of Institutional Support in Protecting Human Research Subjects.” Academic Medicine 75(7):687–692.
Sweeney, L. 1997. “Weaving Technology and Policy Together to Maintain Confidentiality.” Journal of Law Medicine and Ethics
25:98–110.
Valigra, L. 1997. “Rooting Out Rogue Investigators.”
CenterWatch 4(7):1, 5.
Whitaker, R., and D. Kong. 1998. “Doing Harm: Research on the Mentally Ill.” 4 parts. Boston Globe. 15–18 November, A-1.
White, L. 2000. “Colleges Must Protect Privacy in the Digital Age.”
Chronicle of Higher Education. 30 June, B-4.
Wolf, L.E., and B. Lo. 2000. “Ethical Issues in Research: An Issue for All Internists.” American Journal of Medicine 109(1):82–85.
Woodward, B. 1999. “Challenges to Human Subject Protections in U.S. Medical Research.” Journal of the American Medical Association
282(20):1947–1952.
20
Chapter Two 2
A Proposal for Oversight
Introduction
This chapter proposes an enhanced system of U.S. oversight of research involving human participants, specifically addressing its purpose and functions, the parties involved, and its scope and structure. The chapter also focuses on the definitions of research and human participant, terms that are crucial to delineating the scope of the oversight system. Although some of the components of the proposed system already exist in total or in part within the current system, certain alterations and additions are required. Specifically, what is needed is a more effectively coordinated system:
Discussion and recommendations pertaining to each of these criteria are found in this and subsequent chapters of this report. The most significant change proposed is that of consolidation—that is, where there are now multiple sets of overlapping regulations, the National Bioethics Advisory Commission (NBAC) suggests a unified comprehensive policy in the form of a single set of regulations and a single source of guidance and interpretation. This consolidated structure would allow for much needed regulatory flexibility and, in certain areas, a greater use of guidance with less regulation. Of course, an effective oversight system consists of more than governmental regulation. Indeed, it should be devoid of excessive regulation, relying, when appropriate, on the ethical behavior and commitments of all individuals and entities—federal and nonfederal—engaged in research involving human participants.
Purpose of Oversight
For many involved in the oversight system, the bureaucratic procedural requirements (as opposed to ethical principles) that characterize the current federal regulatory system for human research have become synonymous with the purpose, intent, and implementation of the oversight system itself. As a result, some investigators and Institutional Review Board (IRB) members perceive the regulations as lacking an ethical foundation and view them merely as a set of formal procedural requirements,
21
Chapter 2: A Proposal for Oversight
some of which protect human participants (e.g., informed consent), but others of which have little to do with such protection (e.g., assurances of compliance and record keeping).1,2 Unless the regulations and guidance appear ethically substantive, investigators will view them simply as irritating obstacles to doing important, socially valuable work, while IRB members, who are responsible for applying the rules, will become frustrated with investigators’ resistance. In such cases, when the largely procedural requirements are perceived as ineffective in improving protections, some investigators might avoid submitting research protocols to IRBs or fail to disclose certain aspects of the protocol (Ferraro et al. 1999; Liddle and Brazalton 1996), behavior that could place participants at risk and compromise public trust in the oversight system.
Some of this frustration could be alleviated if all who are involved in the human research enterprise recognize that the overarching purpose of the system should be to ensure the protection of the participants in a manner that encourages and facilitates research that is consistent with accepted ethical principles. There is no necessary conflict between the goal of promoting research and its benefits and that of protecting human research participants. In previous reports, NBAC has reinforced the importance of these dual goals (NBAC 1999a, 1999b). In a paper prepared for NBAC, for example, Harold Vanderpool articulated this perspective:
The purposes of protecting human subjects and enhancing the benefits of biomedical and behavioral research are fully justifiable, although their balance should be critically and periodically reassessed. It is a mistake to assume that the relationship between these purposes has to be a zero sum game, whereby increased protection for subjects subtracts from the conducting of research.3
As discussed in Chapter 1, a credible system for the protection of research participants promotes research by encouraging public support and participation. In addition, by requiring careful consideration of the implications of study design on the ethical acceptability of a protocol and independent review prior to the involvement of human participants, the oversight system often leads to improvements in research design. The system also can
promote research by providing a prospective mechanism for discussion and review of controversial research, moving many innovative research studies forward.
Protection of human participants should never be an afterthought. Rather, it should be a factor in decision-making by everybody involved throughout the conception, design, review, and performance of human research. The oversight system should act to forbid research designs that are clearly unethical. In areas of less ethical certainty, the system should ensure protection of research participants by providing mechanisms for applying accepted ethical principles and seeking independent review and public input to resolve issues of uncertainty. But the system should allow the majority of research, for which there is clear ethical acceptance, to proceed with minimal delay. An oversight system succeeds to the extent that it encourages the protection of participants in all aspects of research, but does not create any unnecessary barriers to the conduct of research.
Although the need to protect human research participants is a well-accepted norm, controversy arises over the amount of protection that should be required. Jonathan Moreno provides a useful framework for considering the levels of protectionism that are needed. Weak protectionism requires that participants be provided at least the protection of informed consent, but it relies on the virtue and commitment of the investigators to provide that protection without any external involvement by an outside entity. In moderate protectionism, the investigators remain responsible for providing protection, including obtaining informed consent from participants, but the research study undergoes an independent external review. A system of strong protectionism would greatly reduce reliance on the investigator for providing substantive protections to participants by, for example, increasing the use of third parties for monitoring the conduct of the research.4 The current system of requiring prior approval of research by IRBs and informed consent by participants is a moderately protectionist approach that is appropriate because it can protect participants while not imposing inappropriate delays in initiating ethically sound research.
22
Functions and Responsibilities of the Oversight System
The oversight system should include several interrelated but distinct functions and responsibilities. It should establish clear and effective policies in the form of regulations or guidance; provide education and assistance to all parties; require review of research to determine whether it is ethically acceptable; monitor ongoing research and the operation and behavior of institutions, IRBs, and investigators; enforce requirements; and maintain public accountability.
Policy: Regulation and Guidance
Establishing policy by providing regulations or guidance provides a necessary framework for the entire oversight system. It also provides leadership by setting the direction, approach, and tone. Policies must be established by the federal government in order to ensure uniform protection of research participants regardless of the source of the research funding. In addition, whether issued as regulations or nonbinding guidance, policies must be presented in an understandable form.
Regulations alone are unlikely to be sufficient to address all types of research (e.g., clinical trials, surveys, records review studies, studies using stored human biological materials, behavioral experiments, or oral history studies). Therefore, guidance, because it establishes standards of acceptable practice while allowing for reasoned deviations, can be used to supplement regulations by indicating how substantive ethical principles can be implemented in particular circumstances.5 Moreover, guidance can be particularly helpful in areas of emerging research where the ethical standards and associated procedures are not entirely clear or well developed. In such situations, thoughtful discussion within the framework of existing guidance may be sufficient to allow innovative research to move forward, and if not, new guidance can be developed promptly.
No matter how well regulations and guidance are written, situations will arise in which it is unclear how ethical principles should be applied or regulations should be interpreted.
Policy interpretation and consultation, provided by a federal oversight office, would be particularly helpful to IRBs struggling with especially complex or difficult research studies.
National Bioethics Advisory Commission
Education
Understanding basic ethical principles and how they apply in various research situations is the best way to avoid inadvertent harm to the rights and welfare of participants. Education can also prepare individuals to understand and resolve ethical issues, especially novel ethical problems, as they arise in specific research studies. (Educational programs are discussed in more detail in Chapter 3.) An effective oversight system helps make education available to all relevant parties. Investigators, IRB staff and members, contract research organizations (CROs), institutional officials, and sponsors (public and private) are the primary audiences for education. Educational activities, however, should be tailored to the particular role of each party in the research enterprise and be relevant to different types of research.
It is also important to educate prospective participants and the general public. Education directed at these audiences should strive to increase the public’s awareness of the system’s functions and operations, which can encourage informed decisionmaking by prospective participants, increase public input regarding policies for the protection of research participants, and increase and sustain the willingness of the public to participate in and support the research enterprise.
Review of Research
In the United States, independent review of proposed research to determine whether it is ethically acceptable is largely performed by local IRBs and is one of the primary means by which the current system provides protection to research participants.6 Such review should be responsive to the nature of risk and commensurate with the level of risk involved. For example, the risks and potential benefits arising in a clinical trial are generally different from those arising in a study using existing data. Likewise, social science and humanities research raises risks and potential benefits that differ from those raised by clinical research. Harms may vary from physical to psychological, social, or legal. Within each of these domains, the risk of harm may range from low to high. Review criteria and mechanisms should be matched to the ethical issues arising from the research. IRBs, as full
23
Chapter 2: A Proposal for Oversight
committees, should review research involving more than minimal risk; however, IRBs should be able to develop procedures other than full board review for research involving no more than minimal risk.
Research involving difficult ethical considerations, such as highly innovative interventions or technologies, should be reviewed by a body with particular expertise and experience in these special issues, in addition to the local IRB, unless the local IRB is especially qualified to review such research. Several options should be available for providing such heightened review, including specially trained and accredited local IRBs or specially created regional or national review bodies. Often, these special bodies would be reviewing classes of research rather than individual research protocols in order to assist in the development of guidance to local IRBs regarding the criteria for approval. For example, NBAC previously recommended the use of a special standing panel to review research studies involving persons with mental disorders that may affect their decisionmaking capacity and a national-level review for certain types of stem cell research (NBAC 1998; NBAC 1999a). However, because national or special review panels could increase delays or even prevent some research from being conducted, steps must be taken to ensure that the system does not impose undue delays on the approval of ethically sound research.
IRB Review of Research Involving No More Than Minimal Risk
IRBs should be allowed to use procedures that are more expeditious than full board review for research that involves no more than minimal risk. Such procedures should also be permitted for handling other research-related matters, such as determining whether an activity is research and subject to oversight, reviewing minor proposed changes to an approved protocol, conducting some types of continuing review, and reviewing some reports of unanticipated problems. Under the current system, the expedited review process is akin to the procedures proposed here, which are intended to replace the expedited review process and to expand the activities that could be handled by the IRB more expeditiously than through a full board meeting. Generally, IRBs should be able to use more efficient procedures to handle most, if not all, research-related issues, with the exception of protocols involving more than minimal risk.
Such procedures would differ from the current expedited review in several ways:
Monitoring
The monitoring of activities is an important component of an effective oversight system. One type of monitoring should occur at the federal level, with the federal oversight office (see discussion later in this chapter) monitoring the implementation of oversight activities by federal agencies. Such monitoring should include, for example,
24
ensuring that all federal agencies sponsoring or conducting research follow appropriate policies, regulations, and guidance and report violations, noncompliance, and other unanticipated problems. The federal oversight office should also ensure that accrediting or certifying bodies apply standards applicable to assessing a core set of competencies that has been approved by the federal oversight office and should also play a role in monitoring sponsors and institutions from the private sector when these entities are not monitored either by another federal agency (for example, the Food and Drug Administration [FDA], which oversees research involving investigational new drugs) or by an accrediting body. The goal is to ensure that all parties involved in the various aspects of research protect research participants, both at the time studies are initially approved and throughout the research process. The efforts of the federal oversight office should complement but not duplicate the activities of the accrediting and certifying bodies.
A second type of monitoring involves establishing mechanisms to ensure that institutions, IRBs, and investigators are following regulations and guidance. This type of monitoring is described further in Chapter 3 as verification of compliance. Responsibility for verifying compliance should be shared by different parties. For example, accrediting bodies should be responsible for monitoring accredited institutions and independent IRBs. However, institutions should also establish procedures to monitor their IRBs. They could, for example, have an internal audit process for reviewing, on a sample basis, whether IRB decisions are consistent with regulatory requirements. Institutions also should implement such procedures to determine whether investigators are complying with regulatory requirements and conducting research according to IRB-approved protocols.
The third type of monitoring assesses the progress of research through conducting continuing review of ongoing research and monitoring the safety of research participants and whether unanticipated problems or events occur. Investigators, IRBs, sponsors, or those acting on behalf of sponsors, such as CROs or Data and Safety Monitoring Boards, generally carry out these activities. Monitoring should provide the information needed to evaluate whether the oversight system is achieving its
National Bioethics Advisory Commission
purpose. If gaps exist between purpose and performance, monitoring should help to identify their nature, magnitude, and location and should ensure the development of measures to close them. The results of monitoring might also show a need for changes in policy, additional or enhanced education, or enforcement measures.
Enforcement
The provision of enforcement measures makes all parties aware that the system must be taken seriously and ensures the public’s continuing trust. Monitoring is used as an additional mechanism to prevent harm to participants, to promote improved practices, and to assess whether any violations are inadvertent or intentional. When investigators are unwilling or unable to provide appropriate protection to participants, enforcement should stop individuals and possibly their institutions from conducting human research. Enforcement should complement policy, education, and monitoring of compliance to ensure that research participants are protected; however, it should not be the primary focus of an oversight system.
Sanctions are necessary in serious cases of noncom-pliance or in the case of repeat offenses. Most sanctions involve administrative action by a government agency against an individual or organization for failure to comply with regulatory requirements. The oversight system should include a range of sanctions, including halting research at an institution; de-accrediting an IRB or institution; requiring special training and probation accompanied by mentoring; disqualifying or debarring an investigator from conducting research involving human participants; removing the investigator from a particular research study; issuing letters of reprimand to investigators, IRBs, or institutions; conducting special monitoring of investigators or IRBs; refusing to publish the research results that do not meet all the ethical obligations to human participants (NBAC 1999b); and levying civil monetary penalties. However, enforcement also might include judicial action. Regulatory agencies have the authority to initiate and conduct such actions, but in a broader sense, enforcement includes all the ways in which the oversight system induces actions that support the purposes of the system. Thus, many parties carry responsibility for
25
Chapter 2: A Proposal for Oversight
enforcement, including regulators, sponsors, institutions, IRBs, professional societies, editors of scientific publications, and accrediting and certifying bodies.
Accountability
The proposed oversight system is strengthened when its policies, interventions, and outcomes are open to public view and input. In addition, dissemination of results of research studies and of research protection creates a public record that can form the basis for trust. Easily accessed systems for registering concerns and complaints help to bring problems to the attention of those who can resolve them and offer necessary safeguards to those who believe they have been harmed in the course of research participation.7 See Exhibit 2.1 for a description of the responsibilities of those involved in the oversight system.
Scope and Structure of a Revised Oversight System
Scope of Oversight
In 1997, NBAC unanimously adopted a resolution that “[n]o person in the United States should be enrolled in research without the twin protections of informed consent by an authorized person and independent review of the risks and benefits of the research.” NBAC adopted this resolution based on the conviction that the entitlement to an independent review of risks and potential benefits and the opportunity to exercise informed consent are grounded in civil and human rights. Whether or not participants in currently unregulated research are harmed in greater numbers or to a greater degree than those in regulated research is not and cannot be known because of a lack of data at the national level. Nonetheless, protecting the rights and welfare of all participants is necessary to maintain public trust in the research community as well as to guard against the actual harm of research participants.
However, not all research participants are protected by the Common Rule (see Figures 2.1 and C.1). Some institutions conduct research that is not covered by the Common Rule or regulated as research by FDA, and unless those institutions voluntarily commit to reviewing
Exhibit 2.1: Responsibilities of Those Involved in the Oversight System
Participants: Even though investigators are obligated to adequately inform participants about research, competent participants have a responsibility to become knowledgeable about a prospective research study and to use the information to make an informed decision about participation. During the research study, participants are responsible for staying informed about the research, following the study procedures, reporting problems encountered in the course of research, and informing investigators when they move in order to facilitate additional contact that may be necessary, as long as they voluntarily remain in the study. It is important to remember that participants also have the right to discontinue their participation at any time during the study, so they only have a limited, or qualified, responsibility to follow the study procedures.
Investigators: Investigators have an obligation to ensure that research studies are scientifically and ethically sound, are reviewed and approved by an IRB prior to conducting the study, are conducted in accordance with an approved protocol, and receive appropriate safety monitoring and continuing review. They are also obligated to acknowledge when errors occur or problems develop and to report them to appropriate parties, such as the IRB. The FDA regulations contain a specific definition of investigator, which “…means an individual who actually conducts or leads a clinical investigation…or, in the event of an investigation conducted by a team of individuals, is the responsible leader of that team” (21 CFR 50.3 (d)).
Institutions: Institutions engaged in research may be public or private entities. They are usually organizations (e.g., universities, medical schools, insurance companies, research firms, pharmaceutical companies, or dedicated research facilities) that employ investigators, but may also be facilities in which research is conducted (e.g., hospitals, prisons, schools, community-based organizations). For some research there may be more than one relevant institution involved—for example, when a university-affiliated investigator conducts research in a prison or in an educational, industrial, or corporate setting, or when a pharmaceutical company conducts a clinical trial at multiple sites. By either employing investigators
26
Exhibit 2.1 continued
or allowing research to be conducted in its facilities, each institution engaged in research must take responsibility for the ethical conduct of that research. Moreover, each institution has an obligation to ensure that the research is conducted in accordance with all applicable federal and state laws, regulations, and procedures. Institutions that employ investigators also have responsibilities for education, monitoring, and enforcement. Moreover, institutions in which research is conducted often have other obligations to participants—for example, as patients or students.
IRBs: IRBs serve as a principle component of that part of the research enterprise charged with protecting research participants. IRBs are responsible for reviewing human participant research to ensure that the proposed study is ethically acceptable prior to initiation of the study. Once research begins, IRBs have a responsibility to monitor the progress of the approved research with the primary goal of protecting participants, but also to ensure that the research is proceeding according to the approved proposal. This is usually accomplished through the process of periodic review.
Sponsors: Sponsors are the governmental and private organizations that fund (through grants and contracts) research. Sponsors have responsibility for the manner in which studies are conducted. When sponsors are federal agencies, they share responsibilities with the federal oversight office for guidance development, education, monitoring, and enforcement. However, all sponsors share responsibilities for educating their employees and the investigators who conduct their research. Sponsors are responsible for ensuring that their research has scientific merit and is ethically justifiable prior to funding. For certain types of research (e.g., high-risk research), sponsors are responsible for ensuring that additional oversight protections are provided that allow expert and public vetting of the proposed research. They are also responsible for ensuring that investigators and relevant institutions monitor ongoing progress, and they may have monitoring responsibilities of their own. In conjunction with the federal oversight office, sponsors also may have some enforcement responsibilities. In contrast to the use of terms in this report, FDA regulations use the term sponsor in a more narrow sense. It “…means a person who initiates a clinical investigation, but who does not actually conduct
National Bioethics Advisory Commission
the investigation…. A person other than an individual (e.g., corporation or agency) that uses one or more of its own employees to conduct a clinical investigation it has initiated is considered to be a sponsor (not a sponsor-investigator), and the employees are considered to be investigators” (21 CFR 50.3(e)). In contrast, the FDA regulations state that the term sponsor-investigator “…means an individual who both initiates and actually conducts, alone or with others, a clinical investigation…. The term does not include any person other than an individual, e.g., corporation or agency” (21 CFR 50.3 (f)).
Professional Societies and Advocacy Organizations:
These organizations are valuable resources in the education process that should encourage their members to seek out educational opportunities, inform them of professional codes relating to the ethical conduct of research, and provide relevant materials to them.
Accrediting and Certifying Organizations: Although there are many accrediting and certifying organizations, programs for accreditation and certification of those involved in human research are new or just developing, although the need for such programs has long been recognized (IOM 2001; President’s Commission 1983, 114–121).
Editors and Publishers: As arbiters of scientific quality, scientific editors and publishers are obligated to ensure that the data they publish were collected and analyzed according to the ethical standards set forth by the oversight system (NBAC 1998, NBAC 1999b).
Congress: Congress is responsible for providing the authorizing language and appropriations for research programs and agencies that support the oversight system. Through its governing authorities, Congress can monitor the performance of federal officials and institutions conducting federally regulated research.
State Regulators: Although some states have separate statutes and regulations governing human research,8 federal and state regulatory systems are not structurally connected. When federal and state regulations are applicable to a research study, the more protective provisions must be met (45 CFR 46.101(f); 21 CFR 56.103(c)). A state may require greater protection, but it cannot allow less protection than is mandated by the federal requirements.
27
Chapter 2: A Proposal for Oversight
such research according to the Common Rule, participants might not be protected. The commitment to protect participants should not be voluntary, nor should requirements be adhered to for only some human research (e.g., private organizations following federal requirements for FDA-regulated research, such as an investigational new drug study, but not following federal requirements when conducting an epidemiological study). All research participants should be protected by the oversight system, and the system should apply to all U.S. investigators and sponsors who conduct research domestically and internationally.
Extending current protections to all privately funded research and making uniform all federal regulations and guidance cannot be accomplished within the current oversight system, which has no authority to act on behalf of the entire federal government and for the benefit of research participants. A unified, comprehensive federal policy promulgated and interpreted by a single office would facilitate the extension of the same protections to all humans participating in research.
Recommendation 2.1: The federal oversight system should protect the rights and welfare of human research participants by requiring 1) independent review of risks and potential benefits and 2) voluntary informed consent. Protection should be available to participants in both publicly and privately sponsored research. Federal legislation should be enacted to provide such protection.
The Need for a Unified, Comprehensive Federal Policy
A credible oversight system requires that all federal agencies and private organizations follow the same federal policy in the forms of regulation and guidance. Institutions should not be forced to comply with inconsistent regulations when research falls into the jurisdiction of multiple agencies, such as when an a National Institutes of Health (NIH)-sponsored clinical trial involves an investigational new drug regulated by the FDA. Currently, under this circumstance, institutions must follow the Department of Health and Human Services (DHHS) regulations
(45 CFR 46) and the FDA regulations (21 CFR 50, 56), which are not entirely consistent with each other.
Because research inevitably raises new issues that require responsive policy development and revision, amending multiple sets of regulations, as would be required in the Common Rule, is not a viable long-term option. To achieve consistency and flexibility, there must be a single set of regulations promulgated and interpreted by a single office.
The legal authority of the current oversight system is not sufficient to expand coverage to all research conducted by the private sector as proposed here or to promulgate a unified policy, specifically as one set of regulations. Recent actions by the Office for Human Research Protections (OHRP) reflect both the initiatives of an office committed to improving human participant protections and its limitations in implementing broad, consistent change across departments and agencies. However, no matter how beneficial or needed the new initiatives may be in order to improve protections, OHRP does not have the authority to function as the lead organization for other departments, and it cannot require other departments to adopt new initiatives or ensure consistency in oversight across departments and agencies.
The creation of such an oversight system would require legislation that expands the authority of the current system to cover the private sector beyond the areas now regulated by the FDA and that creates a single office to oversee the system. Although an executive order could extend protections to all departments and agencies, it could not extend protections beyond federally sponsored or regulated research.
To ensure a uniform, comprehensive system of protection, a single, independent office should be created. Such an office is referred to in this report as the National Office for Human Research Oversight (NOHRO). Once established, NOHRO should be given authority to be the lead office for implementing and enforcing the oversight system for research involving human participants.
Recommendation 2.2: To ensure the protection of the rights and welfare of all research participants, federal legislation should be enacted to create a single, independent federal office, the National Office for Human Research Oversight (NOHRO),
28
National Bioethics Advisory Commission
Figure 2-1. Involvement of Human Subjects in Research
Federal Protection of Human Subjects in Research
FEDERALLY FUNDED
Food, Drug, and Cosmetic Act
Food and Drug Administration
Informed consent
Institutional Review Boards
Public Health Service Act
DHHS
45 CFR Part 46
Subpart A
Institutional Assurances Informed consent Institutional Review Boards
16 Departments and Agencies
1991 Federal Policy for the Protection of Human Subjects
=
“COMMON RULE”
Subpart B Pregnant women; fetuses Subpart C Prisoners Subpart D Children
Site visits
IRB inspections
Not federally funded and not subject to FDA
Not federally funded and not subject to FDA
| • |
Assured institutions that do not voluntarily pledge all activity to 45 CFR Part 46 |
Source: Office for Protection from Research Risks
29
Federally funded and not subject to FDA
Chapter 2: A Proposal for Oversight
to lead and coordinate the oversight system. This office should be responsible for policy development, regulatory reform (see Recommendation 2.3), research review and monitoring, research ethics education, and enforcement.
Recommendation 2.3: A unified, comprehensive federal policy embodied in a single set of regulations and guidance should be created that would apply to all types of research involving human participants (see Recommendation 2.2).
If these particular recommendations are not implemented, the federal government should find alternative approaches for realizing the intention of these and subsequent recommendations in this report.
Advantages and Disadvantages of a Single, Independent Office
Advantages
Elevating the government entity that oversees research involving human participants to agency status with government-wide authority gives the federal commitment to protect research participants high public visibility. Empowered with the authorities mentioned above, the office would be more likely to achieve the desired outcomes. Clear, consistent regulations and guidance tailored to various types of research would be developed and modified when needed. Most important, the office would be capable of prompt, decisive action in response to new research issues. NBAC envisions the office implementing a dynamic, evolving, and flexible system of oversight. Even on its first day of operation, the new office would be an improvement over the current system, because it will provide a focal point for oversight and a source for information that does not now exist. It also could begin promptly to develop policy, both regulation and guidance.
NOHRO would also eliminate the inertia that occurs in the current system because of the lack of a lead office with authority to direct federal agencies to carry out a task. One way in which NOHRO could facilitate both protection of research participants and the promotion of research is by introducing mechanisms for review of
research that involves complex ethical issues. These mechanisms might include requesting a federal agency to set up a special review body or working with local institutions in conjunction with a federal agency that is sponsoring a particular research program.
If the proposed office were located within a department rather than given agency status, special provision would need to be made to force other departments and agencies to follow the policies set by that lead agency. Empowering such a lead agency within one department with authority over operations of other departments at the same administrative level presents a difficult arrangement at best. For example, staff of the other agencies may be subject to competing expectations and demands from their own agencies as well as the proposed office.
Another concern of locating NOHRO within a single department is that it could be perceived both externally and within the department as responsive primarily to that department’s interests, views, and the type of research it sponsors or conducts, to the exclusion of other types of research carried out primarily by other departments. For example, OHRP, located in DHHS, has interpreted regulations and developed policy generally in response to the conduct of biomedical research. Furthermore, housing an oversight office within a department that conducts or supports human research could create a conflict of interest. Such a potential conflict provided at least part of the reason for the recommendation to relocate the Office for Protection from Research Risks from NIH to the DHHS Office of the Secretary (OPRR Review Panel 1999). But this degree of separation may still be insufficient, because NIH is part of DHHS, and the new office is still regulating an organization on which it relies for budgetary and other support. The potential conflict of interest is thus attenuated, but not eliminated.9,10 The ability to act without approval from the agencies it regulates is essential to both the perception and reality of NOHRO’s effectiveness.
Disadvantages
Establishing an independent office might have disadvantages. Most notably, an independent office would necessarily be smaller than the departments and agencies affected by the policies it develops and thus might lack some of the political support it would need to be effective.11
30
Creating an independent office could also lead to increased bureaucracy by adding another layer of oversight. NBAC opposes the creation of another office that merely adds regulations that do not provide additional important protections. However, the new office should reduce unnecessary bureaucratic burden by eliminating some duplicative procedures currently existing under the Common Rule. For example, issuing assurances of compliance and registering IRBs should be centralized in the new office (see Chapter 4). The use of such centralizing rulemaking procedures eliminates the requirement that every federal agency bound to the Common Rule institute its own rulemaking procedures in order to revise the Common Rule or make new regulations.
The scarce resources that are currently available to support the oversight system would be insufficient to create and operate the new office. As discussed in Chapter 7, new funds should be appropriated to support the office. Although there might be short-term costs, in the long run, the investment in oversight could ultimately make more funds available for research by reducing the duplication in the current oversight system and streamlining a number of functions.
Another potential disadvantage of an independent office is that its staff may be isolated from those who are involved in the daily conduct of research. However, mechanisms could be established that would make an independent office accountable to the research community, including the IRB community, participants, and the public. First, the office should establish a standing advisory committee to assist in policy development and interpretation with representation from the appropriate federal agencies; the private sector; the research community, including all the major disciplines (e.g., clinical, public health, social sciences, humanities) that conduct such research; the IRB and ethics communities; and the public. Ad hoc advisory committees should be established to deal with specific issues of ethics and review of research. Interacting with the research community will help the office understand and appreciate the nuances in conducting research and deal effectively with emerging ethical issues. At the same time, this office should be closely tied to the IRB community so that it can address issues, problems, or challenges facing IRBs. NOHRO also
National Bioethics Advisory Commission
should develop mechanisms for public input, not only into rulemaking procedures, but also into guidance development procedures. This might include establishing a process for seeking input into decisions involving interpretations of regulations or guidance. The office should also be required to make annual reports to Congress on the status of oversight for research involving human participants.
Two examples of this model of independent, government-wide regulatory bodies, both of which were formerly located within agencies performing the activities they regulated, are the Office of Government Ethics (OGE) and the Nuclear Regulatory Commission (NRC). OGE was formerly part of the Office of Personnel Management and has authority over all federal agencies. NRC was part of the Department of Energy and has authority over both the public and private sector. The creation of OGE and NRC corrected some problems that NBAC has also observed occurring in the current oversight system for research.12 These offices act independently and are free from the interests of other agencies. They are able to make rules and promulgate guidance, visit and audit those they oversee, govern pertinent activity within other federal departments and agencies, and operate with adequate resources, including staff.13
Operating Mode of NOHRO
A lead federal office should not carry out the day-today work of protecting research participants. Once created, it would be imperative for the office to review with all federal agencies their current responsibilities and tasks so that overlapping activities could be avoided or harmonized. Individual federal offices would be responsible for interacting with the lead office and for implementing the oversight system within their departments or agencies. When appropriate, the office should delegate authority to departments or agencies, especially when the implementation of the function will vary by department. For example, departments must have authority to manage the implementation of the human research oversight system in their extramural programs. OHRP should continue to monitor compliance and investigate allegations of noncompliance, provide education, and work with the DHHS agencies in administering their extramural
31
Chapter 2: A Proposal for Oversight
research programs. In fact, OHRP should be able to expand its activities to cover the 11 agencies within DHHS in which they have not had much of a presence.
Generally, NOHRO should be directly responsible for policy development and interpretation through regulation and guidance. Rulemaking and guidance development should occur with input from federal departments, the private sector, academic and research institutions, IRBs, and the public. Education is a shared function, best carried out by other parties in the oversight system, such as academic and other research institutions, public and private sponsors, and professional societies. However, NOHRO should serve a coordinating role, ensuring consistency in educational standards and acting as a catalyst for educational program development.
NOHRO would not only ensure that review of research continues to occur through IRBs, but would also provide the leadership and policy needed to ensure consistency across IRBs. Its role in this function would be to 1) register those institutions and IRBs authorized to review and conduct research using human participants and work with credentialing bodies to develop mechanisms to accredit institutions and certify investigators, IRB members, and IRB staff and 2) coordinate reviews by specially accredited IRBs or specially created regional or national review bodies.
Other responsibilities would include the following:
Monitoring is another shared function that is best carried out by those entities closest to the conduct of research, such as sponsors and institutions or accrediting
bodies. NOHRO’s role should be to set policy and institute procedures for monitoring the federal agencies that sponsor research.
Although the lead office should have final authority for enforcement, this function should be shared with federal departments, institutions, and accrediting bodies. NOHRO should set policy describing the types of non-compliance, types of sanctions, and use of various types of sanctions to address noncompliance. It should have the authority to conduct investigations to detect regulatory noncompliance and should delegate authority to federal agencies to investigate allegations of noncompli-ance. Federal agencies, accrediting bodies, and institutions should be held accountable to report violations to NOHRO, which should investigate serious violations and repeat offenders.
Regulation of Research Involving Human Participants Sponsored by the Private Sector
NOHRO’s authority would extend to research conducted or sponsored by the private sector. Although FDA would continue to regulate research conducted in support of licensure of drugs, medical devices, biological products, or food and color additives, private institutions conducting human research and IRBs would be required to register with NOHRO and to be accredited, and investigators, IRB staff, and IRB members would be required to be certified. Sanctions would apply to both the public and private sectors.
Definition of Research Involving Human Participants
Terminology for Those Who Participate in Research
Thus far, NBAC has defined the scope of the oversight system in terms of the parties that should be covered and the functions to be carried out. This section addresses another aspect regarding scope—that is, which activities should be covered by the oversight system. This involves careful consideration of how to define the terms human participant and research. Various terms are used to describe individuals who participate in research, including subjects, respondents, observed, interviewees, informants, participants, and volunteers.
32
The term subject is generally used in scientific disciplines in which studies involve experimenting on humans. It connotes the fact that the individual is “subjected” to an action by the investigator and highlights an important aspect of the nature of the relationship between the investigator and the individual who is the subject of the experiment. No other term more accurately portrays the relationship and the unequal balance of power between the investigator and the individual in the research. Subject sometimes portrays another aspect of the relationship between the investigator and the individual, referred to as passivity.
The term subject traditionally is used in clinical research and was previously used in psychology (it continues to be used by some of the other social science disciplines). Disciplines in which the primary scientific method is the survey tend to use the term respondent or interviewee, while disciplines employing purely observational methods may use the term observed (as a noun).
The current federal regulations for protecting those who volunteer for research use the term subject, because at the time the regulations were drafted in 1981, the term was commonly used in clinical research, it had validity in describing the relationship between the investigator and the individuals being studied, and sensitivities regarding the term were less recognized. In the past two decades, two other terms have been used—participants and volunteers. In fact, the American Psychological Association began using the term participant in the 1970s (Faden and Beauchamp 1986, 184–185). Both terms reflect the commitment to the ethical principle of respect for persons and the aspiration to make “subjects” more fully protected in the process. Individuals who participate in research are assumed to do so based on their own free will. Thus, the term participant is used. Volunteer is even more specific because it makes explicit that participation is voluntary and frequently unpaid.
Neither of these terms is completely satisfactory in describing those who participate in research. Volunteer cannot readily be applied to children or others who may have diminished decisionmaking capacity and may not be able to give informed consent. Participant may seem overly broad and referring to anyone involved in the research. However, the term is likely to be confusing only
National Bioethics Advisory Commission
to those unfamiliar with conducting research. In the literature on this topic, only those individuals who are participating in research as subjects or respondents, for example, are called participants.
Perhaps more important, some individuals who participate in research find the term subject offensive. The term can be perceived as dehumanizing (Ferré and Jones 1997), and several professional organizations have stopped using it. At least two professional societies representing the social sciences use the term participant,14 as do Public Responsibility in Medicine and Research and the Applied Research Ethics National Association. Internationally, there is a trend toward using the term participant. France,15 Uganda (National Consensus Conference on Bioethics and Health Research in Uganda 1997), the United Kingdom (MRC 1998), Australia (NHMRC 1999), and the Joint United Nations Programme on HIV/AIDS (UNAIDS 2000) have adopted ethics guidelines, statements, or codes that refer to human subjects as participants. Others have suggested that subjects should not be thought of as research material or passive individuals but “simply as persons” and should be encouraged to be active in their participation in research (Veatch 1987, 4).
NBAC believes that the terminology should be changed to human participant.* Even though the term subject has validity, it seems appropriate to change it in order to be more respectful to the individuals who participate in research and to emphasize that individuals should be active, not passive, in the decision to enroll in research studies.
*Commissioner Capron, joined by Commissioners Backlar and Cassell, offers the following observation: “Although I do not believe that we should employ the word participant in place of the traditional term, subject, I recognize that no term is perfect. The traditional term carries a connotation of being under the rule of (and being obedient to) the investigator, which seems to deny a central tenet of research ethics, namely that patients and volunteers are autonomous agents, free to choose whether to participate and to withdraw from a study. Human participant, the term chosen by the Commission, overcomes that problem because it suggests a willing partner in the enterprise, but it fails to convey the fact that among those who participate in the research enterprise, those being termed participants are the ones subject to manipulation and observation and are usually the ones bearing the risk of the research. Participant might be fine as a term of aspiration, but it is premature to adopt the term, because today too many patients and volunteers who are enrolled in research studies are still not free and equal participants in the research; indeed, changing the term could send a false signal that less vigilance is needed to protect human subjects or that investigators and IRBs need not expend further effort to move to a system in which the people being studied are truly research participants.”
33
Chapter 2: A Proposal for Oversight
Defining Research Involving Human Participants and Determining What Is Covered by the Oversight System
Many activities involve interactions between people in which one party does something to another party (manipulates) and in the process of that interaction obtains or analyzes data. For example, physicians diagnose and treat patients, public health officials investigate disease outbreaks in the population, journalists conduct interviews, teachers test students, credit card companies obtain credit information, police officers question suspects and witnesses, the Internal Revenue Service (IRS) audits citizens’ tax returns, and investigators conduct various activities with participants in research. All of these activities involve not only human interaction, but also the collection or analysis of data about individuals. Yet, it is intuitively if not explicitly clear that only some of these activities should be regulated under the proposed system of oversight.
The proposed oversight system is not intended to protect the rights and welfare of individuals in all activities involving interactions between people in which one party manipulates another party and in the process of that interaction obtains or analyzes data. To do so would be impractical and inappropriate, because other systems of protection exist to provide those protections in particular circumstances (e.g., state laws related to disease reporting, the legal system for suspects and witnesses in court proceedings, or ethics codes for the practice of medicine).
Research Versus Practice
Drawing the line between research that should be regulated and that which should not be can be difficult. During each of the town meetings conducted by NBAC, questions were raised about the definition of research. For certain professions—such as medicine, public health, psychology, social work, and education—both practice and research activities are conducted, and the line between the two types of activities can be easily blurred, leading to genuine disagreement or difficult judgments. In other professional areas, there may be only practice activities, but the methods used in these activities are similar to those used in research. For example, the survey
methods used in political polls are the same as those used in health research surveys.
As discussed in Appendix C, the United States began to seriously focus on regulating research after the Nazi war crimes trials and the opening of the NIH Clinical Center. Given the historical context, the focus was on differentiating clinical research from clinical practice. Thus, the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission) was charged16 with distinguishing between research and practice in medical, behavioral, and social interventions and produced eight papers addressing this issue (Campbell and Cecil 1979; Gallant 1979; Goldiamond 1979; Levine 1979; London and Klerman 1979; Robertson 1979; Sabiston 1979; Tropp 1979). Because the National Commission focused its attention on medicine, it used a framework of “the individual versus the group.” The basic element of this distinction was that the practice of medicine referred to activities designed solely to enhance the well-being of the specific patient, while research, often using many individuals, was designed to develop or contribute to generalizable knowledge.
Of great interest to the National Commission were ways in which to classify innovative therapies or practices (London and Klerman 1979; Robertson 1979; Sabiston 1979). It was widely recognized that a common practice in medicine involved physicians trying therapies or administering drugs in a manner that differed from generally accepted practice standards. Such innovative practices were considered not to be research because they are generally not carried out in a systematic fashion and they do not generate generalizable knowledge. In addition, there is no built-in conflict of interest between the physician and the patient, because it is presumed that the sole interest of the physician is to provide potential benefit to the patient. However, the National Commission recommended that innovative practices be studied under a research protocol as soon as it became appropriate to study them systematically (National Commission 1979). NBAC agrees with the views and recommendations of the National Commission regarding the handling of innovative therapies.
34
Missing from the National Commission’s deliberations, however, were activities outside clinical or behavioral settings. For example, public health and certain types of research in the humanities and social sciences (e.g., oral history) were only minimally addressed in the National Commission’s reports. Thus, a definition of research emerged that differentiated clinical practice from clinical research, but did little to help differentiate practice from research activities in other areas.
The National Commission offered its definition of research in the Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research (Belmont Report) as an “activity designed to test an hypothesis, permit conclusions to be drawn, and thereby to develop or contribute to generalizable knowledge (expressed, for example, in theories, principles, and statements of relationships).” The Belmont Report states further that research is “usually described in a formal protocol that sets forth an objective and a set of procedures designed to reach that objective.” Practice is defined as “interventions that are designed solely to enhance the well-being of an individual patient or client and that have reasonable expectation of success” (National Commission 1979, 2). Although the definition of practice is explicit that interventions are undertaken to benefit individuals, in the definition of research, it is implicit that some, if not all, benefit is to the investigator and to society through the potential advancement of scientific understanding or knowledge.
When the federal regulations were revised following the publication of the Belmont Report, the definition in the Belmont Report was incorporated, but only to a limited degree. Since 1981, the regulatory definition of research in 45 CFR 46.102 has been as follows: “Research means a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge.” This regulatory definition has been used to identify the universe of activities covered under the Common Rule, but does not include the important distinguishing concept of who benefits from the activity. Clinical practice activities directly benefit those receiving them; the relationship between the professional and the individual is presumed
National Bioethics Advisory Commission
to be unambiguous with the intent presumed to be that of benefiting the individual. In research, however, the relationship between the investigator and the individual is different; investigators use those participating in research to generate knowledge that is of primary benefit to society. Thus, a potential conflict of interest always exists between investigators’ desire to pursue knowledge and their obligation to protect the rights and welfare of research participants.
Another shortcoming of the current definition is the use of the term generalizable. By not including a fuller definition of generalizable knowledge that would include, for example, theories, principles, statements of relationship, or attempts to develop answers to a particular question, the term is left to be defined by those attempting to apply the research definition to their activities. For example, without clarifying what is meant by generalizable knowledge, journalistic activities, marketing surveys, political polls, or police investigations could be included in the definition of research because they involve using individuals to gain information for the benefit of those who are gathering the information. However, although these activities generate useful, important information that is of general interest, they normally do not produce new knowledge that can be used to develop or revise theories or principles. NBAC does not consider these activities to fall within the scope of research involving human participants that is subject to oversight.
The current definition of research, though, is even more difficult to use in sorting activities at the boundary between research and practice (e.g., surveillance, program evaluation, quality improvement, innovative therapy, or medical monitoring) or for activities that focus on populations (e.g., public health and health services) rather than on individuals. Public health activities and health services studies are two examples for which the current definition of research is problematic.
Public Health Activities
State and local health departments have broad, legally mandated roles to protect the health of the community through data collection and analysis that target public health actions for disease prevention and control. For many public health practice activities, systematically
35
Chapter 2: A Proposal for Oversight
collected data and scientific methods are used to enhance knowledge. In some cases, the knowledge gained from public health practice could be used to develop or refine knowledge. Although the individuals who participate in these activities rarely benefit directly, the intent of public health practice is always to prevent or control disease and improve health or to improve a public health program or service in a specific population (i.e., the “public health patient“) (CDC 1999). In public health practice, immediate action is taken based on the analysis of the data to improve the health of the specific population in question. Indeed, carrying out such practices is usually done as part of activity mandated by state statute. These activities are similar to certain medical interventions in which the individual participant does not directly benefit, but others do (e.g., blood donation, skin grafts, organ transplants). In addition, the role of the public health professional usually can be considered more akin to that of the physician than that of the investigator, because the public health professional performs the activities for the benefit of the population being served, just as the physician performs activities for the benefit of the patient.
One could argue that individuals who participate in public health practice activities should be protected because in some sense they are being used just as individuals who participate in research. However, other mechanisms exist to protect the interests of individuals who participate in public health activities. State and local laws address the requirements for informed consent, protections for privacy and confidentiality, procedures for collecting and handling information (e.g., disease reporting and surveillance systems), and penalties for public health professionals when they do not comply with legal requirements.17 Any remaining deficiencies in these protection mechanisms should be evaluated and addressed through state and local laws.
On the other hand, public health professionals also conduct research in conjunction with routine public health practice activities. To provide clarification on applying the current definition of research to public health, the Centers for Disease Control and Prevention (CDC) has developed specific guidance on this matter (CDC 1999), much of which is devoted to clarifying the
term designed in a context in which data on populations are routinely and systemically collected and analyzed with the intention of providing an immediate benefit to individuals or their community (practice). This contrasts with activities that are involved in generating knowledge that would have no direct or immediate benefit for the participants or their community, but that would be applicable elsewhere (research).
Public health activities that meet the definition of research involving human participants must be subject to the proposed oversight system; however, public health practice (as defined above) would not be considered research for this purpose. For example, outbreak investigations that do not provide information beyond the scope of the investigation are public health practice and not human participant research. In addition, requirements for disease reporting, monitoring, and other data collection activities conducted under state statute or under recognized public health authority are not research that would fall under the oversight system. However, such data may be used for research purposes, in which case the use, not the initial collection of the data, constitutes research involving human participants and is subject to the oversight system (e.g., use of state cancer registry data for research).
Health Services Research and Program Evaluation
Another area in which it is difficult to apply the definition of research is the delivery of health services. A recent Institute of Medicine report defined health services research as “the study of the effects of using different modes of organization, delivery and financing for health care services” (IOM 2000, 1). Health services research differs from clinical research in that it concentrates on the study of intervention effectiveness in applied settings, while clinical research concentrates on studying the efficacy of interventions in controlled settings. Eisenberg describes health services research as part of a broader continuum of health-related research, in which the boundaries between biomedical and health services research are blurred (Eisenberg 1998). Yet, some data collection and analysis activities in health services are not intended to generate scientific knowledge, but rather are used as management tools to improve the provision of
36
services to a specific health care population (IOM 2000). These activities, generally referred to as program evaluation or quality improvement, are not intended to have any application beyond the specific organization in which they are conducted. As is true in the area of public health, because populations are the subject of study and because the methods used in program evaluation or quality improvement are the same as those used in research, it is often difficult to determine whether an activity is research that falls under the oversight system.
Definitional issues regarding program evaluation or quality improvement are not limited to health care delivery. They also occur in industrial or educational settings and in social science and operations research. However, if the purpose is to assess the success of an established program, and the information gained from the evaluation will be used to improve that program, the activity should not be considered research involving human participants. Evaluation is a program monitoring tool, and the information gained will immediately benefit the program and/or the individuals involved.
However, when quality improvement involving human participants is undertaken to test a new, modified, or previously untested intervention, service, or program to determine whether it is effective and can be used elsewhere, the activity is human participant research and subject to the oversight system.
Certain Types of Research That Might Not Require Federal Oversight
In some disciplines, especially the social sciences and humanities, it is unclear whether it is appropriate to include certain types of research in the federal oversight system.18 Although no one would question that the systematic investigations undertaken in these disciplines are research, the interpretation of the current regulations and the requirements that IRBs impose on such research may raise concerns.19 The burden on investigators coupled with the lack of any perceived benefit in added protection to participants have led some to suggest that certain types of research in these areas should not be covered under the oversight system.
In fact, intending to avoid unnecessarily burdensome review, the current federal regulations specify six categories
National Bioethics Advisory Commission
of research as exempt from the federal regulatory system. A review of the rulemaking process that occurred when the exemptions were codified indicates that DHHS assumed that most educational, behavioral, and social science research involves little or no risk to research participants and should not be regulated.20 Although most educational and social science research involves no more than minimal risk to participants, some of these research studies present a significant risk. Because the current regulations do not limit the exemptions only to research involving little or no risk, any kind of research that meets the criteria can be exempted, including, for example, a survey involving sensitive questions. Moreover, some of the exemption categories are used for biomedical research that incorporates the study of behavioral processes and employs behavioral and social science methods.
Further, current regulations specify that when research is exempt from the Federal Policy, the research is exempt from all requirements, not simply from IRB review. It is clear, however, that although requiring IRB review might be not be appropriate, it might be appropriate to require informed consent.
Delineating the criteria for exclusion from coverage is challenging. For the reasons mentioned above, the criteria should not be based on research methods alone. Instead, they should include characteristics of research design that ensure that there is little, if any, risk to participants and that there is a clear and easy opportunity for participants to make an informed refusal to participate. For example, research involving no intervention, the collection of no sensitive information, and no need by investigators to act upon the collected information (e.g., mandatory disclosures) should not be included in the system.
In summary, the definition of covered research can be improved by avoiding the use of terms that are difficult to understand or apply. A clear and accurate definition will help ensure that the federal oversight system will cover activities within its scope. These activities should include those involving the systematic collection or analysis of data with the intent of generating new knowledge, principles, or theories and studies that generate information that revises or improves on an existing principle, theory, or body of knowledge. Some research
37
Chapter 2: A Proposal for Oversight
activities that are conducted in fields that do not rely solely on clinical scientific procedures—such as some types of anthropological or humanities research—should be included under this definition. Among those activities that are considered research, however, some should not be covered under the federal oversight system, especially when they pose little or no risk to participants and allow them to make an informed refusal to participate. Furthermore, although the definition of research need not explicitly state the nature of the relationship between investigators and participants, it should be clear that in those activities defined as research, participants are used at least to some extent by investigators to achieve their research objectives, which might or might not benefit the participants.
In developing one set of regulations and a definition of research, it will be important to harmonize that definition with FDA’s definition of regulated activities to ensure that FDA-regulated activities are included. For example, FDA regulations relating to the use of test articles involving experiments with one or more human participants require protections for human participants even if the activity does not involve “research” (21 CFR 56.102(c); 21 CFR 312.3(b)).
Who Should Be Protected by the Oversight System?
Under the Common Rule, sequential decisions must be made to determine whether an activity constitutes human participant research. That is, one must first determine whether the activity is research; then, if it is research, one must determine whether it involves human participants. In making this determination, one must establish whether private information is obtained and whether it is individually identifiable (NBAC 1999b), meaning that the “identity of the subject is or may be readily ascertained by the investigator or associated with the information” (45 CFR 46.102(f)(2)). No definition of “readily ascertained by the investigator” is provided. Private information includes “information about behavior that occurs in a context in which an individual can reasonably expect that no observation or recording is taking place, and information which has been provided for specific purposes by an individual and which the individual can reasonably expect will not be made public” (45 CFR 46.102(f)(2)).
This algorithm is often poorly understood (NBAC 1999b). A simpler decisionmaking process could be one that describes human participants who should be covered as those who are exposed to manipulations, interventions, observations, or other types of interactions (e.g., surveys) with investigators or who are identifiable through research using biological materials, medical or other records, or databases.
Different terms are used by various disciplines to describe what occurs in the relationship between investigators and participants. Manipulation or intervention means physical, psychological, or social procedures by which data are collected or by which the participants or the participants’ environment are affected as part of the research. Interaction means communication or interpersonal contact between the investigator and the participant.
Investigators include all who are involved in the conduct of a research study, such as scientists, study coordinators, research staff, and data abstractors. Student-initiated research in which the intention of the activity may be as much pedagogical as it is to pursue knowledge is often difficult to classify,21 but students conducting research involving human participants should also be considered investigators. Review of student-initiated research should be commensurate with the risks associated with the research; for example much student-initiated research is likely to involve no more than minimal risk and to be eligible for review using procedures other than full IRB review.
NBAC developed a definition of identifiable in its report on the research use of human biological materials that also applies here, in which coded data are considered identifiable (NBAC 1999b, 17). Coded data were defined as linked or identifiable because, even though they do not include any identifying information, such as names, they have codes that allow the data to be linked to identifying information (NBAC 1999b).
All coded data are identifiable regardless of the how difficult it may be to link the data to identifying information. Even when disclosure of the identifying information is protected under federal or state law and access to the identifying information would be extremely difficult to obtain, the coded data remain identifiable. For example, coded data that are protected under federal confidentiality
38
statutes (e.g., statues adhered to by the Departments of Education and Justice and CDC) should be considered identifiable, and uses of such data for research purposes constitute research involving human participants. Sometimes, investigators who hold coded data will release it to another investigator for use in a research study under a written agreement unequivocally prohibiting release of identifying information to the recipient investigator.22 Even in this instance, the coded data are identifiable, and studies involving such data constitute research involving human participants.
Although all research using coded data involves human participants, the potential to identify individuals varies. Some coded data are easily identifiable (i.e., investigators have access to the key linking the code to identifying information), while other coded data are more difficult to identify (i.e., the recipient of the coded data does not have access to the key), and still other coded data are very difficult if not virtually impossible to identify (i.e., the key is legally protected and cannot be disclosed to investigators). IRBs should consider, when reviewing a particular research study, how easily coded data can be identified and conduct a review that is commensurate with the risks associated with that data. For example, if the coded data are part of a data set that is available for public access, such as those from the National Center for Health Statistics or the Bureau of the Census, the IRB review may consist of nothing more than ascertaining that the data are available to the public and that the owner of the data has taken appropriate steps to protect the coded data (e.g., the confidentiality of the data is protected under federal statute or under state law or a data repository requires the recipient to sign a legally binding agreement stipulating permitted uses of the data and that no identifying information will be released to the recipient).
Research involving the analysis of existing data that are unidentifiable does not involve human participants. NBAC has previously defined two types of unidentifiable data. One type is unidentified, also referred to as anonymous, which are collected from individuals who were not identified at the time of collection. The second type of data is unlinked or anonymized data. Although identifiers were available when the data were collected or stored, at some point, the identifiers were unlinked.
National Bioethics Advisory Commission
Thus, unlinked data lack identifiers or codes that can link the data to identifying information or particular individuals (NBAC 1999b).
Information Revealed About Others
Some research involves interaction with the participant that reveals information about other individuals (e.g., family histories and research on familial interactions). Generally, these other individuals are not considered research participants, although there have been recent discussions about how to consider the rights and welfare of these individuals when constructing appropriate protections (Botkin 2001). NBAC concludes that, in general, these individuals should not be considered human participants in research. Nevertheless, there might be circumstances in which it is appropriate for an IRB to consider whether their rights and welfare are being protected, particularly with regard to confidentiality of data.
Children
Inclusion of children in research raises special issues. Although research with children is not addressed in this report, the proposed system could apply to this population as well. Should this report and its recommendations be implemented, special attention should be given to the ethical standards and regulatory requirements associated with providing adequate protections to children who are research participants.
Embryos and Fetuses
Research involving human fetuses and embryos raises distinct issues. Additional protection exists in the DHHS regulations pertaining to research and related activities involving fetuses, pregnant women, and in vitro fertilization (Subpart B). In 1993, DHHS convened a working group to revise Subpart B. These revisions were published in the Federal Register on January 17, 2001, but are not yet effective.23
Deceased Individuals
Current federal regulations do not cover deceased individuals, which includes cadavers, biological materials from cadavers, and data from cadavers. NBAC concurs with this position and has in part addressed this issue previously (NBAC 1999b, 29).
39
Chapter 2: A Proposal for Oversight
As discussed in Chapter 4, when living persons may be affected by research involving the deceased in such a way that the living might experience social, physical, psychological, economic, or legal harm, these risks should be considered by an IRB. Although deceased individuals do not have independent rights and interests that need to be protected, living family members might claim such rights and interests, especially in research in which the living family members may be contacted or in which they or others become aware of the research findings (NBAC 1999b, 49, 73). The burden is on investigators to be sensitive to these issues.
Materials from deceased individuals that are used in a current research study may be obtained from previous research or practice (e.g., specimens collected as part of a surgical procedure). The understanding of individuals at the time they are living with respect to future uses of such materials should be honored following their death (NBAC 1999b, 49). Consent documents should be reviewed to ensure that currently proposed research is not violating the participant’s understanding at the time consent was obtained, regardless of whether the materials or data were collected as part of a research study or practice.
This same principle holds true for other types of data. For example, oral histories are often archived, with informed consent from the participant for future research uses, and the data could be used for future research.24 So that data can be used in an ethical manner after a participant’s death, investigators should construct informed consent processes to describe as many potential uses of specimens or data after death that can be anticipated at the time of agreement to participate in the research. In this way, investigators can respect the wishes of participants and at the same time use valuable data repositories.
Decisions Regarding Whether an Activity Is Research and Subject to Federal Oversight
This report does not attempt to offer a regulatory definition of research involving human participants that would be subject to the oversight system that clearly and easily identifies all possible activities that must be included. No regulatory definition of covered research can be written with the sensitivity and specificity needed to ensure that all research activities that should be subject
to oversight are always included and all activities that should be excluded from oversight are always excluded. Clarification and interpretation will invariably be required, and ambiguous cases will always arise.
Recommendation 2.4: Federal policy should cover research involving human participants that entails systematic collection or analysis of data with the intent to generate new knowledge. Research should be considered to involve human participants when individuals 1) are exposed to manipulations, interventions, observations, or other types of interactions with investigators or 2) are identifiable through research using biological materials, medical and other records, or databases. Federal policy also should identify those research activities that are not subject to federal oversight and outline a procedure for determining whether a particular study is or is not covered by the oversight system.
One of the important leadership roles NOHRO should fulfill is to provide guidance on determining whether an activity is research involving human participants and thus subject to oversight. In addition, it should generate a list of activities that are normally treated as human participant research and are therefore subject to oversight as well those that are not. For example, the list of activities not covered by the federal oversight system should include journalism, market surveys, political polls, police investigations, IRS audits, academic examinations, athletic competitions, mandatory disease reporting, outbreak investigations, quality improvement or program evaluation when used as management tools, other defined practice activities in public health, medicine, psychology, and social work, and certain types of research activities posing little or no risk to participants and offering the opportunity for participants to make an informed refusal. NOHRO should initiate a process in which representatives from various disciplines and professions (e.g., social sciences, humanities, business, public health, and health services) contribute to the development of the definition and the list of research activities subject to the oversight system. Based on the recommendations in this report, it is likely that some research that is currently exempt would be covered
40
under the federal oversight system while other, currently exempt research would not be covered.
Moreover, at the local level, institutions should develop policies and procedures for ensuring that a disinterested third party, who is not the investigator, determines whether an activity is research involving human participants and subject to oversight. The third party should be sufficiently knowledgeable about the institution’s mission and overall activities to make such determinations, keeping in mind that all disciplines have nuances that make each decision about whether an activity is human participant research unique. Although IRBs or institutional human research oversight offices are obvious locations for this process, other models may be appropriate. In any case, the investigator alone should not make the determination.
Applicability to Various Types of Research
It is NBAC’s intention that the definition of research involving human participants be applied in all disciplines. However, because the risks of research differ both qualitatively and quantitatively across the spectrum of research disciplines, the oversight system should ensure that all covered research is subject to basic protections, such as a process of informed consent, except in the specified conditions in which it can be waived; protection of privacy and confidentiality; and minimization of risks. Having potentially included more research under this proposed system, it is more critical than ever that review mechanisms and criteria for various types of research are suited to the nature of the research and the likely risks. This chapter, for example, has described the use of procedures that could accelerate, streamline, or harmonize review, such as less than full board review for minimal risk research and use of specially accredited IRBs or special regional or national review of novel or controversial research to supplement local IRB review. Moreover, more specific guidance for review of different types of research is needed, including guidance regarding review criteria and IRB composition.
Under the proposed oversight system, a wide range of research activities would be covered, from clinical trials to observational studies, with a continuum between them that includes psychological experiments, studies using existing data, and surveys, with the types and degree of
National Bioethics Advisory Commission
risks varying substantially among the different types of research. Thus, review criteria and mechanisms must be appropriate for the particular type of research. To be effective and efficient, the oversight system should encourage flexibility in IRB review; however, the local IRB should implement a common set of ethical principles and standards.
All research studies that are judged to involve no more than minimal risk should be eligible for IRB review using more efficient procedures than a full review, although full board review could be required if deemed necessary by a local IRB. Most social science and humanities research involves no more than minimal risk and can be reviewed using a more efficacious procedure than full IRB review. In addition, many health services research studies, epidemiological studies, and other studies using existing data involve no more than minimal risk and can be reviewed using a less resource-intensive process. However, even within the IRB review category of no more than minimal risk, the risk level varies from virtually none to minimal, and review within the category should be commensurate with the level of risk.
For example, one significant demarcation along the continuum of research is whether it involves interaction with human participants. In research involving the use of existing data or tissues and no interaction with human participants, the primary ethical concerns are threats to privacy and confidentiality (NBAC 1999b). IRBs should pay attention to these types of ethical concerns and have or find the needed expertise, for example, in data management, storage, and transmission; computer security; and other confidentiality protections (e.g., state law). IRBs that review DNA research on stored tissue samples, for example, might have expertise in genetics. The expertise of the IRB or access of the IRB to such expertise (i.e., use of consultants) should be appropriate for addressing the ethical issues the IRB is likely to face in reviewing research. Chapter 4 further discusses IRB review requirements for different types of research.
Recommendation 2.5: Federal policy should require research ethics review that is commensurate with the nature and level of risk involved. Standards and procedures for review should distinguish between research that poses minimal
41
Chapter 2: A Proposal for Oversight
risk and research that poses more than minimal risk. Minimal risk should be defined as the probability and magnitude of harms that are normally encountered in the daily lives of the general population (see Recommendation 4.2). In addition, the federal government should facilitate the creation of special, supplementary review bodies for research that involves novel or controversial ethical issues.
Summary
A comprehensive and effective oversight system is essential to uniformly protect the rights and welfare of research participants while permitting ethically and scientifically responsible research to proceed without undue delay. To achieve these goals, the oversight system should include several interrelated but distinct functions and responsibilities, including policy development, regulatory reform, research review and monitoring, research ethics education, and enforcement. Many parties are involved in the research enterprise and the system of oversight, including research participants, IRBs, institutions, sponsors, a federal oversight office, state regulators, professional organizations, publishers, Congress, and the public. To function as intended, the system requires that all parties act responsibly, even though the ethical obligation to protect participants lies primarily with investigators, sponsors, and IRBs.
All research participants, regardless of whether the research sponsor is a public or private entity, should be protected by this oversight system, which should apply to both domestic and international research. To accomplish this level and extent of protection, the system must expand beyond its current scope. An independent, single federal office created to lead and coordinate the oversight system would be more effective than the current system and would solve a number of problems. Such an office should promulgate a unified and comprehensive federal policy embodied in a single set of regulations and guidance that can be applied to all types of research involving human participants. Federal protections should be uniform across all sectors of government, academe, and the private sector, but should be flexible enough to be
applied in widely different research settings or to emerging areas of research.
Finally, the newly created office should include in its policy a definition of research involving human participants that is subject to federal protections and develop a range of review mechanisms for the various types of covered research. To meet the goal of protecting those who participate in research, the scope and relevancy of protections must be clear to all. Investigators, IRBs, sponsors, and institutions require and must receive unambiguous guidance on the extent to which their research activities are covered by the oversight system.
Notes
1 OPRR/FDA Town Hall Meeting: June 16, 2000, Chapel Hill, North Carolina; Vanderpool, H., Testimony before NBAC. June 6, 2000. San Francisco, California.
2 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
3 See Vanderpool, H., “Unfulfilled Promise: How the Belmont Report Can Amend the Code of Federal Regulations Title 45 Part 46—
Protection of Human Subjects.” This background paper was prepared for NBAC and is available in Volume II of this report.
4 See Moreno, J., “Protectionism in Research Involving Human Subjects.” This background paper was prepared for NBAC and is available in Volume II of this report.
5 See Chalmers, D., “Research Ethics in Australia.” This background paper was prepared for NBAC and is available in Volume II of this report.
6 See Moreno, J., “Protectionism in Research Involving Human Subjects.” This background paper was prepared for NBAC and is available in Volume II of this report.
7 See Chalmers, D., “Research Ethics in Australia.” This background paper was prepared for NBAC and is available in Volume II of this report.
8 See Schwartz, J., “Oversight of Human Subject Research: The Role of the States.” This background paper was prepared for NBAC and is available in Volume II of this report.
9 See McCarthy, C., “Reflections on the Organizational Locus of the Office for Protection from Research Risks.” This background paper was prepared for NBAC and is available in Volume II of this report.
10 See Fletcher, J., “Location of the Office for Protection from Research Risks within the National Institutes of Health: Problems of Status and Independent Authority.” This background paper was prepared for NBAC and is available in Volume II of this report.
42
11 See McCarthy, C., “Reflections on the Organizational Locus of the Office for Protection from Research Risks.” This background paper was prepared for NBAC and is available in Volume II of this report.
12 Flack, D., Testimony before NBAC. March 1, 2000. Herndon, Virginia; Ley, J., Testimony before NBAC. March 1, 2000.
Herndon, Virginia. See McCarthy, C., “Reflections on the Organizational Locus of the Office for Protection from Research Risks.” This background paper was prepared for NBAC and is available in Volume II of this report.
13 See Fletcher, J., “Location of the Office for Protection from Research Risks within the National Institutes of Health: Problems of Status and Independent Authority.” This background paper was prepared for NBAC and is available in Volume II of this report.
14 All codes available at American Psychological Association, http://www.apa.org/ethics/code.html; American Sociological Association, http://www.asanet.org/members/ecostand.html. Both sites last accessed December 12, 2000.
15 Law 88-1138 Regarding the Protection of Persons Agreeing to Biomedical Research.
16 Pub. Law 348, 93rd Congress, Title II.
17 NBAC Town Meeting: August 14, 2000, Portland, Oregon.
18 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
19 Wax, M., Testimony before NBAC. April 6, 2000. Washington, D.C.; Sieber, J.E., Testimony before NBAC. April 6, 2000.
Washington, D.C.; Abowd, J.M., Testimony before NBAC. April 6, 2000. Washington, D.C.; Shopes, L., Testimony before NBAC. April 6, 2000. Washington, D.C.
20 46 Fed. Reg. 8366–8391.
21 Public Responsibility in Medicine and Research conference on human subjects protections, November 1998, “IRBs and the Shifting Sands of Public Opinion.” San Diego, California.
22 OHRP Guidance: “Engagement of Institutions in Research.” January 26, 1999; “Issues to Consider in the Research Use of Stored Data or Tissues.” November 7, 1997; “Protections for Human Subjects in the NIGMS Human Genetic Mutant Cell Repository: Submission of Non-Identifiable Materials to the Repository.” May 22, 1997; “Guidance on Protections for Human Subjects in the National Institute of General Medical Sciences Human Genetic Mutant Cell Repository.” May 21, 1997; “Operation of Human Cell Repositories Under HHS Regulations at 45 CFR 46.” August 19, 1996. Available at http://ohrp.osophs.dhhs.gov/humansubjects/guidance/reposit.htm. Last accessed December 15, 2000.
23 38 Fed. Reg. 3878–3883.
24 K. Ewing and L.K. George, Duke University, to E.M. Meslin, NBAC. February 16, 2001. Letter relating to draft of this report.
National Bioethics Advisory Commission
References
Botkin, J.R. 2001. “Protecting the Privacy of Family Members in Survey and Pedigree Research.” Journal of the American Medical Association 285(2):207–211.
Campbell, D.T., and J.S. Cecil. 1979. Protection of the Rights and Interests of Human Subjects in the Areas of Program Evaluation, Social Experimentation, Social Indicators, Survey Research, Secondary Analysis of Data from Administrative Records. In Appendix to The Belmont Report, vol. 2, 12-1–12-23. Washington, D.C.: Department of Health, Education, and Welfare (DHEW).
Centers for Disease Control and Prevention (CDC). 1999. Guidelines for Defining Public Health Research and Public Health Non-Research.
October 4. Available at http://www.cdc.gov/od/ads/opspoll1.htm. Last accessed December 1, 2000.
Eisenberg, J.M. 1998. “Health Services Research in a Market-Oriented Health Care System.” Health Affairs 17(1):98–108.
Faden, R.R., and T.L. Beauchamp. 1986. A History and Theory of Informed Consent. New York: Oxford University Press.
Ferraro, F.R., E. Szigeti, K.J. Dawes, and S. Pan. 1999. “A Survey Regarding the University of North Dakota Institutional Review Board: Data, Attitudes, and Perceptions.” Journal of Psychology 133:272–280.
Ferré, C., and L. Jones. 1997. Processes of Community Partnerships: The Healthy African American Families and Harlem Birthright Projects. 17 May. Paper presented at Community Partners for Prevention Research: Implications for Science and the Practice of Public Health Symposium. Centers for Disease Control and Prevention. Atlanta, Georgia.
Gallant, D. 1979. Response to Commission Duties as Detailed in P.L. 93-348, Sec. 202 (a)(1)(B)(i). In Appendix to The Belmont Report, vol. 2, 13-1–13-22. Washington, D.C.: DHEW.
Goldiamond, I. 1979. On the Usefulness of Intent for Distinguishing Between Research and Practice, and Its Replacement by Social Contingency. In Appendix to The Belmont Report, vol. 2, 14-1–14-73. Washington, D.C.: DHEW.
Institute of Medicine (IOM). 2000. Protecting Data Privacy in Health Services Research. Washington, D.C.: National Academy Press.
———. 2001. Preserving Public Trust: Accreditation and Human Research Participant Protection Programs. Washington D.C.: National Academy Press.
Joint United Nations Programme on HIV/AIDS (UNAIDS). 2000.
Ethical Considerations in HIV Preventive Vaccine Research. Geneva: UNAIDS.
Levine, R.J. 1979. The Boundaries Between Biomedical or Behavioral Research and the Accepted and Routine Practice of Medicine. In Appendix to The Belmont Report, vol. 1, 1-1–1-44. Washington, D.C.: DHEW.
43
Chapter 2: A Proposal for Oversight
Liddle, B.J., and E. Brazalton. 1996. “Psychology Faculty Satisfaction and Compliance with IRB Procedures.” IRB: A Review of Human Subjects Research 18(6):4–6.
London, P., and G. Klerman. 1979. Boundaries Between Research and Therapy, Especially in Mental Health. In Appendix to The Belmont Report, vol. 2, 17-1–17-15. Washington, D.C.: DHEW.
Medical Research Council (MRC). 1998. Guidelines for Good Clinical Practice in Clinical Trials. London: MRC.
National Bioethics Advisory Commission (NBAC). 1998.
Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity. 2 vols. Rockville, MD: U.S. Government Printing Office.
———. 1999a. Ethical Issues in Human Stem Cell Research. 3 vols. Rockville, MD: U.S. Government Printing Office.
———. 1999b. Research Involving Human Biological Materials: Ethical Issues and Policy Guidance. 2 vols. Rockville, MD: U.S. Government Printing Office.
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission). 1979.
Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research. Washington, D.C.: U.S. Government Printing Office.
National Consensus Conference on Bioethics and Health Research in Uganda. 1997. Guidelines for the Conduct of Health Research Involving Human Subjects in Uganda. Kampala, Uganda.
National Health and Medical Research Council (NHMRC). 1999.
National Statement on Ethical Conduct in Research Involving Humans.
Australia: NHMRC.
Office for Protection from Research Risks Review Panel. 1999.
Report to the Advisory Committee to the Director, NIH. 3 June. Available at http://www.nih.gov/about/director/060399b.htm. Last accessed December 2, 2000.
President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission). 1983. Implementing Human Research Regulations: Second Biennial Report on the Adequacy and Uniformity of Federal Rules and Policies, and of Their Implementation, for the Protection of Human Subjects. Washington, D.C.: U.S. Government Printing Office.
Robertson, J. 1979. Legal Implications of the Boundaries Between Biomedical Research Involving Human Subjects and the Accepted or Routine Practice of Medicine. In Appendix to The Belmont Report, vol. 2, 16-1–16-54. Washington, D.C.: DHEW.
Sabiston, D. 1979. The Boundaries Between Biomedical Research Involving Human Subjects and the Accepted or Routine Practice of Medicine, with Particular Emphasis on Innovation in the Practice of Surgery. In Appendix to The Belmont Report, vol. 2, 17-1–17-9. Washington, D.C.: DHEW.
Tropp, R.A. 1979. What Problems Are Raised When the Current DHEW Regulation on Protection of Human Subjects Is Applied to Social Science Research? In Appendix to The Belmont Report, vol. 2, 18-1–18-17. Washington, D.C.: DHEW.
Veatch, R.M. 1987. The Patient as Partner: A Theory of Human-Experimentation Ethics. Bloomington: Indiana University Press.
44
Chapter Three 3
Methods for Ensuring Protection: Education,
Certification, and Accreditation
Introduction
Protecting the rights and welfare of research participants is the major ethical obligation of all parties in the oversight system, who, to meet this obligation, must demonstrate competence in research ethics by being able to conduct, review, or oversee research involving human participants in an ethically sound manner. Such competence entails not only being knowledgeable about relevant research ethics issues and federal policies but also ensuring that conflicting interests for institutions, investigators, or Institutional Review Boards (IRBs) are disclosed and managed so that there is no question that research participants are being protected. Finally, the oversight system must include a robust monitoring process so that lapses by institutions, IRBs, and investigators can be remedied.
This chapter addresses measures needed to ensure that all parties involved in the research enterprise are competent with regard to protecting research participants and are reliably carrying out their responsibilities in this area. They include measures:
Education and Competence in Conducting Research Involving Human Participants
Education is an essential ingredient for developing competence in the ethical conduct of research with human participants; it can assist not only in the identification of ethical problems, but also in their resolution. Through well-designed ongoing educational programs, one can learn, for example, the most practical and effective steps for protecting confidentiality or improving the quality of the informed consent process and its documentation. Tailored education programs can help prevent routine ethical issues from becoming needless impediments to research and can provide basic skills to assist investigators and IRB members in dealing with new or complex ethical issues.
The need for education was a major focus of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission), the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission), and the Advisory Committee on Human Radiation Experiments (ACHRE). It also has been central in National Bioethics Advisory Commission (NBAC) reports.
The National Commission focused on education of IRB members and proposed that the federal government and individual institutions play a role in that effort (National Commission 1978, 10, 15). The President’s Commission recommended a broad educational program targeted to investigators, IRB members, and research administrators, including site visits to institutions by
45
Chapter 3: Methods for Ensuring Protection: Education, Certification, and Accreditation
experienced IRB members and administrators (President’s Commission 1983, 135–136). ACHRE highlighted the importance of education by linking the protection of the rights and interests of research participants to the ability of investigators to “appreciate sufficiently the moral aspects of human…research and the value of institutional oversight” (ACHRE 1995, 817). In two previous reports, NBAC recommended that professional associations develop topic-specific educational materials (NBAC 1998; NBAC 1999).
Despite this enduring recognition of the important role of education, the educational function of the oversight system has been only minimally implemented through federal programs. The former Office for Protection from Research Risks (OPRR) devoted most of its limited resources to negotiating assurances and overseeing compliance. Aside from the educational activities associated with obtaining an assurance, regional workshops sponsored in conjunction with the Food and Drug Administration (FDA) were OPRR’s only other sustained educational program. The lack of focus on education at the federal level was repeated at the local level, with institutions often failing to provide educational programs to their investigators, research staff, and IRB members. It should not be surprising, therefore, that the lack of appropriate educational programs and the resulting lack of knowledge about the ethical principles and procedures relevant to protecting research participants are among the deficiencies identified in audits and suspensions of research programs by OPRR, now the Office for Human Research Protections (OHRP) (OHRP 2000). A National Institutes of Health (NIH) survey of IRB chairs, members, administrators, institutional officials, and investigators at institutions holding Multiple Project Assurances (MPAs) found strong support for additional, improved education (Bell et al. 1998).
Some efforts are currently under way to increase educational opportunities. In 1997, two NIH-sponsored educational programs were initiated as part of the government’s apology to the victims and families of the Tuskegee Syphilis Study. In one program, investigators receive grants to participate in ethics training programs tailored to meet their research needs; in another, program grants are awarded to individuals to develop courses in
research ethics.1 In addition, in 1998 OPRR hired its first director of education to develop computer-based education programs for IRB administrators and institutional officials. Outside government, organizations, such as Public Responsibility in Medicine and Research (PRIM&R) and the Association of American Medical Colleges, have traditionally provided education to their constituents. Recently, PRIM&R developed a new “IRB 101” course, which is offered before its annual meeting and at the request of institutions throughout the year.2 In addition, some academic institutions have developed their own courses (Dunn and Chadwick 1999; Sugarman 2000).
Recently, NIH implemented a funding eligibility requirement that all individuals submitting grant applications provide evidence that they have been trained in research ethics.3 This requirement has increased the number of education programs academic institutions offer to faculty and staff with the goal of raising the level of knowledge about and awareness of research ethics among investigators. Although NIH provides a model education program on its website, no requirements or guidance are provided regarding what constitutes appropriate training. Investigators and institutions have considerable discretion in fulfilling the NIH requirement. This activity has generated a beneficial exchange of ideas on how best to teach research ethics to different audiences.
Educational programs should focus on ethical principles, their relationship to ethical standards, and the procedures necessary to implement them. It is critical to emphasize the links among these three components. Because each research study is different and some raise new ethical issues, investigators and IRBs must be able to refer to and apply general ethical principles. Yet, it is equally true that research ethics cannot be taught or learned entirely in the abstract. As one expert cautioned: “rote memorization of guidelines, principles, and regulations, absent the ability to apply such concepts in practical situations, will not ensure the protection of [research participants].”4 Educational programs should include problem-solving components in which investigators and IRB members learn ways in which to identify, discuss, and resolve ethical issues pertinent to specific types of research studies.
46
For the behavioral and social sciences, IRB members and investigators should be educated in the ethical standards and associated procedures that are appropriate to human participant research in these disciplines. In addition, IRB members should be familiar with standards in professional societies’ codes of ethics, particularly for fieldwork in anthropology or survey research in sociology or psychology. Such standards address issues of privacy and confidentiality, informed consent, and community protection. Complex questions concerning deception or stigmatization of individuals or communities may need particular attention, particularly in the context of research involving individuals with vulnerabilities.
NBAC supports the independence of institutions and other entities that would enable them to design their own education programs. However, it is important that all educational programs provide appropriate coverage of the ethical principles and standards and current federal regulations and guidance related to human participant research.
Although the primary focus of education is on those parties involved in the daily conduct of research (e.g., investigators and IRBs), other groups that carry important ethical responsibilities are also important targets for education. For example, sponsors—whether public or private—often are directly involved in providing advice and supervision to grantees and contractors. Federal agencies that have their own oversight offices (see Chapter 2) also should be competent in research ethics, regulations, and guidance.
If investigators are to conduct research ethically and research participants are to be able to make informed decisions about participation, education about the ethical conduct of research needs to be an integral part of science education in the United States (Pritchard 1999). Even for those students who do not pursue careers in science or research, education in research ethics could make them better informed in terms of future research participation or in terms of understanding the nature of the research enterprise. The American Association for the Advancement of Science (AAAS 1993) and the National Academy of Sciences (National Research Council 1995) have produced documents outlining content standards for science education, including standards for the ethical treatment of research participants.
National Bioethics Advisory Commission
While some curricula include courses or lectures on research ethics, such education generally is missing from most undergraduate and graduate school science curricula and is seldom required, including for medical and other health professions education (Coughlin et al. 1999; Mastroianni and Kahn 1998). This phenomenon is particularly disturbing because many students conduct research involving human participants as part of their graduate educational experience (Gunsalus 1997). Moreover, a large number of scientists and many health professionals expect to conduct human research. For example, in a survey of all graduating medical students conducted in 1996, 78.3 percent responded that they expect to be involved in research at least somewhat during their medical career, and 11.9 percent responded that they expected to be involved exclusively or significantly involved during their medical career.5 Given the role of physician-investigators within academic medical centers and the growing number of community physicians conducting research or referring patients to research, education in research ethics and human research protection should be a meaningful component of medical education. Professional societies should assist institutions in developing educational programs and curricula.
The need for education in research ethics in graduate curricula extends beyond the health fields. Graduate programs in social sciences and the humanities also should incorporate curricula relevant to the specific discipline, which could take advantage of the fact that professional societies have developed codes of ethics that deal specifically with conducting research involving human beings. Education also can be extended to other groups likely to be involved in research. Patient advocacy organizations can, and often do, provide education to their members. Informing groups about rights of research participants, protections afforded, and points to consider in deciding whether to enroll in research can empower a large number of prospective research participants.
Recommendation 3.1: All institutions and sponsors engaged in research involving human participants should provide educational programs in research ethics to appropriate institutional officials, investigators, Institutional Review Board members, and Institutional Review Board staff. Among other
47
Chapter 3: Methods for Ensuring Protection: Education, Certification, and Accreditation
issues, these programs should emphasize the obligations of institutions, sponsors, Institutional Review Boards, and investigators to protect the rights and welfare of participants. Colleges and universities should include research ethics in curricula related to research methods, and professional societies should include research ethics in their continuing education programs.
Recommendation 3.2: The federal government, in partnership with academic and professional societies, should enhance research ethics education related to protecting human research participants and stimulate the development of innovative educational programs. Professional societies should be consulted so that educational programs are designed to meet the needs of all who conduct and review research.
The federal government should use a variety of incentives to encourage the enhancement of research ethics education.
Demonstrating Competence
Certification of Individuals
Educating all parties in research ethics and human research protections is effective only when it results in competence to design and conduct ethically sound research, including analyzing, interpreting, and disseminating results in an ethically sound manner. Such competence, however, cannot be assumed to follow from exposure to an educational course or program. As the complexion of research continues to change, new and challenging ethical dilemmas emerge with advances in technology, and as more people become involved in research as investigators or in roles particularly related to oversight, it becomes increasingly important for all parties to demonstrate their competence in the ethics of research involving human participants.
Currently, there is no regulatory requirement to demonstrate competence, even when the level of risk is high or the participants are vulnerable. In recent years, there has been much discussion about the need to implement regulations consistently, set standards for IRBs and measure performance in meeting those standards (OIG 1998a), and measure the competency of investigators (Cohen 2000).
Many IRBs and investigators have suggested that investigators should be required to demonstrate a certain level of competence in order to conduct human research.6 NBAC supports this view. All individuals directly involved in the protection of human research participants should be certified, including investigators and research staff, IRB members and staff, and sponsors. Individuals employed by sponsors also should be certified when they are directly involved in the conduct or oversight of research—for example, grants or contracting officers who might make decisions relevant to human participant protections. Further details regarding just which individuals require certification will, of course, be context dependent.
Because roles vary substantially and among investigations the diversity in research design and subject matter is great, several certification options should be available. For example, certification of investigators may need to be specialized for particular disciplines or in relation to certain subject matter in order to increase its effectiveness. Different mechanisms for certification should be tested and evaluated. Currently, under the NIH requirement, certification of investigators is left entirely up to the institution. Certification of IRB staff and members is not required; however, there is a voluntary certification program for IRB administrators. The Council for Certification of IRB Professionals (CCIP) was formed to promote IRB administration practice and to advance the quality of research participant protection programs through the certification of qualified IRB professionals. CCIP administered the first certification examination in October 2000 and announced recently that 107 professionals were certified based on the results of these exams.7 Certification efforts can surely be improved. Overall, the goal of such programs should be to ensure competence and consistency that is relevant to the performance of the particular individual parties.
Recommendation 3.3: All investigators, Institutional Review Board members, and Institutional Review Board staff should be certified prior to conducting or reviewing research involving human participants. Certification requirements should be appropriate
48
to their roles and to the area of research. The federal government should encourage organizations, sponsors, and institutions to develop certification programs and mechanisms to evaluate their effectiveness. Federal policy should set standards for determining whether institutions and sponsors have an effective process of certification in place.
Accreditation of Institutions and Independent IRBs
Related to measuring competence in individual investigators and IRB members and staff is measuring a core set of competencies in independent IRBs or institutions. In this arena, accreditation programs might be the most effective mechanisms for measuring compliance of such entities to a set of standards (Hamm 1997). In this report, the term accreditation refers to programs directed toward institutions, and the term certification refers to programs directed toward individuals.
Although accreditation and certification do not guarantee desired outcomes, they can be helpful in improving performance. These programs generally involve experts and peers developing a set of standards that represents a consensus of the best practices in the profession. Therefore, the choice of standards and the criteria for evaluating whether an institution has met them are critically important. The emphasis of these programs should be on education, on assuring that appropriate protections are in place, and on avoiding excessively bureaucratic procedures. Accrediting programs may lose their value if they are seen as merely another administrative burden.
Despite concerns that accreditation might add another layer of bureaucracy to an already regulated environment, several advantages of accreditation programs have been noted. They are generally voluntary and represent a profession’s desire to self-regulate. Many accrediting programs strive to meet higher standards than are required by law, so that having the credential implies a higher level of competence than what may be minimally required. Properly governed and organized, accrediting bodies can improve competence (and thus performance) within a profession, which helps to achieve the goals of the profession and build public trust (Hamm 1997). In addition, accreditation serves an educative role.
National Bioethics Advisory Commission
Institutions seeking accreditation, for example, usually go through a periodic review process involving a self-study and a site visit from a team of experts in the profession. The self-evaluation of the institution in preparation for a site visit helps it identify strengths and weaknesses in its programs and make improvements. The site visit can be an especially useful learning experience that encourages the institution to sustain best practices.
Accrediting programs are widely used in the fields of health and education. These programs are seen as having a major and generally positive influence (Hamm 1997). One highly regarded accrediting program in research is the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). This private, nonprofit organization “promotes the humane treatment of animals in science through voluntary accreditation and evaluation programs.” AAALAC accreditation is regarded as a validation of having a high-quality operation, and research programs are willing to spend resources to obtain such accreditation. AAALAC accreditation demonstrates that an institution is going beyond the minimum required by law, achieving excellence in animal care and use (Bayne 1998).
Recently, the Association for the Accreditation of Human Research Protection Programs (AAHRPP) was incorporated to “provide a process of voluntary peer review and education among organizations concerned with research involving humans.” The overall goal of this accrediting organization is to improve protection of human research participants by developing “best practice” performance standards and by recognizing institutions that meet those standards. AAHRPP expects to begin accrediting institutions in the near future.8 The Department of Veterans Affairs (VA) requires all institutions involved in conducting research sponsored by the VA to be accredited (VA 2000), but it is the only federal agency that requires institutions or IRBs to become accredited. OHRP is also moving in the direction of accreditation. Commissioned by OHRP to conduct a study on accreditation and other human participant research issues, the Institute of Medicine issued a report on the use of accreditation programs and offered recommendation regarding standards (IOM 2001).
49
Chapter 3: Methods for Ensuring Protection: Education, Certification, and Accreditation
Moving toward accreditation and certification is a positive step in ensuring improved oversight of human research. As the proposed oversight system evolves to include accreditation and certification programs, flexibility in testing different models and methods of accreditation and certification should be allowed to ensure that when these programs become fully functioning they are sensitive to the needs of the system and responsive to the various entities seeking accreditation certification. For example, institutions that do not have IRBs but that are conducting research involving human participants should be eligible for accreditation because they have education, monitoring, and accountability responsibilities; and independent IRBs that might not perform all the functions of an institution conducting research also should be eligible for accreditation to carry out their more limited responsibilities.
Given the diversity among institutions that might seek accreditation and the fact that there are likely to be several accrediting organizations, the proposed federal oversight office should set criteria for government approval of accrediting bodies. In addition, the federal oversight office should develop a mechanism for monitoring accrediting bodies, and accrediting bodies, in turn, should be monitoring the institutions they accredit for continued competency. In the end, the office should be able to evaluate the effectiveness of accreditation.
Although the establishment of accreditation programs is likely to improve protections to human participants, such programs will be costly. In addition to the cost of accreditation itself, all institutions seeking accreditation will need to devote additional resources and staff to support new functions or strengthen existing functions. For example, some institutions might need to add educational programs or add monitoring functions. However, the potential improvements in the system justify these additional costs.
Recommendation 3.4: Sponsors, institutions, and independent Institutional Review Boards should be accredited in order to conduct or review research involving human participants. Accreditation should be premised upon demonstrated competency in core areas through accreditation programs that are approved by the federal government.
Mechanisms to Ensure Institutional, IRB, and Investigator Compliance
Assessing institutional, IRB, and investigator compliance can help to ensure that standards are being followed consistently. Current mechanisms for assessment include assurances of compliance issued by the Department of Health and Human Services (DHHS) and several other federal departments, site inspections of IRBs conducted by FDA, other types of site inspections, and institutional audits. In addition, some institutions have established ongoing mechanisms for assessing investigator compliance with regulations.9 However, institutions vary considerably in their efforts and abilities to monitor investigator compliance, from those having no monitoring programs to those conducting random audits.10
Institutional/IRB Compliance
Assurances of Compliance
Assurances of compliance (referred to as assurances) were developed in the late 1960s when the NIH Division of Research Grants was assigned responsibility for implementing the federal requirements for protecting human research participants. (See Appendix C.) Assurances were negotiated with each institutional grantee, with the negotiations allowing each institution to create its own policies and procedures for protection as long as they were fully consistent with federal regulations. The negotiation process also allowed federal officials to educate institutions about requirements and procedures for participant protection. Because the assurance indicated what an institution intended to do to protect research participants, it was essentially a pledge or commitment on behalf of the institution to comply with all appropriate regulations and guidance.
The National Commission believed that there should be uniform implementation of the federal regulations and recommended that each institution engaged in regulated research provide assurance to a single office that all research would be conducted in accordance with federal regulations (National Commission 1978). This perspective was reinforced by the President’s Commission, which suggested further steps to ensure that federal monitoring would be coordinated to minimize the bureaucratic burden imposed on institutions.
50
The President’s Commission argued that both assurances and on-site evaluations were needed to monitor implementation of the federal regulations and that a single office should coordinate such procedures for all regulated research (President’s Commission 1983, 133). A single office was never created. Instead, each federal department and agency may issue its own assurance, although many rely on DHHS assurances. (See Table 3.1.) The assurance process provided a means of identifying certain problems and correcting them before research was conducted. However, it provided only a limited indication of how an IRB would actually work. The value of the recommended site visits was the opportunity to learn
National Bioethics Advisory Commission
how IRBs were actually functioning and for institutions to receive feedback on their IRBs’ performance. However, to rely only on after-the-fact, “for-cause” site visits to determine whether an IRB met basic regulatory requirements is not the most effective strategy for ensuring that research protocols are being reviewed by a properly constituted and procedurally sound IRB (President’s Commission 1983).
Although DHHS and some other departments that adopted the Common Rule continue to issue assurances, site visits as envisioned by the President’s Commission were never implemented. In addition, for the most part, the negotiation process for assurances developed into a
Table 3.1: Federal Agency Practices Regarding Assurances of Compliance
| Agency | Issues Assurances | Relies on Other |
| of Compliance | Agency Assurances | |
| Central Intelligence Agency | No | No |
| Department of Commerce | ||
| National Telecommunications and Information Administration | No | No |
| Department of Health and | ||
| National Institute of Standards and Technology | No | |
| Human Services (DHHS) | ||
| Department of Defense | Yes | DHHS |
| Department of Education | Yes | DHHS |
| Department of Energy | Yes | DHHS |
| Department of Health and Human Services | Yes | No |
| Department of Housing and Urban Development | No | Not reported |
| Department of Justice | Some components | DHHS |
| Department of Transportation | No | DHHS |
| Department of Veterans Affairs | Yes | DHHS |
| National Aeronautics and Space Administration | Yes | DHHS |
| National Science Foundation | Yes | Yes |
| Social Security Administration | No | Yes |
| Agency for International Development | Yes | DHHS, Department of Defense |
| Consumer Product Safety Commission | Yes | DHHS |
| Environmental Protection Agency | Yes | DHHS |
Source: NBAC, “Federal Agency Survey of Policies and Procedures for the Protection of Human Subjects in Research.” This staff analysis is available in Volume II of this report.
51
Chapter 3: Methods for Ensuring Protection: Education, Certification, and Accreditation
routinized and somewhat tedious procedure. Standardized documents that mirror the federal regulations substitute for the independently negotiated assurances specific to the institution’s culture, policy, and procedures. The educational function of assurances was lost.11 Currently, few seem to view assurances as meaningful commitments or pledges by institutions to sustain a broad-based program of review and monitoring.12 Instead, they are viewed as unnecessary, additional bureaucratic paperwork with limited, if any, value in protecting research participants.13 Additional problems reported to NBAC were long delays in processing assurances (between 6 and 12 months in some cases), when the expectation is that OHRP should be able to process assurances in a much shorter period, confusion about which institutions required assurances, and OHRP’s review of consent forms submitted by institutions seeking an assurance when the same consent form was reviewed and approved by another collaborating institution with an MPA. Problems with the assurance process are not unique to domestic research. In the context of international research, the assurance process was most often criticized because it requires foreign institutions to follow rigidly U.S. requirements and procedures. The need for greater flexibility by the United States in conducting international research would allow for procedural variations that are responsive to local culture and practice while still ensuring compliance with substantive ethical principles (NBAC 2001).
OHRP revised the assurance process effective December 2000 and is currently testing the new procedures.14 The revised process entails the use of one Federalwide Assurance (FWA) document for domestic institutions and another assurance document for foreign institutions. Each legally separate institution must obtain its own FWA, and assurances approved under this process will cover all the institution’s federally supported research involving human participants. The domestic assurance replaces MPAs, Single Project Assurances, and Cooperative Project Assurances. Individual federal agencies can still choose whether to use the new assurance or issue their own. The assurance document is similar to previous assurance documents in content. However, institutions must meet additional requirements, such as
that certain institutional staff complete OHRP’s computerized educational training. The new assurance process is intended to reduce burden on institutions by allowing all institutions to qualify for the one FWA that may be renewed every three years. The revised process also includes registration of IRBs, regardless of whether they review research sponsored or regulated by a federal agency that follows the Common Rule.15 Modification to the revised system will be made based upon feedback and experience during the first few months of implementation.
FDA Site Inspections of IRBs
FDA conducts site inspections under its own regulations. In 1976, FDA developed its Bioresearch Monitoring Program in response to Congress’ instruction to expand its monitoring activity. The program encompasses monitoring of investigators, sponsors, program monitors (individuals selected by the sponsor to oversee the clinical investigation), and IRBs. The purpose of the Bioresearch Monitoring Program is to ensure the quality and integrity of data submitted to FDA to “demonstrate the safety and efficacy of regulated products, and to determine that human rights and the welfare of human and animal research subjects are adequately protected” (FDA 1998a). FDA conducts inspections of sponsors to determine how they ensure the validity of data submitted to them by investigators and to determine compliance of sponsors, contract research organizations, and monitors with applicable regulations (FDA 1998b). The purpose of FDA inspections of investigators is to help ensure compliance of investigators with the regulations (including the regulations to protect research participants) and to assess through audit procedures whether records substantiate data submitted to FDA (FDA 1998a). These inspections also include assessment of whether the investigator obtained appropriate IRB approval of research studies and whether participants have signed consent forms. However, FDA’s own findings for fiscal year 1998 show that for studies involving medical devices, over 50 percent of sponsors may not be monitoring trials adequately (OIG 2000). Most FDA inspections of investigators are conducted after the trial is complete. Thus, any detected violations of regulations to protect research participants are found after the point when participants in the particular trial could have received adequate
52
protections. However, the inspections are helpful in improving compliance of investigators and, therefore, protection of participants in future research. More recently, FDA’s inspection process has focused more on real-time complaints.16 FDA conducts surveillance (routine) and directed (when information “calls into question” regulated practices) inspections of IRBs. Usually IRB inspections are scheduled every five years, although if there are major problems, inspections can occur more frequently (FDA 1994). During an inspection, an FDA field investigator (inspector) chooses a few studies that received initial IRB review within the past three years and follows them through the IRB review process. Inspectors look at IRB policies and procedures; minutes; membership; and records of studies, including protocol, consent form, investigator’s brochure, and correspondence between the IRB and investigator. IRBs that are found to be out of compliance may be subjected to sanctions ranging from a warning letter to rejection of the data from the trial to prosecution (FDA 1994). FDA conducts approximately 300 IRB inspections annually as part of its Bioresearch Monitoring Program.17 FDA does not require its sponsors to provide assurances of compliance; however the agency does require investigators to provide a written commitment that, before initiating an investigation subject to an institutional review requirement under 21 CFR 56, an IRB will review and approve the investigation in accordance with 21 CFR 56 (21 CFR 312.53(c)(1)(vi)(d); 312.53(c)(1)(vii); 21 CFR 812.43(c)(4)(i)). The sponsor makes similar commitments (21 CFR 312.23(a)(1)(iv); 21 CFR 812.20(b)(6)).
Other Site Inspections
Some other federal departments also conduct site visits. For example, the Department of Energy (DOE) routinely conducts educational site visits at its contract facilities. A team of experienced IRB professionals reviews policies and procedures at individual facilities and makes recommendations for improving protections tailored to the operation at the local facility.18 The DOE human subjects program manager makes determinations of noncompliance.
The Department of Education authorizes its extramural research director to investigate allegations of noncompliance. At the National Science Foundation,
National Bioethics Advisory Commission
allegations of noncompliance are referred to the Office of Inspector General for investigation. The Department of Veterans Affairs uses several new mechanisms to monitor compliance. For example, the Multi-Assessment Program is a systematic, prospective approach to assist VA medical center research programs and investigators fulfill their responsibilities to conduct research so that there is adequate protection of human participants. The VA Office of Research Compliance and Assurance also uses Special Inquiry Force Teams to investigate allegations of research noncompliance, and if warranted, may initiate a Comprehensive Research Integrity Program review.
OHRP/OPRR also conducts both educational and “for-cause” site visits. In 1999, OPRR reported conducting “between zero and one not-for-cause site visits a year” to monitor IRB and institutional compliance with the federal regulations (Foubister 1999). In 1998–1999, OPRR’s number of “for-cause” site visits increased, along with suspensions of assurances for several prominent research institutions (Exhibit 3.1).
Institutional Audits
Institutions and IRBs should regularly review policies and procedures and monitor their implementation. Moreover, they should take steps to open their activities to greater public review and accountability. In a previous report, NBAC suggested that IRBs should make descriptions of their policies and procedures public and provide annual summary statistics regarding the nature and scope of the research approved. NBAC also suggested that institutions adopt internal audit procedures to assure themselves that their IRBs are following regulations (NBAC 1998). Institutions can also assume greater responsibility in ensuring that investigators comply with regulations and carry out protocols as approved by the IRB, as discussed below.
Assurances of compliance, site inspections, and institutional audits are all potentially useful mechanisms for improving institutional and IRB compliance. No one mechanism used in isolation of the others will be sufficient to monitor compliance. As certification and accreditation programs become more widespread, the utility of assurances and site inspections should be examined in order to avoid duplication of effort.
53
Chapter 3: Methods for Ensuring Protection: Education, Certification, and Accreditation
Exhibit 3.1: OPRR Compliance Oversight Investigations Resulting in Restrictions/Actions to MPAs, January 1990–June 2000
Year Institution and Action by OPRR
1990
1991
Massachusetts Eye and Ear Infirmary. Certain research was excluded under the existing MPA pending negotiation of a renewed MPA. New participant accruals were suspended pending re-review by appropriately convened IRB and notification of former participants about inadequate informed consent.
NIH. Certain collaborative research was excluded under the existing MPA. Designation of a signatory official with authority to ensure institutional human participant protections, modification of institutional policies and procedures to ensure IRB review of all human participant research, and development of an education program were required.
University of California - San Francisco. New participant accruals in all pediatric oncology research were suspended pending modification of informed consent documents and revised consent for all current pediatric oncology participants, and mechanisms to ensure regulatory compliance in such research were required.
State University of New York College of Optometry. Use of expedited IRB review procedures was suspended. Suspension of human participant involvement in certain research, re-review of research by an appropriately convened IRB, modification of IRB review and record keeping procedures, and designation of a signatory official with authority to ensure institutional human participant protections were required.
Florida State University. Use of expedited IRB review procedures was suspended. Re-review of research by an appropriately convened IRB, modification of IRB review, and record keeping procedures were required.
1992
1993 NIH. Progress reports on implementation of education program were required.
1994
University of California - Los Angeles. Schizophrenia research was excluded under the existing MPA. Modification of informed consent documents, information materials, and record keeping for schizophrenia research, the addition of participant representatives to the IRB, and the establishment of a Data and Safety Monitoring Board for psychiatric disorders research were required.
West Virginia University. Special monitoring of urology department research and modification of IRB review and record keeping procedures were required.
Memorial-Sloan Kettering Cancer Center. Special monitoring of melanoma immunization research, modification of IRB review and record keeping procedures, verification of completeness of informed consent documents, development of handbook of guidelines for investigators, and designation of a signatory official with authority to ensure institutional human participant protections were required.
Medical University of South Carolina. Review of research by appropriately convened IRB, modification of institutional mechanisms to ensure IRB review of human participant research, and development of an education program were required.
University of California - San Diego. Human participant involvement in certain research was suspended, and re-review of research by an appropriately convened IRB, modification of IRB review and record keeping procedures, and designation of a signatory official with authority to ensure institutional human participant protections were required.
University of Florida. Use of expedited IRB review procedures was suspended. Re-review of research by an appropriately convened IRB, modification of IRB review and record keeping procedures, and revision of IRB guidelines for investigators were required.
University of Minnesota. Research conducted by the Department of Surgery was excluded under the existing MPA, and human participant involvement in certain research was suspended. Re-review of research by an appropriately convened IRB, modification of IRB review, and record keeping procedures were required.
54
National Bioethics Advisory Commission
Exhibit 3.1 continued
1995
University of Virginia. Behavioral and educational research were excluded under the existing MPA, and human participant involvement in behavioral and educational research and certain medical sciences research was suspended. Re-review of research by appropriately convened IRBs, modification of IRB review and record keeping procedures, provision of an enhanced educational program and materials, review of staff support and resources for IRBs, designation of a signatory official with authority to ensure institutional human participant protections, and comprehensive review of IRB policies and procedures were required.
Centers for Disease Control and Prevention. Provision of updated information to measles research participants, development of conflict of interest guidelines for IRB members, review of staff support and resources for IRBs, development of an educational program, a proposal for a mechanism to ensure performance site assurances for international research, and a comprehensive review of IRB policies and procedures were required.
University of California - Los Angeles. Development of an educational program, revised IRB guidelines for investigators and the procedures manual, review of staff support and resources for IRBs, provision of a locked filing system and computerized tracking system to ensure confidentiality of IRB records, and development of mechanisms to ensure that all human participant research receives IRB review were required.
Center for Molecular Medicine and Immunology. Modification of IRB review and record keeping procedures were required.
Veterans Affairs Medical Center, West Los Angeles. Modification of IRB review and record keeping procedures were required.
Cook County Hospital Hektoen Institute for Medical Research. Participant enrollment in research not receiving valid initial or continuing IRB review and use of expedited review procedures were suspended. Revision of IRB review procedures was required.
Cornell University Medical Center. Enrollment in research not receiving adequate initial or continuing IRB review was suspended. Modification of IRB review and record keeping procedures, a plan for increased staffing, a plan for education of IRB members and investigators, and a mechanism for investigation of non-compliance were required.
University of Rochester. Enrollment in research not receiving appropriate initial or continuing IRB was suspended. Review and development of written operating procedures and an investigator handbook, a finalized organization structure of institutional human participant protections, and development of an education program for IRB members and investigators were required.
Wayne State University. Enrollment in research not receiving appropriate initial or continuing IRB review was suspended. Increased staffing and resources for the IRB, development of a mechanism for prompt review of adverse event reports, development of an educational program for IRB members and investigators, and review and revision of IRB policies and procedures were required.
City University of New York. Development of an educational program for IRB members and investigators, review and revision of IRB policies and procedures, revision of sample informed consent documents to comply with DHHS regulations, and revision of IRB record keeping procedures were required.
University of Maryland - Baltimore. Enrollment of participants in research not receiving appropriate initial or continuing IRB review was suspended. Review and revision of informed consent documents for psychiatric research to ensure compliance with DHHS regulations, development of an educational program for IRB members and investigators, and revision of IRB review procedures, including procedures for research involving vulnerable participant populations, were required.
University of California - Irvine. Modification of initial review process to ensure compliance with regulations, revision of continuing review and oversight procedures, increased documentation of IRB actions in accordance with regulations, enhanced education for investigators, IRB members, and staff, and increased support for social and behavioral sciences IRB were required.
Western Carolina Center. Due to limited DHHS support to the Western Carolina Center, Single Project Assurances (SPAs) were required for DHHS-supported human participant research, and the MPA was deactivated on October 30, 1998.
55
1996
1997
1998
Chapter 3: Methods for Ensuring Protection: Education, Certification, and Accreditation
Exhibit 3.1 continued
Rush Presbyterian St. Lukes Medical Center. OPRR suspended the MPA for five days pending development of adequate corrective action plans. The MPA was subsequently reinstated with restrictions. OPRR required correction of 17 identified deficiencies in systemic human participant protections, restructuring of systemic human participant protections including changes in leadership, enhanced institutional commitment, and expanded IRB membership, development of an education program for IRB members, IRB staff, and research investigators, suspension of new enrollments in all federally supported research pending review of all such protocols by the reconstituted, convened IRB, and suspension of IRB review of federally supported research by expedited review procedures.
Scripps Clinic and Research Foundation:The Scripps Research Institute. OPRR required correction of 20 identified deficiencies in systemic human participant protections, re-review of all DHHS-supported human participant protocols to include review of complete grant application, development of an educational program for IRB members, IRB staff, and research investigators, and quarterly progress reports.
Duke University. OPRR restricted the MPA and required several corrective actions.
Duke University Medical Center. OPRR restricted the Duke University Medical Center MPA and required several corrective actions.
1999
Friends Research Institute. OPRR removed from coverage under the existing MPA any performance sites outside the Maryland area, removed recognition of one IRB under the MPA, and withdrew approval of all inter-institutional and cooperative amendments to the MPA.
Mt. Sinai School Of Medicine. OPRR restricted the MPA and required satisfactory implementation of a series of corrective action plans.
Veterans Affairs, Greater Los Angeles Health Care System (Formerly Veterans Affairs Medical Center,West Los Angeles). OPRR deactivated the MPA on March 22, 1999. Enrollment of new participants in all federally supported research was suspended, and involvement of previously enrolled participants was allowed only when it was in the best interest of the participant.
Fordham University. Due to the limited number of DHHS-supported projects at Fordham University and persistent concerns about compliance with human participant protections requirements, SPAs were required for all DHHS-supported human participant research, and the MPA was deactivated on May 31, 1999. Duke University Medical Center. OPRR suspended the MPA for five days. OPRR reinstated the Duke University Medical Center MPA with restrictions and required several corrective actions, including re-review of DHHS-supported research by the IRB, implementation of a second IRB, and implementation of appropriate education programs for IRB members, IRB staff, and all investigators.
University of Illinois at Chicago. OPRR restricted the MPA and required corrective actions in response to 29 identified deficiencies, including revision of the MPA to indicate enhanced institutional commitment to the protection of human participants and development of an educational program for IRB members, IRB staff, and research investigators. New enrollments in all federally supported research pending re-review of all such protocols were suspended.
St. Jude Children’s Research Hospital. OPRR restricted the MPA and required submission of a progress report describing implementation of all corrective actions three months after the site visit.
Virginia Commonwealth University. OPRR restricted the MPA and required corrective actions, including development of an educational program for IRB members, IRB staff, and research investigators and submission of a list of all active IRB-approved protocols. New participant enrollments in all federally supported research pending re-review of all such protocols were suspended.
Virginia Commonwealth University. OPRR suspended the MPA and required revised corrective action plans, including designation of a new signatory official and new IRB chairpersons.
University of Alabama at Birmingham. OPRR restricted the MPA and required suspension of new participant enrollments in all federally supported research that had not had appropriate initial or continuing review. Submission of corrective action plans to address all deficiencies and concerns related to systemic human participant protections and submission of an educational plan for all IRB members, all IRB staff, and all research investigators were required.
2000
Adapted from Compliance Oversight Investigations Resulting in Restrictions/Actions to Multiple Project Assurances, 1/90–11/99 (OPRR 2000).
56
Monitoring Investigator Compliance with IRB-Approved Protocols: Verification
Institutional monitoring to verify that investigators are following IRB-approved protocols is an important means of determining whether investigators are actively meeting their ethical obligations to protect research participants while a study is in progress. Such verification should include activities such as observing research, especially the enrollment of participants, auditing research records, and establishing clear procedures for receiving complaints or concerns from participants or staff.
Some argue that as collegial review bodies, IRBs should not question the information provided by investigators.19 On the other hand, others have argued that IRBs should develop mechanisms for continuing review of research that include verification of the investigator’s conduct of the study (Weijer et al. 1995). There are cases in which investigators’ deviations from IRB-approved protocols have affected participant protections (OIG 1995). In one instance, a case of financial fraud by two clinical investigators may also have affected the welfare of research participants (Teegardin and Whitt 1997). Failure to follow IRB-approved protocols may not be limited to serious abuses. One recent survey of clinical research professionals found that as many as 40 percent had encountered cases of questionable consent, and many had encountered cases of inappropriate alteration of study eligibility criteria, alteration of care provided to participants, and breach of confidentiality (Shefrin and Harper 2000).
IRB review alone does not address these types of problems. Although IRBs are not and should not be the research police, protecting participants will sometimes require IRBs to verify at least on a sample basis the conduct of approved research studies. IRBs are not fully meeting their responsibilities when they make detailed changes to the language in a consent form, for example, but have no way to verify how the consent form is discussed with participants.
Under current regulations, IRBs have the authority “to observe or have a third party observe the consent process and the research” (45 CFR 46.109(e); 21 CFR 56.109(f) [FDA regulations differ in reference to regulation]), and institutions are required to have procedures for reporting
National Bioethics Advisory Commission
on “serious or continuing noncompliance with this policy or the requirements or determinations of the IRB” (45 CFR 46.103(b)(5); 21 CFR 56.108(b)(2) [FDA regulations differ in reference to regulation]). Few IRBs carry out this type of monitoring, because they do not have sufficient resources.20 The desire for such verification is not new. In its report on IRBs, the National Commission intended for IRBs to observe investigators and hear directly from research participants: “At the discretion of the IRB, the consent process or the research itself may be observed on a sample or routine basis, subjects may be interviewed about their experience in research, and research records (including consent forms) may be reviewed. Also at the discretion of the IRB, investigators may be required to provide subjects with a form on which they can report to the IRB their experiences in research…” (National Commission 1978, 17). Moreover, noting the difficulty of such observation, the National Commission made it clear that these processes were not supererogatory on the part of IRBs; for at least some research studies, observing the conduct of research to ensure the protection of the research participants was an “obligation” (National Commission 1978, 18).
More recently, the DHHS Office of Inspector General (OIG) criticized IRBs’ “minimal field presence…[for] as a result, IRBs typically have little basis to know for themselves how research teams approach the informed consent process, how well human subjects understand the implications of their participation in research, and how fully teams remain true to the research design set forth in their approved protocols” (OIG 1998b, 8). Nevertheless, there are cases in which IRBs have developed systems for monitoring compliance. For example, one IRB hired an intermediary—a person who observes the consent process in psychiatric research studies. Another IRB randomly selects studies from a list of approved high-risk research and audits research records, concentrating, however, primarily on consent forms.21 The President’s Commission also noted the need for verification and the difficulty it can present for IRBs. It suggested that institutions, rather than IRBs, develop mechanisms for receiving complaints and conducting investigations. As articulated by the President’s
57
Chapter 3: Methods for Ensuring Protection: Education, Certification, and Accreditation
Commission, “the duty ought to be that of the institution itself…but need not necessarily be lodged with the IRB” (President’s Commission 1981, 80). Institutions could choose to delegate these functions to an IRB or to an entity other than the IRB. The results of all investigations, however, should be reported to the IRB.
Another advantage to having someone other than the IRB monitor investigator compliance is that investigators may be reluctant to ask informal questions of the IRB if there is a chance that a question may trigger closer monitoring of a research study. Likewise, IRBs also hesitate to seek advice from OHRP/OPRR on difficult cases, because such an inquiry might trigger an investigation.22 Assessing compliance of investigators is an important part of protecting research participants and should be taken seriously as a responsibility of each institution. Although a number of practical issues arise in monitoring investigators while they conduct their research studies, investigators, IRBs, and institutions should discuss these issues and provide input into the regulatory process.
Recommendation 3.5: The process for assuring compliance with federal policy should be modified to reduce any unnecessary burden on institutions conducting research and to register institutions and Institutional Review Boards with the federal government. The assurance process should not be duplicative of accreditation programs for institutions (see Recommendation 3.4).
Recommendation 3.6: Institutions should develop internal mechanisms to ensure Institutional Review Board compliance and investigator compliance with regulations, guidance, and institutional procedures. Mechanisms should be put in place for reporting noncompliance to all relevant parties.
Conflicts of Interest
Conflicts of Interest Involving Investigators
A research setting that involves human participants necessarily creates a conflict of interest for investigators who seek to develop or revise knowledge by using individuals to obtain that knowledge. In the words of the National Commission:
The Commission’s deliberations begin with the premise that investigators should not have sole responsibility for determining whether research involving human subjects fulfills ethical standards. Others, who are independent of the research, must share this responsibility, because investigators are always in positions of potential conflict by virtue of their concern with the pursuit of knowledge as well as the welfare of the human subjects of their research (National Commission 1978, 1).
Overzealous pursuit of scientific results could lead to harm if, for example, investigators design research studies that pose unacceptable risks to participants, enroll participants who should not be enrolled, or continue studies even when results suggest they should have been modified or stopped. In addition, decisions about study design, inclusion criteria, and adverse events require judgment and discretion on the part of the investigators, who can be influenced, consciously or unconsciously, by conflicting interests. Thus, it is important to address prospectively the potentially harmful effects on participants that conflicts of interest might cause (Emanuel and Steiner 1995; Lo 2000).
IRB review of research studies is one method for identifying and dealing with conflicts of interest that might face investigators. By having IRBs review research studies prospectively and following an IRB-approved protocol, investigators and IRBs together can manage conflict between the investigators’ desire to advance scientific knowledge and the need to protect the rights and welfare of research participants. IRBs should, for example, determine whether investigators are being paid for enrolling participants in a research study or whether they or their family members have a proprietary interest in the outcome of the research. Steps to strengthen the IRB oversight system should reduce the probability that investigators’ conflicts of interest will cause harm to research participants.
It is worth exploring the problems that result when successful research creates benefits, such as prestige, power, and profit, for those who conduct the research studies. Although conflicting interests associated with obtaining promotion and tenure have always been present for academic investigators, recent changes in the research enterprise increasingly have created opportunities for
58
investigators and institutions to profit financially from research (Steiner 1996). Investigators’ financial conflicts of interest include capitated payments or bonuses for enrolling participants, indirect payments through consul-tantships or honoraria, and equity holdings in companies or royalties from patents whose value may be affected by the research. Institutions’ conflicts of interest also include equity holdings in companies and the economic benefits of patents they hold.
As a result, concern is growing that financial conflicts of interest may compromise scientific objectivity and the welfare of research participants, ultimately leading to decreased public trust in the research enterprise (Bodenheimer 2000; Shalala 2000). The factors contributing to these financial conflicts (e.g., erosion of the academic medical center’s revenue base, encouragement by government to transfer technology into the marketplace, and industry’s increasing financial support of research) are unlikely to wane. In fact, these pressures on investigators and institutions are likely to increase.
On the surface, financial conflicts are tangible and extraneous and therefore might be more easily managed or eliminated than some other conflicts (Angell 2000a; Angell 2000b). However, simply prohibiting financial conflicts may not be feasible in an environment in which private funding of research and new strategies for transferring technology to the marketplace are being encouraged (Emanuel and Steiner 1995; Korn 2000). Thus, managing financial conflicts of interest will remain an important part of preserving the public’s trust in research.
IRB review alone, however, is not sufficient to manage financial conflicts, because the options available to IRBs to eliminate such conflicts are limited. IRBs can disapprove a research study in which the financial conflicts of interest of the investigator are perceived to be too high, or IRBs can require that study design or methods be altered, but they cannot dictate directly the conduct of investigators concerning their financial interests. Moreover, although financial conflicts of interest raise concerns about bias and the reliability of research results (Bodenheimer 2000), attention to these concerns should not rest solely on IRB review. Thus, the responsibility for the review of investigators’ conflicts should also lie with sponsors and institutions.
National Bioethics Advisory Commission
Some have proposed disclosure of conflicts of interest to institutions (AAMC 1990; AMA 2000), IRBs (DeRenzo 2000), and participants (Cohen 2000) and subsequent management of these disclosed interests as a mechanism for dealing with financial conflicts (AAMC 1990). Although disclosure might encourage investigators to think carefully before agreeing to arrangements that pose conflicts or might provide others, such as institutional officials, an opportunity to assess the risks and potential benefits of financial arrangements (Blumenthal 1992), it is not a complete solution (Lo 2000; Thompson 1993).
Organizations, particularly academic institutions, should become more actively involved in managing investigators’ financial conflicts and should increase their self-regulation efforts in this area. Several recently published articles indicate that policies governing conflicts of interest at major academic research institutions vary widely and often lack basic safeguards, such as providing clear guidance regarding the types of relationships that are prohibited (Cho et al. 2000; Lo et al. 2000; McCrary et al. 2000). Federal regulations already require recipients of certain federal funds to establish policies for disclosure, reduction, management, and elimination of financial conflicts of interest (42 CFR 50.601–50.607). As a first step, the regulations and the institutional policies responding to them could be improved (Cho et al. 2000; Cohen 2000). FDA also requires financial disclosure by investigators as part of its review of marketing applications (21 CFR 54).
Some financial conflicts may be inappropriate regardless of the amount of oversight provided either by the institution or the IRB. For example, certain per-capita bonuses are designed solely to provide an incentive to recruit participants; when such payments are excessive they serve only to provide an inappropriately strong incentive to investigators or referring physicians (Lind 1990; Wolf and Lo 2000). Or, in a clinical trial, it might be unacceptable for investigators to own stock or options in the company manufacturing the product being tested, because their remuneration might be greatly influenced by whether the trial results are favorable or unfavorable (Healy et al. 1989; Lo et al. 2000; Topol et al. 1992). The concern would be greatest in a start-up company that has not yet made a public offering. However, the lack of
59
Chapter 3: Methods for Ensuring Protection: Education, Certification, and Accreditation
guidance and policy makes it difficult for individual institutions to set strict standards, especially ones that include the prohibition of certain types of financial relationships. Institutions should work with professional societies and sponsors to clarify types of conflicts and develop mechanisms for managing such conflicts through education and oversight.
IRBs should be aware of investigators’ financial arrangements with respect to research under review (DeRenzo 2000). Most academic medical centers, however, require investigators to disclose financial conflicts of interest to a university official or to a committee, but not to the IRB (Lo et al. 2000; McCrary et al. 2000). Payments to the investigator should not, for example, affect an IRB’s evaluation of the risks and potential benefits of a research study or the equitable selection of participants. Rather, the presence of financial conflicts of interest might affect an IRB’s assessment of the protocol in its entirety and whether the research should be approved, or it might affect its assessment with respect to the amount or type of monitoring needed (AAMC 1990; Gottlieb 2000).
One area in which an IRB must be involved is determining what information about financial conflicts of interest should be shared with research participants as part of the informed consent process. Recently, the Association of American Medical Colleges added its support to the policy of disclosing of investigators’ financial conflicts to research participants (Cohen 2000). A number of concerns surround the issue of disclosure, primarily related to the privacy of investigators and the relevance and understandability of the information to participants. Participants clearly need to understand the nature of the research study in which they are participating, including who is likely to benefit from it. Such information may be relevant to an individual who is deciding whether to participate in a research project.23 However, disclosure to participants should not serve as a substitute for the institutional management of conflicts of interest. Disclosure, although necessary, is not sufficient for managing and resolving these issues.
Conflicts of Interest Involving IRBs
Independent review is the primary mechanism for managing conflicts of interest in research, and assuring the independence of IRBs has been a matter of concern since their inception. The federal regulations require two types of independence for IRBs. First, no IRB member should participate in the review of “any project in which the member has a conflicting interest” (45 CFR 46.107(e); 21 CFR 56.107(e)). Second, to prevent the IRB from representing solely an institutional viewpoint, at least one IRB member must be unaffiliated with the institution (45 CFR 46.107(d); 21 CFR 56.107(d)). These two types of independence specified in the federal regulations reflect concerns about the personal conflicts of individual IRB members and the conflicts an IRB may have as part of the institution.
Individual IRB Members
Conflicts of interest for individual IRB members are currently addressed in the federal regulations by a requirement that “no IRB may have a member participate in the IRB’s initial or continuing review of any project in which the member has a conflicting interest, except to provide information requested by the IRB” (45 CFR 46.107(e); 21 CFR 56.107(e)). The most obvious case of conflict would occur when an IRB member reviews a protocol for which he or she is an investigator. However, the regulations are often interpreted to include conflicts that involve finances, collaborations, or even personal relations. NBAC supports this more inclusive interpretation.
Financial and other obvious conflicts of interest for IRB members, such as collaboration in a research study, are often less difficult to identify and manage than some of the more subtle and pervasive conflicts (Francis 1996). Subtle conflicts are often not detected. For example, IRB members may have intellectual or practical commitments related to a research study with which they are not directly involved. IRB members may also have professional or personal connections to the investigators whose research they review. Although familiarity with the research was an intended part of the IRB system, it can also cause IRBs to become enmeshed in institutional, interdepartment, or interpersonal interests. Thus, a trade-off might occur on IRBs between the level of
60
expertise of its members and their independence. In some cases, IRB members may fear that an unfavorable review might result in personal repercussions. Many IRBs try to keep board discussions confidential in order to protect individual members, but some IRB members have expressed discomfort in reviewing the work of direct supervisors, for example.
Resolving these tensions is not always easy. In cases of clear, strong conflict (e.g., an IRB member’s participation in a research study as an investigator), the member with the conflict can recuse him or herself from the review. However, in cases of weaker conflict (e.g., close working relationship with the investigator; particular academic or other commitments, but no actual involvement in the study), disclosure of potential biases to the IRB should be required and should be sufficient. The IRB should be confident that each member voting on a specific protocol provides a knowledgeable, independent assessment of the research under review.
Guidance should be developed to assist IRBs in identifying various types of conflict. In addition, IRBs can reduce conflicts that employees of the institution may have by increasing the number of members who are not otherwise affiliated with the institution and the number of members who are nonscientists and are more likely to represent the views of the prospective participants rather than the investigators.
The IRB as an Institutional Body
IRBs are institutional entities (Annas 1991), a feature that was intended to reflect an institution’s commitment to safe, ethically sound research. An institution designates an IRB and gives it authority to review research, although the institution retains the power to disapprove research that has been approved by an IRB (45 CFR 46.112; 21 CFR 56.112). This structure applies even when an institution relies on an independent IRB or an IRB at another institution. The IRB’s authority still derives from the institution that has delegated the power of review. Because most institutions have an understandable desire to increase their research activities, institutional IRBs may, themselves, face conflicts of interest. And, even independent IRBs have a strong incentive to consider the interests of their institutional contractors. As Leslie
National Bioethics Advisory Commission
Francis notes: “Consistently unfriendly reviews might be thought to threaten ongoing relationships between IRBs and the institution [sic] with which they contract” (Francis 1996, 424).
Some have suggested that the primary function of IRBs has shifted from protecting the participant to protecting the institution (Annas 1991). In general, institutional interests in protecting themselves from liability, bad publicity, and regulatory sanctions are consistent with an interest in the protection of research participants. However, institutions can have other interests that conflict with the goal of participant protection, and such conflicts might increase when institutions hold equity in companies whose products are tested in research by their investigators or when they have patent licensing arrangements whose value may be affected by specific studies. Institutions therefore aim to maintain a research-friendly environment. Institutions can reduce such conflicts by divesting questionable holdings, by not conducting research that may affect those investments, or by conducting research that affects those investments only as part of a multi-site trial or with external monitoring (Emanuel and Steiner 1995). However, the primary strategy for limiting the influence of inappropriate institutional interests on IRBs is to impose requirements on IRB membership. For an IRB, being research friendly can mean not only being efficient in handling paperwork, but also being less stringent in its review than it should be. An IRB that disapproves protocols, that often requires significant modifications, that requires investigators to provide frequent and detailed information on research studies in progress, or that conducts frequent continuing review might be perceived as overly intrusive or bothersome.
The involvement of particular institutional staff on IRBs has also been questioned. Recently, OHRP suspended assurances at some institutions where institutional grants and contracts officials served as IRB members. OHRP argued that staff responsible for bringing research funds into an institution have a fundamental conflict of interest when they serve on the IRB (OHRP 2000). Such strategies challenge the balance between functioning independently and having members who are able to provide relevant information about investigators or their research. Institutional counsel is another example of a potential
61
Chapter 3: Methods for Ensuring Protection: Education, Certification, and Accreditation
source of conflict. An institution’s attorney is generally concerned with protecting the institution from liability and might require that certain language be included in the consent form to protect the institution, even though the language might not be understandable to prospective participants.
IRB membership is one mechanism for reducing institutional conflicts of interest. Current federal regulations require that each IRB have “at least one member who is not otherwise affiliated with the institution and who is not part of the immediate family of a person who is affiliated with the institution” (45 CFR 46.107(d); 21 CFR 56.107(d)). Interpreting the definition of not otherwise affiliated literally can result in only a small degree of separation between the individual and the institution. For example, ex-employees are sometimes chosen to serve as the unaffiliated member. Moreover, unaffiliated is often interpreted as “representing the community or participants,” which is incorrect, because unaffiliated members can be scientists; other professionals, such as lawyers, business people, or clergy; or individuals who represent participants. The only characteristic that unaffiliated members share other than participating on the IRB is lack of ties to the institution.
Concerns have been raised regarding whether having only one unaffiliated member on an IRB is sufficient to avoid institutional influence, especially when IRBs have 15 to 21 members on average.24 One or two “outsiders” might hesitate to mention concerns or challenge the group (McNeill 1993), and unaffiliated members do not need to be present in order for the IRB to conduct review and approve research studies.25 Thus, IRBs can approve research with only institutional representation present as long as a nonscientist and a quorum are also present.
Others have proposed requiring the presence and participation of more unaffiliated IRB members to reduce the influence of institutional interests on IRB decision-making (OIG 1998a). However, finding appropriate unaffiliated members who are willing to serve on an IRB can be difficult. Paying unaffiliated members for their efforts, as originally proposed by the National Commission (National Commission 1978, 15) might improve the yield, but excessive compensation could call their independence into question. Currently, no rules or
guidance are available describing criteria to meet the definition of unaffiliated, how long such members should serve, under what circumstances they may be removed, or what payment they should be provided. Institutions should be careful to select unaffiliated members who are truly separated from the institution, except for their role on the IRB. Procedures for the selection and removal of unaffiliated members should be established in a way that empowers the independent voices of those members, and providing reasonable payment to IRB members who are otherwise unaffiliated with the institution can be a valuable way to strengthen their role.
Another strategy for reducing institutional influence on an IRB is to use a noninstitutional IRB. These IRBs review research studies for a number of institutions and are, therefore, presumed to be less likely to be influenced by any one institution. It is claimed that one of the advantages of this system is the almost complete elimination of influence by individual institutional interests.26 However, others have argued that paying IRBs for review of research, as the United States does when noninstitu-tional (independent) IRBs are used, constitutes a conflict of interest.27 It appears that neither system of review—institutionally based IRBs or fee-for-service independent IRBs—is free from all real or perceived conflicts of interests. However, it does seem that institutionally based IRBs can reduce conflicts of interest by including more members who are not affiliated with the institution. Independent IRBs, like institutions, can also reduce conflicts by separating business functions from review functions and by using agreements that clearly describe what is covered by the fee (i.e., the fee is for review only and clearly does not guarantee approval).
Another way to limit the influence of an institution’s research interests on an IRB is to ensure that the views of both scientists and nonscientists are considered. Current regulations require that each IRB include “at least one member whose primary concerns are in scientific areas and at least one member whose primary concerns are in non-scientific areas.” A scientist is defined by OHRP as a person who has “substantive training or experience in a scientific discipline or in the scientific method.”28 Thus, physicians, nurses, or individuals trained at the bachelor’s,
62
master’s, or doctoral level in the basic sciences or social sciences are all considered scientists. Unlike the requirement for unaffiliated members, at least one nonscientist must be present for an IRB to review research (45 CFR 46.107(c); 21 CFR 56.107(c)).
As with unaffiliated members, it is sometimes assumed that nonscientist members represent the views of the participants. However, nonscientists might or might not be members from the community of participants. Professionals who are nonscientists might be no more able to represent the view of low-income or seriously ill participants, for example, than scientists.
From an ethical perspective, balancing the views of scientists and nonscientists is important, because although IRBs must be technically competent to identify and quantify the risks and potential benefits of a protocol, they must also be able to put those risks and benefits into a social context in order to judge how ordinary citizens would value them. This kind of assessment requires the input of nonscientists because “experts have a unique commitment to research that favours the interests of science” (McNeill 1993, 184). This is not to suggest that scientists and nonscientists have different roles on an IRB, although nonscientists often do tend to focus on the adequacy of consent documents (Porter 1987). It is important to remember that all IRB members should evaluate protocols according to established ethical principles and the federal regulations. It is also important to remember that they cannot avoid doing so from different perspectives and with different emphases.
The current IRB system requires that unaffiliated and nonscientist members serve on these groups. Although each brings valuable experience, knowledge, and insight to the IRB, neither may reflect the views of the research participants. For this reason, IRBs should include members who are specifically chosen because they represent participants’ interests.
IRBs should strive to complement their membership with members who are clearly unaffiliated with the institutions, members who are nonscientists, and members who represent the perspectives of participants. Federal policy should specify membership and quorum require-
National Bioethics Advisory Commission
ments for each of these categories. Individuals who meet the membership criteria in more than one of these three categories should be able to fulfill the requirements for each. For example, a member of the clergy can fulfill both the nonscientist and unaffiliated membership requirements. NBAC recommends that an IRB have collectively at least 25 percent of its membership comprised of individuals representing these three categories.
Other IRB Composition Issues
Scientific or discipline expertise on an IRB is clearly needed to judge the risks and potential benefits of the research protocols it reviews. However, scientific expertise is to a large extent discipline specific, meaning that it is essential that at least some IRB members have expertise and experience that is directly related to the research being reviewed. Indeed, federal regulations require that IRBs possess “the professional competence necessary to review specific research activities” (45 CFR 46.107; 21 CFR 56.107). NBAC heard repeatedly from investigators conducting research in the social sciences and humanities that IRBs with primarily medical expertise sometimes impose unreasonable, and even irrational, requirements that are inappropriate for nonbiomedical research (AAUP 2000). Although ethical principles for review should be consistent for all research, factors such as standards for research design, participants’ expectations of privacy and confidentiality, and the appropriate consent processes to be used can, and often should, vary by type of research. Appropriate expertise and experience are essential both for the protection of participants and the promotion of ethical research.
At the same time, however, IRBs are intended to be interdisciplinary and may not “consist entirely of members of one profession” (45 CFR 46.107(b); 21 CFR 56.107(b)). Thus, an appropriate balance must be found, which for large institutions might mean multiple IRBs with specific areas of expertise and which for smaller institutions might mean one IRB with a varied membership and a roster of consultants. In either case, institutions should have discretion in determining the most appropriate arrangement.
63
Chapter 3: Methods for Ensuring Protection: Education, Certification, and Accreditation
Recommendation 3.7: Federal policy should define institutional, Institutional Review Board, and investigator conflicts of interest, and guidance should be issued to ensure that the rights and welfare of research participants are protected.
Recommendation 3.8: Sponsors and institutions should develop policies and mechanisms to identify and manage all types of institutional, Institutional Review Board, and investigator conflicts of interest. In particular, all relevant conflicts of interest should be disclosed to participants. Policies also should describe specific types of prohibited relationships.
Recommendation 3.9: Federal policy should establish standards and criteria for the selection of Institutional Review Board members. The distribution of Institutional Review Board members with relevant expertise and experience should be commensurate with the types of research reviewed by the Institutional Review Board (see Recommendation 3.10).
Recommendation 3.10: Institutional Review Boards should include members who represent the perspectives of participants, members who are unaffiliated with the institution, and members whose primary concerns are in nonscientific areas. An individual can fulfill one, two, or all three of these categories. For the purposes of both overall membership and quorum determinations 1) these persons should collectively represent at least 25 percent of the Institutional Review Board membership and 2) members from all of these categories should be represented each time an Institutional Review Board meets (see Recommendation 3.9).
Summary
This chapter discussed the important role that education plays in protecting research participants. Individuals should be able to demonstrate that they understand their ethical obligations and how to carry them out. To help investigators and IRBs fulfill their responsibilities, the federal government should promote the development of education and certification programs that apply to all investigators as well as all IRB members and staff.
Institutions and independent IRBs must do a better job of meeting their ethical obligations to research participants. Accreditation programs are one way in which all organizations involved in conducting or reviewing research can develop highly efficient and effective protection programs.
A key protection is that of ensuring institutional, IRB, and investigator compliance. At all levels in the system, various mechanisms, including assurances of compliance, site inspections, and internal audits, should be used. Regular periodical monitoring of compliance is the most likely way to detect and remedy problems.
Institutions and IRBs increasingly are confronting conflicts of interest as more investigators and institutions enter into financial arrangements in which they stand to benefit from the results of their research. Conflicts of interest are not new, but they have changed and intensified as the research enterprise has evolved. Disclosure and management of investigator and institution conflicts still seem to be the best strategies. Conflicts affecting the IRB can be handled by increasing the percentage of members who do not have any direct interests in the institution or its research program (unaffiliated members). Increasing the percentage of nonscientists and members who represent the views of participants can also reduce conflicts. Ultimately, clear policies and guidance will provide the best way for IRBs to fulfill their responsibilities and meet their obligations in educating their members, monitoring for compliance, and avoiding conflicts of interest.
Notes
1 NIH Guide for Grants and Contracts, “Short Term Courses in Research Ethics.” PA-99-051. January 22, 1999; NIH Guide for Grants and Contracts, “Mentored Scientist Development Award in Research Ethics.” PAR-98-006. November 7, 1997. Both available at http://grants.nih.gov/grants/guide. Last accessed December 4, 2000.
2 PRIM&R, “IRB 101 On the Road.” Available at http://www.primr.org/ 101road.html. Last accessed December 4, 2000.
3 NIH Guide for Grants and Contracts, “Required Education in the Protection of Human Research Participants.” Notice OD-00-039. June 5, 2000. Available at http://grants.nih.gov/grants/guide. Last accessed December 4, 2000.
4 See Peckman, S., “Local Institutional Review Boards.” This background paper was prepared for NBAC and is available in Volume II of this report.
64
5 Association of American Medical Colleges Medical School Graduation (GQ) Questionnaire, 1980–1996.
6 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
7 See http://www.ptcny.com/PDF/ccip2000.pdf. Last accessed May 1, 2001.
8 See http://www.primr.org/aahrpp.html. Last accessed May 1, 2001.
9 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
10 Ibid.
11 See McCarthy, C.R., “Reflections on the Organizational Locus of the Office for Protection from Research Risks.” This background paper was prepared for NBAC and is available in Volume II of this report.
12 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
13 Ibid.
14 OHRP Guidance: “Procedures for Registering Institutional Review Boards and Filing Federal Wide Assurances of Protection for Human Subjects.” December 3, 2000. Available at http://ohrp.osophs.dhhs.gov/irbasur.htm. Last accessed December 8, 2000.
15 See http://ohrp.osophs.dhhs.gov/humansubjects/assurance/faq.htm.
16 Letter from D.A. Lepay, Food and Drug Administration, to E.M. Meslin, NBAC. February 20, 2001. Providing comments on a draft of this report.
17 Lepay, D., 2000, FDA’s Program for Human Subject Protection. 16 October. Presentation to ORCA Teleconference.
18 NBAC, “Federal Agency Survey on Policies and Procedures for the Protection of Human Subjects in Research.” This staff analysis is available in Volume II of this report.
19 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
20 Ibid.
21 NBAC Town Meeting: April 5, 2000, Pittsburgh, Pennsylvania.
22 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
National Bioethics Advisory Commission
23 Moore v. The Regents of the University of California et al.
793 P.2d 479. (Cal. 1990).
24 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
25 39 Fed. Reg. 18915.
26 See Holm, S., “The Danish Research Ethics Committee System—Overview and Critical Assessment”; Chalmers, D., “Research Ethics in Australia”; and Heath, E., “The History,
Function, and Future of Independent Institutional Review Boards.” These background papers were prepared for NBAC and are available in Volume II of this report.
27 See Heath, E., “The History, Function, and Future of
Independent Institutional Review Boards.” This background paper was prepared for NBAC and is available in Volume II of this report.
28 “Instructions for Registering an IRB or IEC.” Available at http://OHRP.osophs.dhhs.gov. Last accessed March 7, 2001.
References
Advisory Committee on Human Radiation Experiments (ACHRE). 1995. Final Report. Washington, D.C.: U.S. Government Printing Office.
American Association for the Advancement of Science (AAAS). Project 2061. 1993. Benchmarks for Science Literacy. New York: Oxford University Press.
American Association of University Professors (AAUP). 2000.
Protecting Human Beings: Institutional Review Boards and Social Science Research. Available at http://www.aaup.org/irbdoc.htm. Last accessed April 30, 2001.
American Medical Association (AMA). Council on Ethical and Judicial Affairs. 2000. “Conflicts of Interest: Biomedical Research.” Opinion 8.031. Updated June 1999. In Code of Medical Ethics: Current Opinions with Annotations. Chicago, IL: AMA.
Angell, M. 2000a. “Is Academic Medicine for Sale?” [Editorial.]
New England Journal of Medicine 342(20):1516–1518.
———. 2000b. “Reply to Letters.” New England Journal of Medicine
343(7):508–510.
Annas, G.J. 1991. “Ethics Committees: From Ethical Comfort to Ethical Cover.” Hastings Center Report 21(3):18–21.
Association of American Medical Colleges (AAMC). 1990.
Guidelines for Dealing with Faculty Conflicts of Commitment and Conflicts of Interest in Research. Washington, D.C.: AAMC.
Bayne, K. 1998. “Developing Guidelines on the Care and Use of Animals.” Annals of the New York Academy of Sciences 862:105–110.
65
Chapter 3: Methods for Ensuring Protection: Education, Certification, and Accreditation
Bell, J., J. Whiton, and S. Connelly. 1998. Final Report: Evaluation of NIH Implementation of Section 491 of the Public Health Service Act, Mandating a Program of Protection for Research Subjects.
Contract N01-OD-2-2109. Arlington, VA: James Bell Associates.
Blumenthal, D. 1992. “Academic-Industry Relationships in the Life Sciences.” Journal of the American Medical Association
268(23):3344–3349.
Bodenheimer, T. 2000. “Uneasy Alliance—Clinical Investigators and the Pharmaceutical Industry.” New England Journal of Medicine 342(20):1539–1544.
Cho, M.K., R. Shohara, A. Schissel, D. Rennie. 2000. “Policies on Faculty Conflicts of Interest at U.S. Universities.” Journal of the American Medical Association 284(17):2203–2208.
Cohen, J.J. 2000. Trust Us to Make a Difference. 29 October. Presentation to Association of American Medical Colleges Annual Meeting. Chicago, Illinois.
Coughlin, S.S., W. H. Katz, and D. R. Mattison. 1999. “Ethics Instruction at Schools of Public Health in the United States.”
American Journal of Public Health 89(5):768–770.
Department of Veterans Affairs (VA). 2000. “VA Raises Standard for Protecting Human Research Participants.” [Press Release.] 2 May. Available at http://www.va.gov/opa. Last accessed December 8, 2000.
DeRenzo, E.G. 2000. “Coercion in the Recruitment and Retention of Human Research Subjects, Pharmaceutical Industry Payments to Physician Investigators, and the Moral Courage of the IRB.” IRB: A Review of Human Subjects Research 22(2):1–5.
Dunn, C.M., and G. Chadwick. 1999. Protecting Study Volunteers in Research: A Manual for Investigative Sites. Boston, MA: CenterWatch, Inc.
Emanuel, E., and D. Steiner. 1995. “Institutional Conflict of Interest.” New England Journal of Medicine 332(4):262–267.
Food and Drug Administration (FDA). 1994. Bioresearch Monitoring: Institutional Review Boards. Compliance Program Guidance Manual 7348.809. Issued 18 August. Available at http://www.fda.gov/ora/compliance_ref/bimo/default.htm. Last accessed December 8, 2000.
———. 1998a. Bioresearch Monitoring: Clinical Investigators.
Compliance Program Guidance Manual 7348.811. Issued
2 September. Available at: http://www.fda.gov/ora/compliance_ref/ bimo/default.htm. Last accessed December 8, 2000.
———. 1998b. Bioresearch Monitoring: Sponsors, Contract Research Organizations and Monitors. Compliance Program Guidance Manual 7348.810. Issued 30 October. Available at http://www.fda.gov/ora/compliance_ref/bimo/default.htm. Last accessed December 8, 2000.
Foubister, V. 1999. “More Centers Cited for Ethics Lapses in Research.” American Medical News Nov. 1.
Francis, L. 1996. IRBs and Conflict of Interest. In Conflicts of Interest in Clinical Practice and Research, eds. R.G. Spece, D.S. Shimm, and A.E. Buchanan, 418–436. New York: Oxford University Press.
Gottlieb, J. 2000. Human Subject Protection and Financial Conflict of Interest: An Institutional Perspective. 15 August. Conference presentation, Human Subject Protection and Financial Conflicts of Interest. National Institutes of Health, Bethesda, Maryland.
Gunsalus, C.K. 1997. “Ethics: Sending Out the Message.” Science 276(5311):335.
Hamm, M.S. 1997. The Fundamentals of Accreditation. Washington, D.C.: American Society of Association Executives.
Healy, B., L. Campeau, R. Gray, J.A. Herd, B. Hoogwerf, D. Hunninghake, G. Knatterud, W. Stewart, and C. White. 1989. “Conflict-of-Interest Guidelines for a Multicenter Clinical Trial of Treatment After Coronary-Artery Bypass-Graft Surgery.”
New England Journal of Medicine 320(14):949–951.
Institute of Medicine (IOM). 2001. Preserving the Public Trust: Accreditation and Human Research Participants Protection Programs.
Washington D.C.: National Academy Press.
Korn, D. 2000. “Conflicts of Interest in Biomedical Research.”
Journal of the American Medical Association 284(17):2234–2237.
Lind, S. “Finder’s Fees for Research Subjects.” 1990. New England Journal of Medicine 323(3):192–194.
Lo, B.L. 2000. Resolving Ethical Dilemmas: A Guide for Clinicians,
2d ed. Philadelphia: Lippincott, Williams, and Wilkins.
Lo, B.L., L.E. Wolf, and A. Berkeley. 2000. “Conflict-of-Interest Policies for Investigators in Clinical Trials.” New England Journal of Medicine 343(22):1616–1620.
Mastroianni, A.C., and J. P. Kahn. 1998. “The Importance of Expanding Current Training in the Responsible Conduct of Research.” Academic Medicine 73(12):1249–1254.
McCrary, S.V., C.B. Anderson, J. Jakovljevic, T. Kahn, L.B.
McCullough, N.P. Wray, and B.A. Brody. 2000. “A National Survey of Policies on Disclosure of Conflicts of Interest in Biomedical Research.” New England Journal of Medicine 343(22):1621–1626.
McNeill, P.M. 1993. The Ethics and Politics of Human Experimentation. Cambridge University Press: New York.
National Bioethics Advisory Commission (NBAC). 1998.
Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity. 2 vols. Rockville, MD: U.S. Government Printing Office.
———. 1999. Research Involving Human Biological Materials: Ethical Issues and Policy Guidance. 2 vols. Rockville, MD: U.S. Government Printing Office.
———. 2001. Ethical and Policy Issues in International Research: Clinical Trials in Developing Countries. 2 vols. Bethesda, MD: U.S. Government Printing Office.
66
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission). 1978.
Report and Recommendations: Institutional Review Boards.
Washington, D.C.: U.S. Government Printing Office.
National Research Council. 1995. National Science Education Standards. Washington, D.C.: National Academy Press.
Office for Human Research Protections (OHRP). 2000. OHRP Compliance Activities: Common Findings and Guidance–9/21/2000. Washington, D.C.: Department of Health and Human Services. Available at http://ohrp.osophs.dhhs.gov./references/findings.pdf. Last accessed November 7, 2000.
Office of Inspector General (OIG), Department of Health and Human Services (DHHS). 1995. Investigational Devices: Four Case Studies. OEI-05-94-00100. Washington, D.C.: DHHS.
———. 1998a. Institutional Review Boards: A Time for Reform.
OEI-01-97-00193. Washington, D.C.: DHHS.
———. 1998b. Institutional Review Boards: Their Role in Reviewing Approved Research. OEI-01-97-00190. Washington, D.C.: DHHS.
———. 2000. FDA Oversight of Clinical Investigators.
OEI-05-99-00350. Washington, D.C.: DHHS.
Office for Protection from Research Risks (OPRR). 2000.
Compliance Oversight Investigations Resulting in Restrictions/Actions to Multiple Project Assurances, 1/90–11/99. NBAC archives.
Porter, J.P. 1987. “How Unaffiliated/Non-Scientist Members of Institutional Review Boards See Their Roles.” IRB: A Review of Human Subjects Research 9(6):1–6.
President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission). 1981. Protecting Human Subjects. Washington, D.C.: U.S. Government Printing Office.
———. 1983. Implementing Human Research Regulations: Second Biennial Report on the Adequacy and Uniformity of Federal Rules and Policies, and of their Implementation, for the Protection of Human Subjects. Washington, D.C.: U.S. Government Printing Office.
National Bioethics Advisory Commission
Pritchard, I.A. 1999. Education and the Common Rule: Comments for the National Bioethics Advisory Commission. NBAC archives.
Shalala, D. 2000. “Protecting Research Subjects—What Must Be Done.” New England Journal of Medicine 343(11):808–810.
Shefrin, A.P., and B. Harper. 2000. “Current Ethical Climate in Clinical Research, Part 1: ACT Ethics Survey Report Card.”
Applied Clinical Trials 9(2):28–40.
Steiner, D. 1996. “Competing Interests: The Need to Control Conflict of Interests in Biomedical Research.” Science and Engineering Ethics 2(4):457–468.
Sugarman, J. 2000. “The Role of Institutional Support in Protecting Human Research Subjects.” Academic Medicine 75(7):687–692.
Teegardin, C., and R. Whitt. 1997. “VA Patients Exploited, Doctors Say.” Atlanta Journal and Constitution, 7 June, 8-A.
Thompson, D. 1993. “Understanding Financial Conflicts of Interest.” New England Journal of Medicine 329(8):573–576.
Topol, E.J., P. Armstrong, F. Van de Werf, N. Kleiman, K. Lee, D. Morris, M. Simoons, G. Barbash, H. White, and R. M. Califf. 1992. “Confronting the Issues of Patient Safety and Investigator Conflict of Interest in an International Clinical Trial of Myocardial Reperfusion.” Journal of the American College of Cardiology
19(6):1123–1128.
Weijer, C., S. Stanley, A. Fuks, K.C. Glass, and M. Skrutkowska. 1995. “Monitoring Clinical Research: An Obligation Unfulfilled.”
Canadian Medical Association Journal 152(12):1973–1980.
Wolf, L.E., and B.L. Lo. 2000. “Ethical Issues in Clinical Research: An Issue for All Internists.” American Journal of Medicine 109(1):82–85.
67
Chapter Four 4
Assessing Risks and Potential Benefits and Evaluating Vulnerability
Introduction
This chapter addresses two issues central to the ethical analysis conducted by Institutional Review Boards (IRBs)—analysis of risks and potential benefits and the protection of vulnerable individuals—and recommends policy in the form of regulation and guidance to govern the review of research.
The current regulatory requirements relating to these ethical issues reflect the principles underlying the recommendations of the Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research (Belmont Report) (National Commission 1979). For example, the application of the ethical principle of respect for persons gives rise to the concern for vulnerability; the application of the principle of beneficence leads to the necessity of assessing risks and potential benefits; and the principle of justice requires investigators to attend to the process of recruiting research participants, particularly as it relates to the inclusion of individuals categorized as vulnerable.
In protecting the rights and welfare of participants in research, it is especially important to protect them from avoidable harm. An IRB’s assessment of risks and potential benefits is central to determining that a research study is ethically acceptable and would protect participants, which is not an easy task, because there are no clear criteria for IRBs to use in judging whether the risks of research are reasonable in relation to what might be gained by the research participant or society.
When an IRB determines that risks are reasonable, it must also ensure that all segments of society have the opportunity to participate, as appropriate. But some individuals might be considered vulnerable because of
either intrinsic characteristics (e.g., mentally or terminally ill) or situational factors (e.g., living in poverty, employees of the institution conducting the research) that render their fully informed and voluntary participation more susceptible to coercion or exploitation. Recognizing the various types of vulnerability and providing adequate safeguards can also prove challenging for IRBs.
IRB Approval of Research
Current regulations instruct IRBs in approving research studies as follows:
In order to approve research…the IRB shall determine that all of the following requirements are satisfied:
1. Risks to subjects are minimized: (i) by using procedures which are consistent with sound research design and which do not unnecessarily expose subjects to risk, and (ii) whenever appropriate, by using procedures already being performed on the subjects for diagnostic or treatment purposes.
2. Risks to subjects are reasonable in relation to anticipated benefits, if any, to subjects, and the importance of the knowledge that may reasonably be expected to result. In evaluating risks and benefits, the IRB should consider only those risks and benefits that may result from the research (as distinguished from risks and benefits of therapies subjects would receive even if not participating in the research). The IRB should not consider possible long-range effects of applying knowledge gained in the research (for example, the possible effects of the research on public policy) as among those research risks that fall within the purview of its responsibility.
69
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
3. Selection of subjects is equitable. In making this assessment the IRB should take into account the purposes of the research and the setting in which the research will be conducted and should be particularly cognizant of the special problems of research involving vulnerable populations, such as children, prisoners, pregnant women, mentally disabled persons, or economically or educationally disadvantaged persons.
4. Informed consent will be sought from each prospective subject or the subject’s legally authorized representative, in accordance with, and to the extent required by 46.116.
5. Informed consent will be appropriately documented, in accordance with, and to the extent required by 46.117.
6. When appropriate, the research plan makes adequate provision for monitoring the data collected to ensure the safety of subjects.
7. When appropriate, there are adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data.
8. When some or all of the subjects are likely to be vulnerable to coercion or undue influence, such as children, prisoners, pregnant women, mentally disabled persons, or economically or educationally disadvantaged persons, additional safeguards have been included in the study to protect the rights and welfare of these subjects (45 CFR 46.111; 21 CFR 56.111 [FDA regulations differ only in regulation citations]).
Scrutinizing research studies according to these criteria is not always a straightforward process, because it is difficult for any set of regulations to fully anticipate the variety of research situations that might arise and because the federal regulations often lack specificity with respect to a particular requirement (e.g., protecting privacy or confidentiality). Moreover, little guidance is available to assist IRBs in their reviews. For example, the IRB Guidebook, published by the former Office for Protection from Research Risks (OPRR), has not been updated since 1993 (OPRR 1993). Consequently, IRBs often tend to focus more on matters where clearer regulatory direction is available—for example, reviewing the language used for consent forms—rather than on areas that require more judgment. Furthermore, investigators have a tendency to
mimic their IRBs in this respect and focus on the more straightforward ethical issues (e.g., avoiding easily recognizable physical harms) and easily defined bureaucratic requirements (e.g., a signed consent form).
This pragmatic focus is understandable, given the limited guidance provided for interpreting many aspects of the regulations. For example, with the exception of those populations that are protected by additional regulatory requirements (i.e., fetuses and pregnant women, prisoners, and children), the federal oversight system offers no direction regarding the types of additional protections that are needed for other vulnerable individuals, even for those identified in the regulations—such as mentally disabled persons and economically or educationally disadvantaged persons. In addition, investigators and IRBs often struggle with the meaning of crucial terms such as minimal risk, minor change, and minor increase over minimal risk, on which key ethical and regulatory decisions rest.1 Applying these regulatory requirements to nonclinical research is even more difficult and cumbersome, because the limited regulatory detail provided is written in the context of clinical research (i.e., “that the research presents no more than minimal risk of harm to subjects and involves no procedures for which written consent is normally required outside of the research context” (45 CFR 117(c)(2)).
Aside from the challenges involved in understanding and applying the federal regulations, changes in the research environment have influenced how investigators conduct research and, to some degree, how regulatory requirements are applied. For example, growth of Internet-accessible health information databases has raised novel concerns about protecting privacy and confidentiality;2 increased interest in the study of social problems that may uncover illicit behavior has heightened awareness regarding the importance of confidentiality protections (Fitzgerald and Hamilton 1996; Fitzgerald and Hamilton 1997); and increases in overall educational attainment for Americans (Census Bureau 2000) have raised questions concerning which groups should be labeled educationally disadvantaged. In addition to the effects that these types of social and technological changes can have in the research environment, 10 years of experience with the Common Rule and 20 years of
70
experience with the Department of Health and Human Services (DHHS) and the Food and Drug Administration (FDA) regulations suggest that it is time to re-examine the criteria for research approval as described in the regulations (45 CFR 46.111; 21 CFR 56.111). Clarity in both the regulations and guidance would help investigators and IRBs more effectively and efficiently carry out their responsibilities, thereby improving protections, increasing public trust in the enterprise, and promoting research involving human participants.
Risks of Harms and Potential Benefits to Participants and to Society
The principle of beneficence states that persons should be “treated in an ethical manner not only by respecting their decisions and protecting them from harm, but also by making efforts to secure their well-being” (National Commission 1979, 6). Therefore, the principle requires that investigators attempt to maximize possible benefits and minimize possible harms. In research, however, the process of gathering data to gain knowledge of benefit to society may expose some individuals to harm, and IRBs must determine, therefore, “when it is [ethically] justifiable to seek certain benefits despite the risks involved, and when the [potential] benefits should be foregone because of the risks” (National Commission 1979, 7). In the simplest terms, IRBs assess risks and benefits. However, risk refers both to the probability that harm may occur and its magnitude. Thus, the term small risk is ambiguous, for it could mean that the probability of the harm is small, or that the magnitude (i.e., seriousness) of the possible harm is insignificant, or both. On the other hand, benefit refers only to the magnitude of the positive outcome, not to its probability. Thus, harms are properly contrasted with benefits, and risks (i.e., possible harms) with potential benefits.
Federal regulations incorporate the obligation of beneficence by requiring IRBs to ensure that risks are minimized to the extent possible, given the research question, and are reasonable in relation to potential benefits (45 CFR 46.111(a)(1)-(2); 21 CFR 56.111(a)(1)-(2)). Such an analysis of risks and potential benefits often will be complex, because IRBs are called on to assess the balance between any number and type of risks and potential
National Bioethics Advisory Commission
benefits. The current regulations do not further elaborate how risks and potential benefits are to be assessed, and little additional guidance is available to IRBs. The National Bioethics Advisory Commission (NBAC) discussed these issues at some length in two previous reports (NBAC 1998; NBAC 1999b) and has made specific recommendations in this area. Among the important considerations NBAC has raised is how IRBs and prospective participants might not evaluate risks and potential benefits in the same way. To account for this factor, in making assessments of risks and potential benefits, IRBs must find ways to be sensitive to the perspectives of prospective participants (NBAC 1998).
Types of Harms
Harms can be categorized broadly as physical, psychological, social, economic, legal, or dignitary; these categories are not mutually exclusive, however, and more than one type of harm might be present in a given research study. The terms harm and injury have distinct meanings in law, but as in the work of the Health Education and Welfare Task Force (DHEW 1977) and the President’s Commission (1982), the terms are used interchangeably in this report.
Physical harms include injury, illness, pain, suffering, or discomfort. This category encompasses such diverse harms as death in a cancer study, myocardial infarction related to a maximal exercise treadmill test, and discomfort related to the requirement to lie still in an MRI machine for an extended period during an imaging study.3 Psychological harms include the research participant’s negative perception of self, emotional suffering (e.g., anxiety or shame), or aberrations in thought or behavior. Examples of such harms were found in Milgram’s “Obedience to Authority” experiments (Milgram 1974). Psychological harms also include distress, anger, or guilt related to the disclosure of sensitive or embarrassing information (NBAC 1999a) and distress and fear upon learning of one’s likelihood of developing a disease for which there is no treatment or cure (Glass et al. 1997; Marteau and Croyle 1998; Meiser and Dunn 2000). Psychological harms also could be associated with refusal to participate in a research study, such as the guilt that arises when a prospective participant does not wish to
71
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
donate allogeneic bone marrow to be used in an experimental manner to treat a patient with AIDS.4 Social harms involve the negative effects on one’s interactions or relationships with others.5 For example, research findings or even study participation itself may expose participants to insurance or employment discrimination or other forms of social stigmatization (NBAC 1999b). Research findings may also damage a participant’s relationship with others—for example, when paternity status is disclosed in genetic or child development studies.6 Economic harms involve the imposition, direct or indirect, of financial costs on participants.7 For example, participants in clinical research may incur financial obligations for treatments that are higher than those associated with standard therapies. In some studies, participants may need to take time off from work or pay for transportation or childcare to enable them to take part in the research. They also may be faced with loss of health insurance, or even loss of employment.8 Legal harms are legal actions that could be taken against the participant, such as arrest, conviction, incarceration, or lawsuits. Such harms could occur, for example, in studies of possession or use of illicit drugs, sexual abuse, or workplace theft.9 Dignitary harms are those incurred when individuals are not treated as persons with their own values, preferences, and commitments,10 but rather as mere means, not deserving of respect.11 Such harms occur in failures to respect personhood or in violations of justice. These types of harms might be present, for example, in research studies in which informed consent is not obtained or in genetic research that results in the discrimination against the individual participant (NBAC 1999b, 45–46). Determinations concerning the probability of physical harms are often easier to make than those involving the probability of nonphysical harms. For example, the magnitude and probability of harms associated with a blood draw are well known and can be objectively quantified. This is generally not the case for psychological, social, economic, and legal harms. Although IRBs are able to identify such nonphysical potential harms, it is often difficult to determine the probabilities of their occurrence. IRBs, therefore, can err in either direction, by assuming a higher probability and recommending
unnecessary protections or preventing research from being conducted or by assuming a lower probability and allowing research to occur without all the appropriate protections. Although a good deal of information has been gathered about some nonphysical harms—for example, the risks from disclosures associated with transmitting or storing certain types of information—the possibility of such harms is not widely appreciated. Efforts should be made to ensure that IRBs are sensitive to the possibility that nonphysical harms might occur and that they better understand how to consider such risks in their assessments.
Risks to Those Other Than Participants
Risks may accrue not only to research participants, but also to persons not directly involved in research, such as to the participants’ family, loved ones, other contacts, social groups, and to society in general (National Commission 1979; NBAC 1999b). For example, in a previous report, NBAC recommended that to the extent that risks of harm to groups can be anticipated, investigators should design their research studies to minimize these risks and should consult with representatives of relevant groups regarding study design (NBAC 1999b). This recommendation was directed to investigators as one important measure that could be taken without revising the federal regulations. However, encouraging investigators may not be sufficient. Both individuals and other groups experience research risks, so it is appropriate for IRBs to be given explicit authority and guidance in how to consider them.
All of the types of risks described above could accrue to others affected by the research. For example, in a study of a new live virus vaccine, there may be risks to participants’ family members or other contacts who could contract the attenuated disease. In some states, there may be legal risk to parents whose minor children participate in a study of illegal activity.12 Genetic research may result in certain groups being associated with certain diseases, thus exposing members of those groups to the possibility of stigmatization or discrimination in insurance or employment.13 Society in general may incur harm related to research involving procedures, such as xenotransplan-tation or studies of viruses in which there may be some potential of releasing pathogenic organisms.14
72
Current regulations focus only on individual participants. However, other individuals and communities not directly involved in research can also bear risks that are associated with a research study, but that are not acknowledged or mentioned in the regulations. If IRBs focus only on risks to individual research participants, they fail to apply fully the principle of beneficence, because the scope of this principle can extend beyond the individual participants in the research.15
Minimizing Risks
Once risks have been identified, IRBs and investigators must ensure that they are minimized to the extent possible within the limitations imposed by the nature of the research study. Risks may be reduced in a variety of ways—for example, by assuring that the study design is valid; the investigator and research study personnel are qualified; the necessary infrastructure is in place to conduct the research and deal with any harmful sequelae; participant privacy and confidentiality are adequately protected; participants are properly monitored; criteria for participant enrollment and withdrawal are appropriate; a timely treatment plan is in place; and prospective participants at undue risk of harm are excluded.16
Types of Potential Benefits
IRBs should identify potential benefits associated with a research study. Generally, IRBs consider potential benefits as accruing either to society or to participants. Just as research studies might involve risks to individuals other than the participants, they might also provide potential benefits to others, especially the participants’ community.
Potential Benefits to Society
The goal of research is to develop knowledge that is beneficial to society. In considering whether to approve a proposed research study, IRBs must make judgments about the importance of the knowledge that is likely to result from research studies. The importance of the knowledge to be gained may increase when significant new findings are expected; when it may result in new products, treatments, or cures; or when it is applicable to many different social groups.
The assessment of whether risks are reasonable in relation to the potential gain in knowledge is difficult to
National Bioethics Advisory Commission
make, because those who participate in research and who will be exposed to the risks are often not the same individuals who stand to benefit from the research. Thus, the IRB must be able to clearly identify what might be learned from the research and decide whether the gain in knowledge justifies the exposure of participants to harm. For studies involving little risk, this assessment is not so difficult. However, when the research involves significant risks to human participants—for example, death or disability—the IRB must carefully weigh the risks and the potential gain in knowledge.
Potential Benefits to Participants
Individuals may directly benefit from participation in certain types of research, such as studies designed to offer interventions or procedures that offer a prospect of benefit. For example, potential benefits include receiving clinically significant information that could be used to influence the care provided, receiving standard treatments or interventions as part of the research, such as counseling and testing, or gaining access to experimental therapies that may improve the participant’s health status. These types of benefits are to be contrasted with unplanned or unanticipated benefits that are secondary to the objectives of the study. Sometimes, unanticipated direct benefits can result that are relevant to the research objectives. For example, in a research study on treatment for injection drug users, participants were asked to keep daily diaries as a data collection strategy, which served to raise self-awareness about the effect of daily habits on drug use. As a result, some users sought treatment (Singer 2000; Singer et al. 2000). When identifying potential direct benefits to participants, however, IRBs should consider only those that might result strictly from study participation.
Individuals might also benefit indirectly from participation in certain types of research, from experiencing increased social contact, sharing information with another person, or gaining personal satisfaction from participating in the research (NBAC 1998). Indirect benefits typically are not planned by investigators in their research design and do not relate to the objectives of the research study. In addition, they are likely to vary among research participants. Although these benefits
73
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
should be acknowledged, they should not weigh in the judgment of IRBs regarding the balance of risks and potential benefits to the participant. To the extent that indirect benefits can be anticipated, however, investigators should design research to increase them.
For purposes of IRB review, incentives for and payments associated with participation in research (e.g., remuneration for transportation, childcare, or lost work time) should not be considered potential benefits to research participants (OPRR 1993). Such potential benefits to research participants make it feasible to participate, but they do not result directly from study procedures in which the individuals participate. Including such incentives as potential benefits in the IRB assessment would inappropriately skew judgments concerning risks and potential benefits, because nearly any level of research risk could be offset by such gains if they were significant enough—for example, if participants were promised large sums of money for participating in research. Although IRBs should not consider such incentives or payments as benefits, they should recognize that prospective participants might consider them as such. Thus, IRBs should determine whether incentives or payments are set so high that they induce prospective participants to enroll without carefully considering the risks involved in participation.
Potential Benefits to Others
Research institutions, social groups, or communities also can benefit from research. Institutions benefit, for example, from enhanced capacity to conduct research or by receiving resources to improve a program as a result of a research study. Communities can benefit through improved access to programs or through the emergence of programs targeted to specific groups within the community. Research can result in the preservation of local history, customs, or practices that might otherwise be lost. IRBs should acknowledge that these potential benefits might be present and should consider ways to increase their likelihood as part of the research design. However, they should not weigh them in assessing risks and potential direct benefits to the participants.
Relation of Risks to Potential Benefits
Once risks and potential societal and direct benefits have been identified and ways to minimize risks have been considered, IRBs must judge whether the risks are reasonable in relation to potential benefits. These can be difficult judgments to make, because IRBs might have to compare the significance of different types of risks and potential benefits, which are often difficult to quantify (Martin et al. 1995). Further, IRBs have little guidance on how to classify and compare risks and potential benefits. It would be impossible to provide IRBs with a single algorithm for making such decisions that would do justice to the large variety of cases that might come before them. However, the counsel provided in the Belmont Report should guide IRB deliberations:
It is commonly said that potential benefits and risks must be ‘balanced’ and shown to be ‘in a favorable ratio.’ The metaphorical character of these terms draws attention to the difficulty of making precise judgments. Only on rare occasions will quantitative techniques be available for the scrutiny of research protocols. However, the idea of systematic, nonarbi-trary analysis of risks and benefits should be emulated insofar as possible. This ideal requires those making decisions about the justifiability of research to be thorough in the accumulation and assessment of information about all aspects of the research, and to consider alternatives systematically. This procedure renders the assessment of research more rigorous and precise, while making communication between review board members and investigators less subject to misinterpretation, misinformation and conflicting judgments (National Commission 1979, 16–17).
An IRB may approve a research proposal only if it judges that the risks are reasonable in relation to potential benefits (National Commission 1979). This judgment may be an IRB’s single most important and difficult determination, because it ensures that when research participants voluntarily consent to participate in a research study, they are offered a “reasonable choice.”17 Research with significant risks could be approved after such an analysis if the potential benefit is significant. However, this does not imply that there is no upper limit to the determination of acceptable risk. For any research study involving significant risk, not only should the local IRB
74
find the risks reasonable in relation to the potential benefits, but also so should a larger community of investigators, IRBs, and the public.
Historical Perspective: The Foundation Provided by the National Commission
A coherent conceptual framework is needed for the analysis of risks and potential benefits, and yet there is evidence that this is not provided by current regulations. For example, it has been suggested that the National Commission espoused three distinct views on the analysis of risk: risk assessment based on an analysis of the whole protocol, analysis of risks for whole protocols based on the combined analysis of each of the particular components within a protocol; and distinct analyses of risks for individual components of research protocols without judging the whole protocol as a single unit.18 Each of these views is reflected in the current federal regulations.
In its first report, Research on the Fetus, the National Commission presented a framework for risk analysis that relies on categorizing research studies into two types, therapeutic and nontherapeutic (National Commission 1975) and made separate recommendations concerning the evaluation of risks for each type of research. Current DHHS regulations concerning fetal research (45 CFR 46 Subpart B) incorporate this framework.19 According to some commentators, this “whole protocol” approach, which requires that entire research studies be classified as either therapeutic or nontherapeutic, is flawed (Levine 1986).20 The National Commission characterized therapeutic research as “designed to improve the health condition of the research subject by prophy-lactic, diagnostic, or treatment methods that depart from standard medical practice but hold out a reasonable expectation of success” (National Commission 1975, 6). Levine has argued that the concept of therapeutic research is a contradiction in terms:
There is, of course, no such thing as a ‘systematic collection of data or observations…designed to improve the health condition of a research subject… that departs from standard medical practice.’ Thus, the [National] Commission developed recommendations for the conduct of a nonexistent set of activities… (Levine 1986, 298).
National Bioethics Advisory Commission
Because the purpose of research is to acquire knowledge, no research study as a whole can be accurately classified as therapeutic research. Indeed, for this reason the National Commission later rejected this whole protocol framework. NBAC has also recognized the difficulty of such a framework (NBAC 1998, 46). As a result of this conceptual flaw, the whole protocol framework allows any research study to be classified as therapeutic as long as it contains at least one component that offers the potential of direct benefit to the participant. However, such research might include nontherapeutic components involving significant risk. For example, a pharmacokinetic study might involve the administration of a standard therapy followed by extensive nontherapeutic testing. In the whole protocol approach, the entire protocol would be classified as therapeutic and there would be no limit to the number of nontherapeutic procedures that could be administered to participants with the protocol still classified as therapeutic. Indeed, IRBs might fail to consider the risk associated with the nontherapeutic procedures, because the conceptual framework encourages them to think about the entire protocol as offering the prospect of some direct benefit. Thus, it is difficult to envision research participants being adequately protected under such a framework.21
In its reports Research Involving Prisoners (1976) and Research Involving Children (1977), the National Commission rejected the distinction between therapeutic and nontherapeutic research, recognizing that an entire research study could not be considered fully therapeutic, but that, instead, protocols might contain therapeutic or nontherapeutic components. Thus, the National Commission eliminated the conceptual problem that stems from the category of “therapeutic research.” However, the National Commission did not avoid the ethical problems described above. It adopted the language of components, but retained an analytical model based on assessing the protocol as a whole. For example, in its report on research on children, the National Commission endorsed one set of recommendations for research involving no more than minimal risk, another for research involving therapeutic components that exceed the minimal risk threshold, and yet another for research involving nontherapeutic components that exceed this threshold. It has been argued that this framework also
75
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
suffers from several weaknesses.22 All research studies containing components that offer the prospect of direct benefit to research participants also contain components that do not offer the prospect of benefit and only answer the research question(s). Thus, two different sets of recommendations for assessing risks would apply in a research study involving children that contains both therapeutic and nontherapeutic components in which both components involve more than minimal risk. Because the recommendations that comprise this framework refer to a research study as a whole involving a particular type of component, it is unclear how multiple recommendations could be applied simultaneously to an entire study. Although the model avoids the linguistic confusion associated with a “whole protocol” approach, it still fails to analyze risk and potential benefits separately for therapeutic and nontherapeutic components.23 Nevertheless, current DHHS regulations concerning research involving prisoners (45 CFR 46 Subpart C) and children (45 CFR 46 Subpart D) incorporate this framework.24 Finally, in its report Institutional Review Boards (1978) the National Commission began to move toward a framework that involved the analysis of components of a research study rather than the study as a whole; however, it did not adopt specific recommendations for performing such an analysis of risk. Requirements relating to analysis of risks and potential benefits in the Common Rule (45 CFR 46 Subpart A) seem to be based on this framework.25 The lack of a single coherent, fully developed conceptual framework hinders the efforts of IRBs to assess and evaluate risks and potential benefits of research studies. At best, the terms therapeutic and non-therapeutic are confusing. By labeling research or procedures as therapeutic, it implies that these activities are beneficial when, in fact, the very purpose of research is to make that determination. NBAC has not used these terms in its previous reports, and it does not do so here. Rather, components should be assessed on the basis of whether they offer the prospect of direct benefits to the individuals being studied or general, more indirect benefits to society.
A Framework for the Analysis of Risks of Harms and Potential Benefits
Charles Weijer has proposed a framework based on a “component analysis” that he believes provides a better
ethical analysis of research that contains a mixture of components—some that offer the prospect of direct benefit to research participants and others with the sole intent of answering the research question(s).26 In the context of a research study, all components are designed to answer the research question(s). Components may include one or more procedures. However, some components also offer the prospect of direct benefit to research participants, while others do not; the latter solely offer the potential societal benefit of knowledge. For example, a research study designed to test whether aspirin or aceta-minophen reduces fever more effectively uses a number of procedures, including the administration of aspirin to one group; the administration of acetaminophen to another group; the randomization of participants to these two groups; and the measurement of fever. All of these procedures are included in the study design because they are needed to answer the research question(s). However, some of these procedures are also designed to offer the prospect of direct benefit to participants—for example, the administration of aspirin or of acetaminophen. Others, such as randomization, temperature taking, and chart comparisons, do not offer the prospect of such direct benefits; their sole intent is to answer the research question(s). Although a research study must be examined in its entirety, the two types of components should be judged differently.
The component-based approach to the analysis of risks and potential benefits requires IRBs to sort research study procedures into these two types of components to determine their ethical acceptability. The first type consists of those components containing particular procedures that may offer the prospect of direct benefit to participants. The second type includes procedures that do not. Most procedures in a research study are easily classified into one of these two types of components. However, in some studies, it might not be clear into which component the procedures best fit—for example, a survey involving a questionnaire and measurements of health in which clinically meaningful results (e.g., blood pressure) are reported back to participants, but no treatment is offered. Further, in any research study using diagnostic procedures that might provide useful health information to the participant in addition to the information provided for the study—such as the use of CAT
76
scans to study brain activity that could also identify tumors—the classification of such procedures may be ambiguous and therefore requires careful consideration by the IRB.
IRBs should be able to identify whether a clear and direct benefit to society or the research participants might result from participating in the study. However, IRBs should be cautious in classifying procedures as offering the prospect of direct benefit. In fact, if it is not clear that a procedure also offers the prospect of direct benefit, IRBs should treat the procedure as one solely designed to answer the research question(s). A major advantage of this approach is that it avoids justifying the risks of procedures that are designed solely to answer the research questions based on the likelihood that another procedure in the protocol is likely to provide a benefit.
To the extent possible, IRBs should independently weigh the risks and potential benefits of each type of procedure. The risks associated with individual procedures offering the prospect of direct benefits are justified in relation to their potential to benefit the participant in addition to their potential to generate knowledge, and those procedures designed solely to answer the research question(s) are justified in relation to their potential to generate knowledge.
Recommendation 4.1: An analysis of the risks and potential benefits of study components should be applied to all types of covered research (see Recommendation 2.4). In general, each component of a study should be evaluated separately, and its risks should be both reasonable in themselves as well as justified by the potential benefits to society or the participants. Potential benefits from one component of a study should not be used to justify risks posed by a separate component of a study.
Components Designed Solely to Answer the Research Question(s)
All research involves the administration of procedures and use of methods solely for the purpose of answering the research question(s). These procedures or methods can be questionnaires, interviews, chart reviews and use of existing data, observations, randomization, and clinical procedures such as blood draws. Many health services
National Bioethics Advisory Commission
studies and most epidemiological, behavioral, and social science studies as well as historical research involve only such components.
By definition, risks associated with components containing procedures designed solely to answer the research question(s) can be justified only by the potential benefit associated with the knowledge gained, not by the potential direct benefits to individual participants. Freedman argued that this judgment requires the opinion of both scientific experts and community representatives (Freedman 1987b). Community representatives might come from geographic areas or from affected participant communities (e.g., persons with a particular disorder). Involving community representatives can be challenging, as it is sometimes difficult to define relevant groups or individuals who can represent the community (Davis 2000). As risks increase, scrutiny from the communities that may be affected by the research findings will help to ensure public confidence and trust that participants are offered “reasonable choices.”
Components That Also Offer the Prospect of Direct Benefits
Clinical and other types of research studies might include procedures that, in addition to answering the research question(s), also offer the prospect of direct benefit to participants, such as drug interventions, surgical and medical procedures, or educational, psychological, and behavioral interventions. They might also include diagnostic and other procedures that guide the administration of treatment, even if these procedures are not routinely administered.27 Studies with components containing procedures of this kind pose their own distinctive challenge in the ethical analysis of risks and potential benefits.
Here too, IRBs must determine whether the relation between risks and potential benefits is reasonable. To do so, IRBs should determine whether the procedures meet the criteria of research equipoise (see Exhibit 4.1) in addition to being justified in terms of the potential knowledge gain for society. Investigators and IRBs should understand that the term research equipoise applies to any type of research involving interventions or procedures that offer the prospect of direct benefit to participants, which
77
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
Exhibit 4.1: Research Equipoise
Research equipoise, a new term introduced in this report, describes the state in which genuine uncertainty exists regarding which intervention—experimental or control (including placebos)—is better (Freedman 1987a, 144). Normally, this concept is referred to as clinical equipoise, because it is used in medicine. However, because the concept can be applied to other areas of practice, such as public health or psychology, the term should be broadened.
Accepted practice is defined as practice that falls within the bounds of what is considered standard—that is, what the community of expert practitioners accepts. The standard of research equipoise recognizes explicitly that it is the community of experts that establishes standards of practice (Freedman 1987a). Thus, the judgment that a state of research equipoise exists is an acknowledgement that study interventions are thought to be fully consistent with accepted practice.28
A judgment of research equipoise relies on a comparison of the risks and potential benefits of the proposed study interventions with those of accepted practice. Research equipoise does not require numeric equality of intervention risks or potential benefits. Rather, research equipoise requires approximate equality in the relation between the risks and potential benefits of the study and control interventions. A variety of intervention-related factors are likely to contribute to the determination of this assessment, including, among others, the relative efficacy of the intervention, the probability and magnitude of side effects, ease of administration, and participant compliance.29 An experimental intervention may pose greater risk to participants than accepted practice, as long as it also offers the prospect of greater direct benefit to the participant and the relation between the risks and potential benefits falls within a range of equivalency to accepted practice.30
would include, for example, health education interventions, clinical psychology interventions, and public health interventions.
Clinical trials involving the use of drugs, vaccines, or other biologic products are other examples of research studies in which IRBs would subject the experimental procedures to a test of research equipoise. However, such
a test would also need to be applied to research studies involving the evaluation of any intervention that is designed to benefit those who would receive it. For example, educational interventions, such as teaching reading skills or providing public health interventions, such as smoking cessation and psychological interventions, should also be subject to the test of research equipoise.
Procedure for Assessing the Balance Between Risks and Potential Benefits
Figure 4.1 outlines the framework for risk and potential benefit analysis that IRBs should undertake in reviewing research. The proposed component analysis offers several advantages.
First, the model can be applied to all types of research. Second, it focuses the attention of the IRB appropriately on distinct procedures for which the ethical analysis is different. Components designed solely to answer the research question(s) are judged based on an analysis of whether the risks are reasonable in relation to the potential gain in knowledge, while components that also offer the prospect of direct benefit must meet an additional standard of research equipoise. That is, the balance of risks and potential benefits associated with the study interventions are equivalent to those associated with accepted practice and, therefore, there is genuine uncertainty (among experts) about whether the study intervention or accepted practice is preferred. Third, under this model, components that are designed only to answer the research question(s) cannot be justified based on the inclusion of other procedures that offer the prospect of direct benefit. This aspect of the model prevents investigators conducting clinical trials, for example, from including additional procedures, such as biopsies, blood tests, or scans, that are unlikely to yield any information of benefit to society by justifying them on the basis of including an intervention that holds the prospect of direct benefit, such as an experimental oncology treatment.
The proposed ethical analysis should be performed after risks have been identified and, to the extent possible, minimized by investigators. Only that part of the framework that is relevant to a particular research study should be used. In the case in which a research study
78
National Bioethics Advisory Commission
Figure 4-1. Process of IRB Review Including Analysis of Risks and Potential Benefits
Protocol submitted to IRB for review.
IRB reviews to
IRB’s ethical analysis of risks and potential benefits after risks identified and minimized, possible benefits identified.
COMPONENTS DESIGNED SOLELY
TO ANSWER THE RESEARCH
QUESTION(S)
ARE THE RISKS REASONABLE IN RELATION TO POTENTIAL BENEFITS OF KNOWLEDGE?
COMPONENTS THAT ALSO OFFER
THE PROSPECT OF DIRECT BENEFITS TO PARTICIPANTS
ARE THE RISKS REASONABLE IN RELATION TO POTENTIAL BENEFITS TO PARTICIPANTS?
| n |
Assessment of risks and potential benefits made separately for experimental and control procedures. |
|
| n |
Does study pass test of research equipoise? |
|
| — |
Insufficient data exist to validate whether control or experimental procedure more likely to provide more favorable risk/benefit combination. |
|
| — |
May require input from experts and community representatives. |
|
STUDY ACCEPTABLE ONLY IF ALL COMPONENTS PASS ETHICAL TESTS.
79
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
involves only the use of components designed solely to answer the research question(s), the IRB and investigators would follow only the left side of Figure 4.1. For example, in a study involving the economic analysis of identifiable data, only the part of the framework relating to components designed solely to answer the research question(s) is used. Likewise, for any research study that does not involve components that hold the prospect of direct benefit to participants, the relevant ethical analysis is only that part of the model relating to components designed solely to answer the research question(s). Examples of such procedures are surveys, records review, blood draws, and other procedures used solely to identify health conditions. Procedures administered in experiments as part of data collection, such as using varying stimuli in a psychological perception experiment or using drugs to alter cognitive states, are evaluated only by the components outlined on the left side of Figure 4.1.
For research involving both types of components (those that solely answer the research question[s] and that also offer the prospect of direct benefit to participants), IRBs should follow both sides of Figure 4.1. Whether one side or both sides of the suggested framework are used, each component of the protocol is considered in turn. IRBs should conclude that risks are reasonable in relation to the potential benefits only when all of the individual procedures have met their respective ethical criteria (see examples in Exhibit 4.1). In the end, an IRB must determine that the research study in its entirely is ethically acceptable.
Standard procedures may be used in a research study in at least four ways, and in each case, they are evaluated differently. First, standard procedures might be used as a control. When standard procedures are used as a control to evaluate the experimental intervention, IRBs must assess them as they do experimental procedures by following the right side of Figure 4.1. (See Exhibit 4.2 for an example.) Second, standard procedures might be used solely to answer the research question(s), such as blood draws or CAT scans. IRBs should follow the left side of Figure 4.1 to assess such procedures. Third, standard procedures might be offered in conjunction with a research study, but not as part of the research study. For example, in a study involving known cases of HIV, participants might be offered counseling and testing for
other sexually transmitted diseases (STDs). However, because the STD counseling and testing procedures do not relate to the research question(s) and are instead offered as part of standard practice in a STD clinic, the use of standard procedures in this case should not be factored into the ethical analysis by the IRB. Fourth, some research studies might involve the evaluation of an ongoing standard procedure. That is, the standard procedure would occur or be offered regardless of the existence of a research study. For example, a research study might be designed to evaluate the sensitivity and specificity of a surveillance system to detect cases of disease, with the surveillance system a standard, ongoing procedure, and additional data collection techniques added in order to evaluate the surveillance system. In this scenario, the surveillance system is not part of the research study and should not be part of the ethical analysis of the research study. But the additional data collection is research and should be part of the ethical analysis.
Minimal Risk
Current federal regulations for the protection of research participants call for the classification of research as involving either minimal risk or greater than minimal risk.31 As defined in the federal regulations:
Minimal risk means that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests (45 CFR 46.102(i); 21 CFR 56.102(i)).
This classification is ethically relevant, because it is intended to provide protection to human participants by focusing IRBs’ attention on riskier research. When used as a sorting mechanism, this classification determines the level of review required of an IRB.32 For example, under the current regulations, if a research study is determined to pose only minimal risk and involves a procedure contained on the expedited review list, it may be evaluated using the expedited review process in which the IRB chair or a designee may review the research study in accordance with all the required
80
National Bioethics Advisory Commission
Exhibit 4.2: Using the Framework to Assess Risks and Potential Benefits
The following examples are taken from recently published research to show how research procedures can be separated into those solely designed to answer the research question(s) and those that also offer the prospect of direct benefit to participants. The risk/potential benefit analysis can be used with studies involving no more than minimal risk as well as studies involving more than minimal risk.
Example 1: Analysis of Existing Data
Investigators studied gender differences in industry wages by analyzing data from the 1988 Current Population Survey. Variables in the analysis include hourly wage, level of education, years of work experience, size of the metropolitan area in which workplace is located, whether the workplace is in an urban or rural area, geographical region of the United States, marital status, race, type of occupation, and type of industry.
If the data are unidentifiable, the research should not be covered under the oversight system, because no human participants are involved. If the data are identifiable (i.e., coded), then an IRB should review the research. In this case, risks are associated with breaches in confidentiality, and the IRB should determine whether confidentiality protections are appropriate. Because none of the research procedures offer the prospect of direct benefit, the risk/potential benefit analysis involves judging whether the risks are reasonable in relation to the potential gains in knowledge for society.
Based on this analysis, this study should be eligible for review using procedures other than the full board because it involves no more than minimal risk.
Example 2: Face-to-Face Interviews Involving Sensitive Issues and a Small Sample
Investigators studied the effect of traumatic experiences on memory. Five individuals who were incarcerated between 1942 and 1945 in Auschwitz were the participants. A detailed questionnaire was given concerning personal characteristics (e.g., age, education, and place of work), concentration camp characteristics, daily routine at the camp, and events that occurred while there. They were also shown ten photos of famous Nazis who were either known nationally or active at Auschwitz. Interviewers administered the questionnaires in the participants’ homes and conducted interviews in Hebrew or German. Investigators analyzed responses for accuracy based on historical accounts.
Concerns of the IRB should be threats to privacy, breaches in confidentiality, and psychological risks, such as distress. The sample size may result in individuals being identified even if no direct identifiers are maintained. Because none of the research procedures offer the prospect of direct benefit, the risk/potential benefit analysis involves judging whether the risks are reasonable in relation to the potential gains in knowledge for society. Investigators might argue that participants might feel better after the interview and derive some satisfaction from talking about their experiences. However, these types of indirect benefits should not be taken into consideration in the risk/potential benefit analysis.
Based on the analysis of the components of this study, it should receive full IRB review because some components pose more than minimal risk.
Example 3: Observational Study Using Mail Questionnaires to Evaluate the Effects of State Law on Behavior
Investigators studied the effects of two divorce laws on the functioning of parents and children immediately after separation and two years later. In this state, until 1987 the divorce law was no-fault and permissive of joint custody. In 1988, a new law went into effect that focused on parental functions and responsibilities rather than custody. Investigators identified divorcing couples through their divorce petitions. One hundred couples were identified in 1987 and 100 in 1988 and were sent a questionnaire at the time of separation. If they responded to the questionnaire, they were sent another questionnaire two years later. Individuals were asked to respond to questions about themselves regarding depression, social withdrawal, irritability, work problems, and childrearing problems. They were asked to answer questions about their children regarding depression, anxiety, eating behavior, sleeping patterns, nighttime fears, and withdrawal. They also responded to questions regarding the circumstances of the separation, including parental substance abuse and child abuse.
Primary concerns of the IRB should be threats to privacy and breaches in confidentiality. Privacy and confidentiality risks related to psychological, social, and legal harms are heightened, because divorcing spouses might attempt to obtain through legal mechanisms information about the other or physically harm the other based on information disclosed in the survey. Other issues would include whether cases of child abuse must be reported. Because none of the research procedures offer the prospect of direct benefit, the risk/potential benefit analysis involves judging whether the risks are reasonable in relation to the potential gains in knowledge for society. Thus, the study should receive full IRB review.
81
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
Exhibit 4.2 continued
Example 4: Psychological Experiment to Study Human Cognition and Behavior
Investigators studied the relationship between task difficulty and performance, because performance sometimes improves when tasks become more difficult. In this experiment, motor difficulty and cognitive difficulty were manipulated in a recognition task. Sitting in front of a computer, student participants were instructed to move the cursor using a standard analog joystick as quickly and accurately as possible into contact with an image that they thought matched the sample image. Within the different experimental groups, exposure time of the sample stimulus was altered (fast versus slow), and ease of moving the joystick was altered (easy versus hard). Students were offered course credit for participating in the study.
Primary concerns of the IRB should be whether offering course credit for participation is an undue inducement; whether there are any physical risks associated with operating a hard-to-move joystick; and whether there are any psychological risks associated with task difficulty, such as frustration or loss of self-confidence. Because none of the research procedures offer the prospect of direct benefit, the risk/potential benefit analysis involves judging whether the risks are reasonable in relation to the potential gains in knowledge for society. This study should be eligible for review using procedures other than full board review because it involves no more than minimal risk.
Example 5: Clinical Trial to Determine Safety and Immunogenicity of a Vaccine
Investigators conducted a Phase I, dose-escalating trial of a prophylactic recombinant human papillomavirus vaccine candidate in healthy participants. The study was designed to evaluate the safety and immunogenicity of two dose levels of the vaccine in a double-blind, randomized, placebo-controlled trial. To determine whether the dose of the vaccine or the addition of alum or MF59 adjuvant would influence the reactogenicity or immune response, a dose-escalation design was used, starting with the lower dose of the vaccine alone, then with alum added to the vaccine, then with MF59 added to the vaccine, and the design repeated with the higher dose of the vaccine. Assessments included a physical examination and laboratory tests prior to enrollment and clinical evaluations and clinical and immunologic tests before each injection at 0, 1, and 4 months and 1 month after each injection.
The trial was not designed to offer a potential direct benefit (protection from human papillomavirus) to participants. The vaccine, the design of the trial, and all of the assessments were administered solely to answer the research question(s) related to safety and immunogenicity. Because none of the research procedures offer the prospect of direct benefit, the risk/potential benefits analysis involves judging whether the risks are reasonable in relation to the potential gains in knowledge for society. Thus, the study should receive full IRB review.
Example 6: Clinical Trial to Test Efficacy of a Drug
Investigators conducted a trial to determine whether paroxetine, an antidepressant, would reduce depressive symptoms in patients with malignant melanoma who were treated with a high dose of interferon therapy. The investigators used a double-blind, placebo-controlled trial design. Participants were randomly assigned to either the experimental arm or the control arm of the trial. Paroxetine or placebo was administered for two weeks before initiation of the inter-feron therapy as well as during the treatment period. Participants were evaluated at regularly scheduled intervals for depressive symptoms. Assessments included depression and anxiety rating scales, a neurotoxicity rating scale used in the measurement of psychiatric and physical symptoms related to cytokine therapy, and a psychiatric evaluation.
This trial was designed to offer the prospect of direct benefit to participants in addition to answering the research question(s). In this study, the research (whether paroxetine reduces depression-like symptoms) takes place in the context of providing standard medical care (interferon therapy). The use of standard medical care should not be factored into the risk/potential benefit analysis of the IRB. The research study procedures are of two types. The first type, designed solely to answer the research question, includes the trial design (randomization and double-blindness) and the assessment measures. The risk/potential benefit analysis involves judging whether the risks are reasonable in relation to the potential gains in knowledge for society. The other types of procedures are those that offer the prospect of direct benefits. These procedures are the experimental and control conditions. In addition to determining that these procedures will yield potentially useful societal knowledge, they are evaluated in terms of whether they meet the test of research equipoise—that is, whether there is disagreement among experts as to whether either should be preferred as accepted practice.
This study should receive full IRB review.
82
regulations (45 CFR 46.110(b); 21 CFR 56.110(b)). Research involving more than minimal risk requires full IRB review. As the risk of research increases above the minimal risk threshold, protections for participants become more stringent. For example, with greater than minimal risk research the process of informed consent cannot be waived or altered (45 CFR 46.116(d)).
Definition of Minimal Risk
The definition of minimal risk in federal regulations (45 CFR 46.102(i); 21 CFR 56.102(i)) does not specify an unambiguous standard. That is, risks involved in the research study are compared to those encountered in daily life, but it is unclear whether daily life applies to healthy individuals or the target group of the research. Existing sources of guidance offer conflicting interpretations of the standard to be used in determining level of risk.33 In the context of its discussion of research involving children, the National Commission defined a so-called absolute standard when it defined minimal risk as “the probability and magnitude of physical or psychological harm that is normally encountered in the daily lives, or in the routine medical or psychological examination, of healthy children” (National Commission 1977). This standard was not adopted in the regulations pertaining to research involving children (45 CFR 46 Subpart D). However, DHHS regulations concerning research involving prisoners limit minimal risk to the experience of healthy individuals. In 1993, OPRR endorsed such an absolute standard interpretation for Subpart A (Ellis 1995). OPRR’s interpretation is inconsistent with DHHS’ intention as expressed in the preamble of the 1981 version of 45 CFR 46 which stated that “the risks of harm ordinarily encountered in daily life means those risk encountered in the daily lives of the subjects of the research.”34 If minimal risk is not characterized in terms of the daily life and experiences of healthy individuals, then it might be taken to refer to the daily life and experiences of research participants. If this is the case, then the same intervention could be classified as minimal risk or greater than minimal risk, depending on the health status of the research participants and their particular experiences. For example, a bone marrow aspiration would not be considered minimal risk in relation to the daily life of a healthy individual, but it might well be determined to fall
National Bioethics Advisory Commission
within this category in relation to the experience of a person with acute leukemia. Such an understanding entails a relative standard for minimal risk.
A relative standard for minimal risk would allow ill participants to be exposed to greater risks than healthy participants. Such a standard would impose disproportionate burdens of research on the ill and provide weaker protections for them than for healthy individuals. This would violate the ethical principle of justice.
IRBs should use a standard related to the risks of daily life that are familiar to the general population for determining whether the level of risk is minimal or more than minimal. The standard should not refer to the particular risks encountered by particular persons or groups. It should refer, therefore, to common risks—for example, driving to work, crossing the street, getting a blood test, or answering questions over the telephone.35 Research, then, involves no more than minimal risk when it is judged that the level of risk is no greater than that encountered in the daily lives of those in the general population. The general population standard is less restrictive than the healthy individuals standard; however, the general population standard more accurately captures the risks that are familiar to most persons.
When a research study is determined to involve no more than minimal risk, the IRB should also consider whether the procedures in question pose additional risks to some of the potential research participants. In such cases, additional protections might be required to reduce the level of risk among that subgroup. For example, drawing a small quantity of blood would normally be considered a minimal risk procedure; its risks do not exceed those normally encountered by the general population. However, if a particular research study involved participants with immunosuppressive disorders, a blood draw may present a higher level of risk, and additional procedures to reduce exposure to infections may be required.
It must be stressed that, in making the determination that a research study involves no more than minimal risk, the IRB must take into consideration all types of risk posed. For example, a research study involving a blood draw to study HIV might involve not only the relatively inconsequential physical risks associated with a blood draw, but also all the nonphysical risks mentioned earlier
83
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
that might be associated with, for example, learning one’s HIV status or having that information released to people other than the investigators.
Use of the Concept of Minimal Risk as a Sorting Mechanism
The concept of minimal risk serves to focus the IRB’s attention on riskier research. Minimal risk is used in determining which research studies can be reviewed using procedures other than the full IRB (see Chapter 2). Currently, research studies that are judged to be minimal risk and fall into one of nine categories of study types may be reviewed using the expedited process. For purposes of determining level of review, there are no relevant ethical differences between studies involving minimal risk that do not fall into one of these categories and those that do. Therefore, all studies judged to involve no more than minimal risk should be eligible for review using procedures other than full IRB review.
Limited Utility of the Category of Minimal Risk
The current regulatory system and IRBs place too much attention on determining whether research fits into the minimal risk category. As suggested in the framework proposed above for the analysis of risks and potential benefits, risks fall into a two-dimensional continuum of the likelihood of the harm (probability) and the severity of the harm if it were to occur (magnitude). Likelihood of harm runs from very low to high, and the severity of the harm runs from trivial to fatal. IRBs must judge the combination of these two dimensions. A low likelihood of a trivial harm would engender little concern, a high likelihood of a trivial harm or a low likelihood of a severe harm tends to raise more concern, and a high likelihood of a severe harm is of the greatest concern. Investigators and IRBs can seek to minimize risks by designing the study in such a way that there is a reduced likelihood of possible harms, that the magnitude of possible harms is reduced, or both. To establish norms for separating risks into two categories without acknowledging the degree of variation in those risks might foster a misrepresentation of the nature of risk, which might result in an inadequate appreciation of the IRB’s responsibility to insist on appropriate protections and to evaluate and minimize all the risks involved in a research study.
Indeed, as discussed in Chapter 2, for research studies involving novel or controversial ethical issues, no single local IRB should assume sole responsibility for conducting the analysis of risks and potential benefits. The proposed National Office for Human Research Oversight should work with the research community to define such research. This pool of research is not likely to be large and might include, for example, certain gene therapy studies that present novel issues or significant unknown risk, xenotransplantation studies, studies involving genetically altered vectors to control certain infectious diseases, or some studies involving low doses of toxic substances in humans to test antidotes that present novel issues or significant and perhaps unknown risks. Some of these kinds of studies are ethically acceptable and should be conducted. However, this type of research would benefit from additional scrutiny, such as a national review panel, with public input into the review process. In fact, investigators should engage the target community of participants in discussion regarding this research, because having only one or two members who represent the pool of prospective participants on the IRB might not provide a sufficient perspective. The purpose of additional review would be to provide the expertise and experience for these types of studies and to offer guidance so that they could be reviewed ultimately by local IRBs.
In two previous reports, NBAC has recommended the use of national panels for such review—in some areas of research involving persons with mental disorders that may affect decisionmaking capacity and for research using human embryonic stem cells—and the need to involve the public (NBAC 1998; NBAC 1999b). In both reports, NBAC asserted that oversight at the national level could provide the public and policymakers with additional assurance that research involving high risks is being carefully reviewed. Some public commentators expressed strong concerns that such national review could be burdensome and unnecessarily delay research.36,37,38 These are legitimate concerns. However, with careful planning, national oversight mechanisms could be implemented in ways that are constructive and that ensure efficiency and timeliness.
84
Recommendation 4.2: Federal policy should distinguish between research studies that pose minimal risk and those that pose more than minimal risk (see Recommendation 2.5). Minimal risk should be defined as the probability and magnitude of harms that are normally encountered in the daily lives of the general population. If a study that would normally be considered minimal risk for the general population nonetheless poses higher risk for any prospective participants, then the Institutional Review Board should approve the study only if it has determined that appropriate protections are in place for all prospective participants.
Vulnerability
Because most research involves uncertainty, all research participants are vulnerable in some sense. However, it has been suggested that vulnerability, in the context of research, should be understood to be a condition, either intrinsic or situational, of some individuals that puts them at greater risk of being used in ethically inappropriate ways in research.39 Individuals can be used in ethically inappropriate ways even if they have given their informed consent to participate in a research study. For example, poor individuals are enrolled in research even though the benefits of the research will be used primarily by more economically privileged individuals (Cohen 1996; Kolata and Eichenwald 1999). In addition, numerous studies have revealed specific disease associations in certain groups (e.g., sickle cell disease in African Americans, Tay Sachs disease and breast cancer in Ashkenazi Jews), which has led to discrimination and stigmatization when research results are inappropriately reported (King 1998; Weijer 1999a; Weijer 1999b; Wilkinson 1974). More recently, in a study testing whether an experimental measles vaccine could be used in infants too young to receive the standard vaccine, African American and Latino families were not told that the vaccine was experimental or that it had been associated with increased death rates in Africa (Cimons 1996; Gamble 1997; Glenn 1996).
In general, persons are vulnerable in research either because they have difficulty providing voluntary,
National Bioethics Advisory Commission
informed consent arising from limitations in decision-making capacity (as in the case of children) or situational circumstances (as in the case of prisoners), or because they are especially at risk for exploitation (as in the case of persons who belong to undervalued groups in our society). An adequate characterization of vulnerability must attend to both types of concern.
Current Protections
Subpart A of the federal regulations describes two requirements related to the enrollment of vulnerable persons in research. First, IRBs must determine that “the selection of subjects is equitable,” and, in so doing, should be “particularly cognizant of the special problems of research involving vulnerable populations, such as children, prisoners, pregnant women, mentally disabled persons, or economically or educationally disadvantaged persons” (45 CFR 46.111(a)(3); 21 CFR 56.111(a)(3)). Second, IRBs must ensure that “when some or all of the subjects are likely to be vulnerable…additional safeguards have been included in the study to protect the rights and welfare of these subjects” (45 CFR 46.111(b)); 21 CFR 56.111(b) [FDA regulations differ only in sentence structure]).
Some federal departments have additional regulations that provide protection for specific groups. The Department of Education has additional regulations protecting children, the Department of Justice has regulations protecting prisoners, and DHHS has additional protections for selected vulnerable populations: Subpart B pertains to pregnant women and fetuses, Subpart C to prisoners, and Subpart D to children. The Central Intelligence Agency (CIA) and the Social Security Administration follow the DHHS subparts. From this summary, it is clear that coverage of vulnerable individuals is uneven across the federal government. This lack of additional protection is particularly worrisome because, as shown in Table 4.1, every department (except CIA and the Department of Transportation) responding to a survey conducted by NBAC indicated that it sponsored or conducted research involving individuals who are vulnerable in some way.40 The subparts in the DHHS regulations provide two general types of substantive protections. First, in some
85
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
Table 4.1: Agency Sponsorship or Conduct of Research That Targets Participants with Vulnerabilities*
| Mentally | Economically | Educationally | ||||||
| Pregnant | Disabled | Disadvantaged | Disadvantaged | |||||
| Agency | Children | Prisoners | Women | Fetuses | Persons | Persons | Persons | Other |
| CIA | ||||||||
| DOC | ||||||||
| NTIA | X | X | X | X | X | X | X | |
| NIST | X | X | X | |||||
| Census | X | |||||||
| DOD | X | X | X | X | X | Military | ||
| Students | ||||||||
| ED | X | X | X | X | X | X | ||
| DHHS | ||||||||
| ACF | X | X | X | X | ||||
| AHRQ | X | X | X | X | X | |||
| CDC | X | X | X | X | X | X | X | |
| FDA | X | X | X | |||||
| HCFA | X | X | X | Elderly | ||||
| HRSA | X | X | X | |||||
| IHS | X | X | X | X | X | X | X | X |
| NIH | X | X | X | X | X | X | X | X |
| OPRR | ||||||||
| SAMHSA | X | X | X | X | X | X | ||
| HUD | X | X | ||||||
| DOE | X | X | Workers | |||||
| DOJ | ||||||||
| OJP | X | X | X | X | X | X | ||
| COPS | X | X | X | X | ||||
| BOP | X | X | X | X | ||||
| FBI | X | |||||||
| DOT | ||||||||
| FAA | ||||||||
| USCG | ||||||||
| FHA | ||||||||
| NHTSA | ||||||||
| VA | X | X | X | X | X | X | Veterans | |
| NASA | Workers | |||||||
| NSF | X | X | X | X | X | X | X | |
| SSA | X | X | X | X | ||||
| CPSC | X | |||||||
| EPA | X | X | X | |||||
| USAID | X | X | X | X | ||||
| TOTALS | 13/16 | 6/16 | 8/16 | 4/16 | 9/16 | 10/16 | 10/16 | |
*Categories of participants are based on the vulnerable groups identified in 45 CFR 46.
Source: NBAC, “Federal Agency Survey on Policies and Procedures for the Protection of Human Subjects in Research.” This staff analysis is available in Volume II of this report.
86
instances, the regulations limit the level of risk to which participants with vulnerabilities may be exposed. For example, two of the four categories of research involving prisoners are permitted only if the research is no more than minimal risk and are described as follows:
(A) study of the possible causes, effects, and processes of incarceration, and of criminal behavior, provided that the study presents no more than minimal risk and no more than inconvenience to the subjects;
(B) study of prisons as institutional structures or of prisoners as incarcerated persons, provided that the study presents no more than minimal risk and no more than inconvenience to the subjects (45 CFR 46.306).
Second, in some cases, the subparts impose stricter informed consent requirements for vulnerable individuals as compared to the requirements for competent adult participants. For example, in the regulations governing research involving children, permission of the child’s parent or guardian (in some cases, permission of both parents) and assent of the child (if the child is capable of giving assent) are required before enrollment of the child can proceed (45 CFR 46.408).
The current DHHS regulations regarding vulnerable individuals are beset with various conceptual and practical difficulties. For example, the subparts offer no definition of vulnerability and no analysis of the types of characteristics that render persons vulnerable.41 The list of vulnerable groups (children, prisoners, pregnant women, fetuses, mentally disabled persons, and economically and educationally disadvantaged persons) provided in Subpart A, 45 CFR 46.111 and 21 CFR 56.111 is incomplete on the one hand and overly broad on the other. For example, injection drug users, the seriously ill, the elderly, and undocumented immigrants could, in particular circumstances, also be considered vulnerable.42 On the other hand, vulnerability is sensitive to context, and individuals may be vulnerable in one situation but not in another. For example, people of low income are rendered vulnerable in studies offering large financial incentives to take on research risk. In addition, the regulations provide inadequate guidance about the types of safeguards that would be appropriate to protect against the risks associated with vulnerability. Subpart A of the regulations requires IRBs to ensure that such individuals
National Bioethics Advisory Commission
are protected, but it does not describe how to achieve such protection.43 In addition to the shortcomings in the regulations, at least two aspects of the current research enterprise suggest a need to evaluate the protections for vulnerable research participants. Increases in research funding are leading to more research involving human participants, and as more people become research participants, more individuals with vulnerabilities are likely to be included. Moreover, because clinical research often holds the prospect of direct benefit and may be perceived as a means of access to health care, serious illness or lack of health insurance may significantly intensify a person’s desire to be involved in research.
Although the use of subparts in the DHHS regulations makes it clear which groups should be considered vulnerable, this advantage is outweighed by several disadvantages. Given the limited required safeguards, the current regulations provide insufficient respect for persons and are not sufficiently responsive to the full array of vulnerability experienced by prospective participants. The use of regulatory subparts is not the optimal means by which to protect vulnerable individuals for the following reasons:
87
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
improve the protections found in the current subparts, each subpart would have to be revised based on an analytical understanding of vulnerability in order to fully reflect its nature and variability and to contain appropriate safeguards.
| n |
Group-based subparts classify certain persons as vulnerable, rather than classifying situations in which individuals might be considered vulnerable. For example, persons with severe illnesses for which there are no acceptable treatments may be medically vulnerable in certain kinds of clinical trials, but not vulnerable at all in many other types of research (e.g., survey research). |
An analytic approach provides a context-sensitive understanding of vulnerability that avoids the implementation of unnecessary protections, which not only impede the progress of research, but also fail to respect the per-sonhood of the potential participants. Thus, an analytical approach not only provides for greater regulatory efficiency than group-based categorization, but it provides more appropriate safeguards. Further discussion is needed regarding the issue of whether a subpart approach to regulation provides appropriate safeguards.
Nonetheless, the current system of protections should be revised to accommodate different types of vulnerability. An analysis of these characteristics or circumstances provides a list of at least six types of vulnerability.44 Each type of vulnerability is discussed below, and suggestions are made about the types of safeguards that should be required to prevent harming participants who are vulnerable in these ways. Historically, such safeguards have been designed to be protectionistic, focusing exclusively on the risks involved in research. Thus, vulnerable persons have too often simply been excluded from research that might have yielded benefits to them or the groups to which they belong. In developing appropriate safeguards, a balance should be reached between protecting vulnerable persons from harm in research and allowing them to share in its potential benefits (Backlar 2000; Kahn et al. 1998; NBAC 1998).45
Cognitive or Communicative Vulnerability
Prospective research participants who are insufficiently able to comprehend information, deliberate, and make decisions about participation in a proposed
research study have a cognitive or communicative vulnerability. Prospective participants might be vulnerable in this way for one of three reasons. First, those with capacity-related cognitive vulnerability, such as young children, or adults with cognitive impairments that affect decision-making, to some extent lack capacities to make informed choices. Second, those with situational cognitive vulnerability do not lack capacity, but are in situations that do not allow them to exercise their capacities effectively, such as stressful emergencies.46 Third, those with a communicative vulnerability, such as participants who speak or read different languages than do investigators, do not lack capacity, but are in situations that do not allow them to exercise their capacities effectively. This type of vulnerability heightens the risk that investigators will not fully respect the prospective participants because standard informed consent procedures will not suffice. Because this report focuses on competent adults, only situational cognitive and communicative vulnerability will be discussed.
Every effort must be made to enable prospective research participants to exercise autonomous choice (NBAC 1998, 9, 57), and investigators should make every effort to reduce situational barriers that impinge on the ability of prospective participants to exercise their authority. Sometimes, when a situational barrier is temporary, it might be possible to delay enrollment of prospective participants until the situation has passed. In other cases, it might be best to obtain informed consent from participants before exposure to the situation that limits autonomy, for example, obtaining consent from pregnant women who will be studied during labor and delivery. Sometimes such situations are structural. For example, sometimes investigators and prospective participants speak different languages. Language barriers can be reduced by using translators or translating consent forms.
Institutional Vulnerability
Prospective participants may have an institutional vulnerability when they have the cognitive capacity to consent but are subject to the formal authority of others who may have independent interests in whether the prospective participant agrees to enroll in the research study. The most commonly cited examples of individuals
88
facing such institutional influences are prisoners and enlistees in the military, but the category also includes college students when they are required to be research participants for course credit or when such participation could affect their grades. This type of vulnerability increases the risk that one’s decision concerning participation will not be truly voluntary, consequently increasing the risk that one’s personhood will not be fully respected.47 In addition, it presents the risk that the subordinated status of these individuals will be exploited.
To safeguard against the ethically inappropriate enrollment of institutionally vulnerable persons, special attention should be paid to both participant selection and the voluntariness of the choice of prospective participants. Ways to reduce vulnerability might include working with institutional officials before initiating the research study to ensure that there are no inappropriate incentives or pressures to enroll, and when possible, not informing institutional staff about which individuals are participating. For example, if study participation takes one hour, those who refuse could be offered the option of staying in the study area for an hour. In this way, institutional staff would not know who participated and who did not.
Selection of participants from within the institutional setting should be fair and immune from the influence of institutional authorities. For example, in the informed consent process, investigators should emphasize that participation is voluntary, and protections should be in place to protect prospective participants from possible retaliation for their decisions (e.g., no effect on parole status or grades). In addition, during the informed consent process, no institutional authority should be present, except for possibly an ombudsperson.
Deferential Vulnerability
Prospective participants might have a deferential vulnerability when they have the cognitive capacity to consent but are subject to the authority of others who might have independent interests in whether prospective participants agree to enroll in the research study. This category raises the concern that the interests of the prospective participants could be subordinated to those of others. However, with deferential vulnerability, the subordination is affected not by formal hierarchies (as it is
National Bioethics Advisory Commission
with institutional vulnerability), but instead by informal ones.48 Such informal power relationships can be socially constructed (e.g., based on gender, race, or class inequalities, or they can be inequalities of power and knowledge of the kind that occur in doctor-patient relationships), or they can be more subjective in nature (e.g., parents who regularly defer to the wishes of their adult children regardless of their own concerns).49 In any case, deferential vulnerability may be subtle. Like institutional vulnerability, deferential vulnerability heightens the risk that the prospective participant’s decisions will not be truly voluntary. In addition, it presents the special risk that the subordinated status of these individuals will be used to someone else’s advantage, resulting in their exploitation.
It should be noted that not all deferential behavior is subordinating. For example, some individuals might so trust their physicians’ expertise that they defer to them about enrolling in a research study. Physician-investigators should be especially aware of this vulnerability, because when they approach their patients about enrolling in a research study, they could be concerned that refusing will negatively affect attitudes toward them or the quality of care they will receive. Here, as with institutional vulnerability, care must be taken to design the research study to ensure that the informed consent process is truly voluntary and that investigators do not take advantage of the subordinated status of prospective participants. Safeguards might include employing research staff who are sensitive to such deference and who can assess whether the participant is truly exercising autonomy and who can adjust the informed consent process to the prospective participant. Investigators should consider whether to have discussions with the prospective participant with or without the presence of the party to whom he or she ordinarily defers.
Medical Vulnerability
This category concerns potential participants who have serious health conditions for which there are no satisfactory standard treatments (e.g., metastatic cancer or rare disorders).50 Seriously ill individuals are often drawn to research because they or their physicians believe it is the best alternative to standard treatment. In these dire circumstances it can be difficult for prospective participants to weigh the risks and potential benefits
89
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
associated with the research. This type of vulnerability increases the risk that informed consent might be based on misunderstanding potential benefits or might be motivated by a desire to find a treatment. It also increases the risk that these participants will be exploited, because either they have unreasonable expectations about the potential benefits or investigators mislead them about risks and potential benefits, and the risks are not reasonable in relation to the potential benefits (Brody 1998).51 In research involving the medically vulnerable, every effort must be made to ensure that prospective participants are presented with accurate information (to avoid exploitation) and that they comprehend that information. Assuring appropriate comprehension might require more than attending to the clarity of information provided. Because physician-investigators might harbor unrealistic expectations for their patients-participants, and because patients-participants too easily blur the roles played by physician-investigators, these situations can lead to what has been called the therapeutic misconception (Appelbaum et al. 1987; Fost 1998; Levine 1992). In these cases, medical vulnerability could be reduced by having an impartial third party approach prospective participants about enrollment and conduct the informed consent process or by having investigators make third parties available to discuss the research. Prospective participants could also be given time to consider the risks and potential benefits of the study and make a decision about participation. As much as possible, individuals should not learn of a diagnosis or that a standard treatment has failed at the same time that they are being asked to participate in a research study.
Some safeguards relating to physician-investigators can be used. Separating the multiple roles of physicians can help to reduce the therapeutic misconception. For example, when possible, the treating physician should not be the investigator. In addition, regular and ongoing education of physicians regarding the informed consent process will reduce the likelihood of overemphasizing potential benefits and underemphasizing risks.
To avoid the exploitation of prospective participants, every effort should be made to optimize the balance between risks and potential benefits in such research without compromising its scientific integrity (Brody 1998).52 IRBs can play a significant role in reducing
medical vulnerability by carefully weighing the risks and potential benefits to be certain that procedures that offer the prospect of direct benefit are not used to justify procedures that involve considerable risk and no prospect of direct benefit. Further, in doing so, IRBs would ensure that prospective participants are offered a “reasonable choice.”
Economic Vulnerability
Prospective participants might have an economic vulnerability when they have the cognitive capacity to consent but are disadvantaged in the distribution of social goods and services such as income, housing, or health care. This type of vulnerability heightens the risk that the potential benefits from participation in the research study might constitute undue inducements to enroll, threatening the voluntary nature of the choice and raising the danger that the potential participant’s distributional disadvantage could be exploited. For example, offers of large sums of money as payment for participation or access to free health care services (for conditions not related to the research) could lead some prospective participants to enroll in a research study when it might be against their better judgment and when otherwise they would not do so. To safeguard against this vulnerability, IRBs should make certain that research offers a “reasonable choice” to prospective participants. This might be an easy assessment for the IRB reviewing a research study in which payment is involved, and the amount of payment could be reduced. However, it can be more difficult for the IRB when the potential benefits include access to free medical care or social or other services.
Social Vulnerability
Prospective participants might have a social vulnerability when they have the cognitive capacity to consent but belong to undervalued social groups. The treatment of members of such groups is not simply attributable to their economic vulnerability, although it is true that members of undervalued groups often lack financial resources. Social vulnerability is a function of the social perception of certain groups, which includes stereotyping and can lead to discrimination. In any case, the perceptions devalue members of such groups, their interests, their welfare, or their contributions to society.
90
These social perceptions are pervasive and often insidious and can affect persons’ conceptions of certain groups. Thus, investigators, IRB members, and research sponsors should be sensitive to such social perceptions and their effects, and efforts should be made to allow members of such groups to participate in decisionmaking and oversight processes (CDC et al. 1998; Eckenwiler 1999; Eckenwiler 2000). Involving the community in the various stages of the research process, especially in study planning, can be helpful in reducing stereotyping and stigmatization. Also, investigators and IRBs should consider whether studies can be designed to include participants from all segments of society, rather than only or primarily from socially undervalued segments.
Aligning Protections with Vulnerabilities
Although there may be some overlap among the six distinct, ethically relevant types of vulnerability described above, each depicts a particular susceptibility to be used in ethically inappropriate ways in research. Moreover, many individuals might experience more than one of these vulnerabilities. For example, prospective participants might be poor, seriously ill, and not conversant in English. When multiple vulnerabilities exist, appropriate safeguards to address each vulnerability should be in place.
This model improves on the current DHHS regulations, which focus on groups of vulnerable people rather than on types of vulnerability, in three ways. First, it recognizes a fuller array of vulnerabilities that might be experienced by members of a particular group, while current regulations often fail to recognize that members of a vulnerable group might be vulnerable in more than one way. For example, current regulations recognize the institutional vulnerability of prisoners, but not their economic or social vulnerability. Second, the model portrays certain individuals as vulnerable in certain circumstances, while the current regulations classify entire groups as vulnerable. For example, current regulations classify all pregnant women as vulnerable, even though women are seldom vulnerable because of the pregnancy itself. Rather, pregnant women might at certain times be medically vulnerable (e.g., during labor). In addition, the current regulations classify economically disadvantaged
National Bioethics Advisory Commission
persons as vulnerable, but, in certain situations, they would not be (e.g., certain types of survey research involving no remuneration).
The proposed model better expresses respect for persons by allowing people to be treated as individuals rather than as members of a group. Such an analytical model both challenges investigators and enables them and IRBs to extend their consideration of vulnerability beyond the incomplete list of vulnerable groups provided in the current regulations and to account for variations among prospective participants.
Third, the model also suggests appropriate safeguards for each type of vulnerability. The current system proposes two general sorts of safeguards: limiting the risk to which participants may be exposed and implementing stricter consent requirements. Given the limited variety of substantive safeguards required, current regulations are not sufficiently responsive to the full array of vulnerabilities. Safeguards must be tailored to respond to particular types and should avoid the exclusively protectionistic attitude toward vulnerability inherent in the current regulations. In general, the suggested safeguards have been designed to strike a balance between protecting vulnerable persons from harm and allowing them to reap the potential benefits of participation in research.
For all types of vulnerability, IRBs should ensure that the risks to which vulnerable persons are exposed would be acceptable to all prospective participants, i.e., those who are vulnerable as well as those who are not. Because the perspectives and experiences of vulnerable persons can differ considerably from those who are not vulnerable, vulnerable persons should be encouraged to participate in the study design and oversight processes (CDC et al. 1998; Eckenwiler 1999; Eckenwiler 2000).53 Such participatory processes serve as important safeguards in research involving vulnerable persons and can help to build trust in the research enterprise.
In designing a research study, investigators should consider how they would handle prospective participants who are vulnerable. To the extent possible, investigators and IRBs should try to identify, at the time of the IRB’s initial review of a research study, the types of vulnerabilities of individuals who might be enrolled. In many studies, enrolling prospective participants who might be
91
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
vulnerable can be anticipated by its stated purpose and design or by the inclusion criteria. When such individuals can be identified up front, IRBs can require that appropriate safeguards be in place before the initiation of the study. However, in some studies, it might not be possible to anticipate the inclusion of prospective participants with vulnerabilities until enrollment. In general, such prospective participants should not be enrolled until the protocol has been discussed with the IRB and then potentially revised and approved. IRBs should not halt the study, but should consider delaying enrollment of these individuals. In addition, IRBs should develop mechanisms for reviewing such protocols.
Finally, unless the particular research study requires such an approach (e.g., a research study of the efficacy of a particular drug when used in children), as a matter of justice, it is never appropriate to target persons with vulnerabilities for disproportionate inclusion or exclusion in research. This position is endorsed in a previous NBAC report (NBAC 1998).
Use of Minimal Risk as It Relates to Vulnerability
It has been suggested that research involving no more than minimal risk should receive full IRB board review if it involves vulnerable persons.54 However, this is not necessarily true. If all types of risk are identified and considered and research components are appropriately determined to involve no more than minimal risk, then such research should be eligible for review using procedures other than full IRB review, because such research, by definition, involves no more risk than is normally encountered in the daily life of the general population. To require full board review for research involving such little potential for harm is to adopt an unsupportable protectionist stance that imposes unnecessary bureaucratic procedures. The protection of vulnerable research participants must be balanced with the pursuit of benefits both to those participants and to society. At the same time, any review of research involving vulnerable individuals is responsible for ensuring that the protections include
appropriate safeguards to protect against the particular vulnerabilities involved. However, as the level of risk increases above minimal risk, the level of scrutiny should also increase, as should the protections required by an IRB.
Appropriate type-specific safeguards should be put in place to protect persons with such vulnerabilities. For example, for research involving socially vulnerable persons, it is appropriate to educate investigators, IRBs, and sponsors about stigmatizing and discriminatory social conceptions of these persons and to design studies to avoid the reflection or perpetuation of such problematic social conceptions. Given the existence of such safeguards, investigators should not exclude such persons from research involving greater than minimal risk in any of the components, for to exclude them would be to deprive them of whatever potential direct benefits they might receive from the research and to deprive their communities and society in general from the benefit of knowledge the research might generate. Moreover, to exclude these persons would be to treat them as though they are not autonomous agents, which would be a violation of the principle of respect for personhood. Once safeguards are in place that are tailored to address their particular variety (or varieties) of vulnerability, vulnerable persons who have the cognitive capacity to provide informed consent should be allowed to exercise autonomous choice about their participation in research. IRBs should exclude such persons from research studies only if they determine that appropriate safeguards cannot satisfactorily be put in place. However, efforts must be made to institute these safeguards and to allow for the inclusion of such persons in the research study.
Recommendation 4.3: Federal policy should promote the inclusion of all segments of society in research. Guidance should be developed on how to identify and avoid situations that render some participants or groups vulnerable to harm or coercion. Sponsors and investigators should design research that incorporates appropriate safeguards to protect all prospective participants.
92
Summary
No person should be enrolled in research until an IRB concludes that the risks are reasonable in relation to the potential benefits. Research studies often involve multiple components or procedures. With some components, the procedures that participants will endure will involve some risk with no direct potential benefit accruing to them. In these situations, some limitation should be placed on the amount of risk that can be imposed, regardless of the participants’ willingness to participate. Other components of a study might involve procedures that also offer the prospect of direct benefit—for example, when standard medical interventions are being compared in a patient population. In such cases, it is essential that participants and investigators do not fall into the therapeutic misconception, in which they come to believe that participating in research is tantamount to being in a more traditional therapeutic relationship. Thus, for research that has both types of components, it is important that risks and potential benefits be appropriately evaluated by the IRB. In other words, the possibility that one component of a study will provide benefits should not be used to justify otherwise unacceptable elements of the research, the potential benefits of which (if any) accrue solely to society.
Even when risks are reasonable and informed consent is obtained, it may nonetheless be wrong to enroll certain individuals as participants. Although individuals should not be arbitrarily excluded from research because they are viewed as vulnerable, often it is not any personal characteristic that causes this vulnerability, but certain situations that make individuals prone to exploitation. Often, safeguards can be provided in the research so that it does not create situations in which people are unnecessarily harmed and so that it protects their rights and welfare. Research participants should be treated equally and with respect, and whenever possible, research should be designed to encourage the participation of all groups, while protecting their rights and welfare.
National Bioethics Advisory Commission
Notes
1 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
2 See Goldman, J., “Privacy and Confidentiality in Health Research.” This background paper was prepared for NBAC and is available in Volume II of this report.
3 See Prentice, E.D., and B.G. Gordon, “Institutional Review Board Assessment of Risks and Benefits Associated with Research.” This background paper was prepared for NBAC and is available in Volume II of this report.
4 Ibid. 5 Ibid. 6 Ibid.
7 See Weijer, C., “The Ethical Analysis of Risks and Potential Benefits in Human Subjects Research: History, Theory, and Implications for U.S. Regulation.” This background paper was prepared for NBAC and is available in Volume II of this report.
8 See Prentice, E.D., and B.G. Gordon, “Institutional Review Board Assessment of Risks and Benefits Associated with Research.” This background paper was prepared for NBAC and is available in Volume II of this report.
9 See Prentice, E.D., and B.G. Gordon, “Institutional Review Board Assessment of Risks and Benefits Associated with Research.” This background paper was prepared for NBAC and is available in Volume II of this report.
10 See Buchanan, A., “An Ethical Framework for Biological Samples Policy.” This background paper was prepared for NBAC and is available in Volume II of Research Involving Human Biological Materials: Ethical Issues and Policy Guidance, B-1–B-31, 1999.
11 Ibid.
12 See Prentice, E.D., and B.G. Gordon, “Institutional Review Board Assessment of Risks and Benefits Associated with Research.” This background paper was prepared for NBAC and is available in Volume II of this report.
13 See Weijer, C., “The Ethical Analysis of Risks and Potential Benefits in Human Subjects Research: History, Theory, and Implications for U.S. Regulation.” This background paper was prepared for NBAC and is available in Volume II of this report.
14 See Prentice, E.D., and B.G. Gordon, “Institutional Review Board Assessment of Risks and Benefits Associated with Research.” This background paper was prepared for NBAC and is available in Volume II of this report.
15 IRBs should recognize, however, in their deliberations that some risks to others should not be considered in their assessment—for example, financial losses to a company in a research study involving a licensed product, reputational damage to an institution, or inability of the investigator to publish study results.
93
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
16 See Prentice, E.D., and B.G. Gordon, “Institutional Review Board Assessment of Risks and Benefits Associated with Research.” This background paper was prepared for NBAC and is available in Volume II of this report.
17 See Weijer, C., “The Ethical Analysis of Risks and Potential Benefits in Human Subjects Research: History, Theory, and Implications for U.S. Regulation.” This background paper was prepared for NBAC and is available in Volume II of this report..
18 Ibid. 19 Ibid. 20 Ibid. 21 Ibid. 22 Ibid. 23 Ibid. 24 Ibid. 25 Ibid. 26 Ibid. 27 Ibid. 28 Ibid. 29 Ibid. 30 Ibid.
31 The Commission has discussed the concept of minimal risk at some length (NBAC 1998).
32 See Prentice, E.D., and B.G. Gordon, “Institutional Review Board Assessment of Risks and Benefits Associated with Research,” and Weijer, C., “The Ethical Analysis of Risks and Potential Benefits in Human Subjects Research: History, Theory, and Implications for U.S. Regulation.” These background papers were prepared for NBAC and are available in Volume II of this report.
33 Ibid.
34 46 Fed. Reg. 8366.
35 See Weijer, C., “The Ethical Analysis of Risks and Potential Benefits in Human Subjects Research: History, Theory, and Implications for U.S. Regulation.” This background paper was prepared for NBAC and is available in Volume II of this report.
36 Letter from J.E. Fanale, American Geriatrics Society, to H.T. Shapiro. February 15, 2001. Providing comments on a draft of this report.
37 Letter from M.J. Werner, Biotechnology Industry Organization, to NBAC. February 16, 2001. Providing comments on a draft of this report.
38 Letter from S. McConnell, Alzheimer’s Association, to H. T. Shapiro. February 19, 2001. Providing comments on a draft of this report.
39 See Kipnis, K., “Vulnerability in Research Subjects: A Bioethical Taxonomy,” and Moreno, J, “Protectionism in Research Involving Human Subjects.” These background papers were prepared for NBAC and are available in Volume II of this report. See Fisher, C.B., “Relational Ethics and Research with Vulnerable Populations.” This background paper was prepared for NBAC and is available in Volume II of the report Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity, 29–49, 1998.
40 NBAC, “Federal Agency Survey on Policies and Procedures for the Protection of Human Subjects in Research.” This staff analysis is available in Volume II of this report.
41 See Kipnis, K., “Vulnerability in Research Subjects: A Bioethical Taxonomy.” This background paper was prepared for NBAC and is available in Volume II of this report.
42 Ibid. 43 Ibid. 44 Ibid. 45 Ibid. 46 Ibid. 47 Ibid. 48 Ibid. 49 Ibid. 50 Ibid. 51 Ibid. 52 Ibid.
53 See Fisher, C.B., “Relational Ethics and Research with Vulnerable Populations.” This background paper was prepared for NBAC and is available in Volume II of the report Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity, 29–49, 1998.
54 See Weijer, C., “The Ethical Analysis of Risks and Potential Benefits in Human Subjects Research: History, Theory, and Implications for U.S. Regulation.” This background paper was prepared for NBAC and is available in Volume II of this report.
94
References
Appelbaum, P.S., L.H. Roth, C.W. Lidz, P. Benson, and W.
Winslade. 1987. “False Hopes and Best Data: Consent to Research and the Therapeutic Misconception.” Hastings Center Report 17(2):20–24.
Backlar, P. 2000. “Human Subjects Research, Ethics, Research on Vulnerable Populations.” In Encyclopedia of Ethical, Legal, and Policy Issues in Biotechnology, eds. T.H. Murray and M.J. Mehlman, 641–651. New York: Wiley-Interscience.
Brody, B.A. 1998. Research on the Vulnerable Sick. In Beyond Consent: Seeking Justice in Research, eds. J.P. Kahn, A.C. Mastroianni, and J. Sugarman, 32–46. New York: Oxford University Press.
Census Bureau. 2000. Educational Attainment in the United States
(Update). Available at http://www.census.gov/population/www/ socdemo/educ-attn.html. Last accessed April 24, 2001.
Centers for Disease Control and Prevention (CDC), National Institutes of Health (NIH), Food and Drug Administration (FDA), Health Resources and Services Administration, Substance Abuse and Mental Health Services Administration, and Indian Health Service. 1998. Building Community Partnerships in Research.
Recommendations and Strategies. Report to the President. 10 February.
Cimons, M. 1996. “CDC Says It Erred in Measles Study.”
Los Angeles Times, 17 June, A-11.
Cohen, L.P. 1996. “To Screen New Drugs for Safety, Lilly Pays Homeless Alcoholics.” Wall Street Journal, 14 November, p. 1.
Davis D. 2000. “Groups, Communities, and Contested Identities in Genetic Research.” Hastings Center Report 30(6):38–45.
Department of Health, Education, and Welfare (DHEW). Secretary’s Task Force on the Compensation of Injured Research Subjects. 1977. Final Report. Pub. OS-77-003. Bethesda, MD: DHEW.
Eckenwiler, L.A. 1999. “Pursuing Reform in Clinical Research: Lessons from Women’s Experience.” Journal of Law, Medicine, and Ethics 27(2):158–170.
———. 2000. “Moral Reasoning and Review of Research Involving Human Subjects.” Unpublished Manuscript. NBAC archives.
Ellis, G.B. 1995. Federal Policy for the Protection of Human Subjects. In Report on the Public Forum on Informed Consent in Clinical Research Conducted in Emergency Circumstances. Rockville/Bethesda, MD: FDA/NIH.
Fitzgerald, J.L., and M. Hamilton. 1996. “The Consequences of Knowing Ethical and Legal Liabilities in Illicit Drug Research.”
Social Science and Medicine 43(11):1591–1600.
———. 1997. “Confidentiality, Disseminated Regulation and Ethico-Legal Liabilities in Research with Hidden Populations of Illicit Drug Users.” Addiction 92(9):1099–1107.
National Bioethics Advisory Commission
Fost, N. 1998. “Ethical Dilemmas in Medical Innovation and Research: Distinguishing Experimentation from Practice.” Seminars in Perinatology 22(3):223–232.
Freedman, B. 1987a. “Equipoise and the Ethics of Clinical Research.” New England Journal of Medicine 317(3):141–145.
———. 1987b. “Scientific Value and Validity as Ethical
Requirements for Research: A Proposed Explication.” IRB: A Review of Human Subjects 9(6):7–10.
Gamble, V.N. 1997. “Under the Shadow of Tuskegee: African Americans and Health Care.” American Journal of Public Health
87(11):1773–1778.
Glass, K.C., C. Weijer, T. Lemmens, R.M. Palmour, and S.H. Shapiro. 1997. “Structuring the Review of Human Genetics Protocols, Part II: Diagnostic and Screening Studies.” IRB: A Review of Human Subjects Research 19(3–4):1–13.
Glenn, B. 1996. “Bad Blood Once Again.” St. Petersburg Times, 21 July, 5-D.
Kahn, J.P., A.C. Mastroianni, and J. Sugarman, eds. 1998. Beyond Consent: Seeking Justice in Research. New York: Oxford University Press.
King, P.A. 1998. “Race, Justice, and Research.” In Beyond Consent: Seeking Justice in Research, eds. J.P. Kahn, A.C. Mastroianni, and J. Sugarman, 88–110. New York: Oxford University Press.
Kolata, G., and K. Eichenwald. 1999. “Stopgap Medicine: A Special Report. For the Uninsured, Drug Trials are Health Care.” New York Times, 22 June, A-1.
Levine, R.J. 1986. Ethics and Regulation of Clinical Research. 2d ed. New Haven: Yale University Press.
———. 1992. “Clinical Trials and Physicians as Double Agents.”
Yale Journal of Biology and Medicine 65(2):65–74.
Marteau, T.M., and R.T. Croyle. 1998. “Psychological Responses to Genetic Testing.” British Medical Journal 316(7132):693–696.
Martin, D.K., E.M. Meslin, N. Kohut, and P.A. Singer. 1995. “The Incommensurability of Research Risks and Benefits: Practical Help for Research Ethics Committees.” IRB: A Review of Human Subjects Research 17(2):8–10.
Meiser, B., and S. Dunn. 2000. “Psychological Impact of Genetic Testing for Huntington’s Disease: An Update of the Literature.”
Journal of Neurology, Neurosurgery and Psychiatry 69(5):574–578.
Milgram, S. 1974. Obedience to Authority: An Experimental View. 1st ed. New York: Harper and Row.
National Bioethics Advisory Commission (NBAC). 1998.
Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity. 2 vols. Rockville, MD: U.S. Government Printing Office.
———. 1999a. Ethical Issues in Human Stem Cell Research. 3 vols. Rockville, MD: U.S. Government Printing Office.
95
Chapter 4: Assessing Risks and Potential Benefits and Evaluating Vulnerability
———. 1999b. Research Involving Human Biological Materials: Ethical Issues and Policy Guidance. 2 vols. Rockville, MD: U.S. Government Printing Office.
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission). 1975.
Report and Recommendations: Research on the Fetus. Washington, D.C.: DHEW.
———. 1976. Report and Recommendations: Research Involving Prisoners. Washington, D.C.: DHEW.
———. 1977. Report and Recommendations: Research Involving Children. Washington, D.C.: DHEW.
———. 1978. Report and Recommendations: Institutional Review Boards. Washington, D.C.: U.S. Government Printing Office.
———. 1979. Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research. Washington, D.C.: U.S. Government Printing Office.
Office for Protection from Research Risks (OPRR). 1993.
Protecting Human Research Subjects. Institutional Review Board Guidebook. Washington, D.C.: U.S. Government Printing Office.
President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission). 1982. Compensating for Research Injuries: The Ethical and Legal Implications of Programs to Redress Injured Subjects. Washington, D.C.: U.S. Government Printing Office.
Singer, M. 2000. Qualitative Methods and Preliminary Findings on Sterile Syringe Access and Use Among Injection Drug Users in Three Northeastern Cities. 20 September. Presentation at Centers for Disease Control and Prevention. Atlanta, Georgia.
Singer M., T. Stopka, C. Siano, K. Springer, G. Barton, K. Khoshnood, A.G. de Puga, and R. Heimer. 2000. “The Social Geography of AIDS and Hepatitis Risk: Qualitative Approaches for Assessing Local Differences in Sterile-Syringe Access Among Injection Drug Users.” American Journal of Public Health
90(7):1049–1056.
Weijer, C. 1999a. “Selecting Subjects for Participation in Clinical Research: One Sphere of Justice.” Journal of Medical Ethics 25:31–36.
———. 1999b. “Protecting Communities in Research:
Philosophical and Pragmatic Challenges.” Cambridge Quarterly of Healthcare Ethics 8(4):501–513.
Wilkinson, D.Y. 1974. “For Whose Benefit? Politics and Sickle Cell.” The Black Scholar 5:26–31.
96
Chapter Five 5
Ensuring Voluntary Informed Consent and Protecting Privacy and Confidentiality
Introduction
Two analyses that are central to the ethical review conducted by Institutional Review Boards (IRBs) are an appraisal of the informed consent process to be used in a proposed research study and a determination of the extent to which the privacy and confidentiality of the potential participants will be protected. The current regulatory requirements relating to these ethical issues reflect the concept of respect for persons, as espoused in the Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research (Belmont Report)
(National Commission 1979). Informed consent becomes an essential protection, however, only after research has been determined to be ethically justifiable in terms of beneficence and justice. Adherence to these principles in the review of research serves a gatekeeper function, permitting only ethically acceptable research involving human participants to proceed.1
Voluntary Informed Consent
The principle of respect for persons requires that 1) individuals are treated as autonomous agents and 2) that persons with diminished autonomy are protected (National Commission 1979). (This report addresses informed consent only in the context of research involving competent adults.) An autonomous agent is “an individual capable of deliberation about personal goals and of acting under the direction of such deliberation” (National Commission 1979, 5). Respect for persons requires that prospective research participants “be given the opportunity to choose what shall or shall not happen to them” and thus necessitates adequate standards for informed
consent (National Commission 1979, 10). As Vanderpool notes, the Belmont Report’s insistence on informed consent “accords with the fundamental moral values of a free and democratic society.”2 The informed consent process involves three elements: 1) disclosing information to potential research participants, 2) ascertaining that they understand what has been disclosed, and 3) ensuring their voluntariness in agreeing to participate in research (Faden and Beauchamp 1986). The disclosure of information should be sufficient “such that persons…can decide whether they wish to participate in the furthering of knowledge” (National Commission 1979, 11). Even when some direct benefit to participants may be anticipated, this high standard for disclosure should be met, because research inherently involves uncertainty and is always designed to acquire knowledge. To ensure comprehension, the presentation of information must be adapted to the potential participant’s capacities and characteristics, and great care must be taken to ascertain that the prospective participant understands the information. Extra efforts may be warranted to verify comprehension when risks are especially high or there is uncertainty whether the prospective participants are capable of understanding the risks. For consent to be voluntarily given, the process must be free of coercion and undue influence.
In previous reports, NBAC has focused much of its attention on addressing specific issues related to informed consent that arise from the interpretation of existing regulations. (See Exhibit 5.1.) This chapter looks beyond current regulations to focus on the overall scope and structure of the requirements for informed consent and focuses specifically on four issues: 1) emphasis on
97
Chapter 5: Ensuring Voluntary Informed Consent and Protecting Privacy and Confidentiality
Exhibit 5.1:
Previous NBAC Recommendations Regarding Informed Consent
NBAC has addressed the issue of informed consent in four previous reports (NBAC 1998; NBAC 1999a; NBAC 1999b; NBAC 2001), and, because the topic is central to the protections offered to research participants, recommendations regarding informed consent have played a key role in each of these reports. NBAC, for example:
| 1. |
fully supports the principle of voluntary informed consent as an essential safeguard to protect participants’ rights and welfare (NBAC 1998; NBAC 1999a; NBAC 1999b; NBAC 2001); |
| 2. |
has acknowledged that certain research contexts may require greater flexibility in procedures used to obtain consent, for example, where cultural or language differences exist (NBAC 1999b; NBAC 2001); |
| 3. |
recognizes the importance of appropriate limitations regarding the use of substitute or third-party consent (NBAC 1998); |
| 4. |
recommends that individuals be provided with certain options for providing informed consent regarding future research studies (NBAC 1998; NBAC 1999b); |
| 5. |
acknowledges that it is sometimes difficult to distinguish between research and treatment and has suggested that the distinction needs to be clear to participants (NBAC 1998; NBAC 1999b; NBAC 2001); |
| 6. |
recognizes the importance of limiting coercion or undue influence in obtaining voluntary informed consent (NBAC 1999b; NBAC 2001); |
| 7. |
recognizes that informed consent should be sought from donors who are contributing biological materials to research (NBAC 1999b); and |
| 8. |
emphasizes that informed consent is a process and not merely a form to be signed in a routine manner (NBAC 1998; NBAC 1999b; NBAC 2001). |
disclosure; 2) appropriateness of informed consent processes for different types of research; 3) waiver of the informed consent process; and 4) documentation of the informed consent process.
Deficiencies with the Current Regulations
Federal regulations permit IRBs to approve research when informed consent is sought and documented from each prospective participant (45 CFR 46.111(a)(4)-(5);
21 CFR 56.111(a)(4)-(5)). Requirements for informed consent are further described in two sets of federal regulations in 45 CFR 46.116 and 45 CFR 46.117, and 21 CFR 50.20, 50.25, 50.27 and 56.109. There are substantial differences between the two sets of requirements; the most notable being that the FDA regulations do not contain the criteria for waiver or alteration of informed consent as described in 45 CFR 46.116(d). The current regulatory system specifies eight basic elements of information disclosure that must be provided to prospective participants during the informed consent process, except in cases of an approved waiver or alteration of the consent process by the IRB. (See Exhibit 5.2.) Also specified are six additional elements of disclosure that must be included when appropriate (46 CFR 46.116(b); 21 CFR 50.25(b)). The regulations for documentation require that, except in specified circumstances, “informed consent shall be documented by the use of a written consent form approved by the IRB and signed by the subject or the subject’s legally authorized representative” (45 CFR 46.117(a); 21 CFR 50.27(a) [FDA regulations differ in requiring that the form be dated at the time the consent form is signed]).
Emphasis on Disclosure
Although the regulations pertaining to informed consent (45 CFR 46.116; 21 CFR 50.20) begin with a paragraph from the Belmont Report related to the “three elements” of consent—information, comprehension, and voluntariness—the actual procedures emphasize disclosure requirements, and in so doing may distort the understanding of the ethical principle of respect for persons.3 Especially in the context of requirements that informed consent be documented using signed, written forms, this exclusive emphasis on information leads
98
Exhibit 5.2: Required Elements of Disclosure
| 1. |
A statement that the study involves research, an explanation of the purposes of the research and the expected duration of the subject’s participation, a description of the procedures to be followed, and identification of any procedures which are experimental. |
| 2. |
A description of any reasonably foreseeable risks or discomforts to the subject. |
| 3. |
A description of any benefits to the subject or to others which may reasonably be expected from the research. |
| 4. |
A disclosure of appropriate alternative procedures or courses of treatment, if any, that might be advantageous to the subject. |
| 5. |
A statement describing the extent, if any, to which confidentiality of records identifying the subject will be maintained. |
| 6. |
For research involving more than minimal risk, an explanation as to whether any compensation and an explanation as to whether any medical treatments are available if injury occurs, and, if so, what they consist of or where further information may be obtained. |
| 7. |
An explanation of whom to contact for answers to pertinent questions about research and research subjects’ rights, and whom to contact in the event of a research-related injury to the subject. |
| 8. |
A statement that participation is voluntary, refusal to participate will involve no penalty or loss of benefits to which the subject is otherwise entitled, and the subject may discontinue participation at any time without penalty or loss of benefits to which the subject is otherwise entitled (45 CFR 46.116(a); 21 CFR 50.25(a) [FDA regulations differ in requiring an additional statement that FDA may inspect records]). |
investigators and IRBs to equate informed consent with the information provided in a consent form and to focus mostly on disclosures to participants.4 Thus, these regulations fail to emphasize the informed consent process and to attend to the importance of comprehension and voluntariness, a failure the implications of which have
National Bioethics Advisory Commission
been described in previous NBAC reports (NBAC 1998; NBAC 2001). Moreover, others have noted that the particular emphasis on disclosure in informed consent is a product of legal and regulatory thinking that equates informed consent solely with the requirements to disclose information (Beauchamp and Childress 1994, 145). This emphasis is not, however, fully conducive to the more comprehensive view of informed consent, which emphasizes the aspects of understanding, capacity to consent, voluntariness, and features of decisionmaking, including who may authorize consent (Beauchamp and Childress 1994; Faden and Beauchamp 1986).
Empirical evidence suggests that these regulatory shortcomings all too frequently result in failures to achieve voluntary informed consent (Verheggen et al. 1996; Waggoner and Mayo 1995). For example, a study by the Advisory Committee on Human Radiation Experiments (ACHRE) reviewed materials submitted to and approved by IRBs from a sample of federally funded studies. Generally, consent forms were difficult to understand, requiring an advanced reading level and including technical language and overwhelming level of detail (ACHRE 1995, 707). Difficulty in understanding consent forms has been reported in many research studies (Blenkinsop 1997; Hochhauser 1997; Waggoner and Mayo 1995; Young et al. 1990). Indeed, another study by ACHRE found that at least some individuals failed even to understand whether they had participated in research at all. At least 5 percent of patients who reported they were not research subjects actually were, and at least 6 percent of patients who reported they were in research actually were not (ACHRE 1995, 736). In general, these studies reveal the deficiencies in the standard practice of seeking informed consent. Anecdotal evidence in support of these findings was provided at each of the five town meetings held by NBAC. For example, IRBs are concerned that there is too much emphasis on consent forms, that forms are too long and written at a reading level that is too advanced, and most important, that participants often do not understand to what they are giving their consent.5 Rather than focusing on the ethical standard of informed consent and what that entails for the process of obtaining informed consent, IRBs and investigators have
99
Chapter 5: Ensuring Voluntary Informed Consent and Protecting Privacy and Confidentiality
followed the lead of the regulations and tended to focus on disclosures found in the consent form. However, from an ethics perspective, the informed consent process is the critical communication link between the prospective participant and the investigator, beginning with the initial approach of the investigator to the participant (e.g., via a flyer, brochure, or any advertisement regarding the research study) and continuing until the end of the project. It should be an active process of sharing information by both parties throughout which the participant at any time is able to freely decide whether to withdraw or continue participating in the research. The consent form, if there is one, is intended only to document the interaction between the participant and the investigator, and it is only one part of the informed consent process. It is time to emphasize the process of informed consent, with the goals of ensuring that information is fully disclosed, that competent participants fully understand the research so that they can make informed choices, and that their decisions whether to participate are always made voluntarily.
Informed Consent Processes Appropriate for Various Research Settings
Although placing greater emphasis on the informed consent process is crucial, it is also important to tailor the process to suit the type of research being proposed and the interests and situations of the potential participants. The eight basic elements of disclosure specified in 45 CFR 46.116(a) and 21 CFR 50.25(a) are appropriate for clinical research, but some of them are inappropriate to the point of being nonsensical for other types of research, especially behavioral and social science or historical research. For example, the basic element requiring “a disclosure of appropriate alternative procedures or courses of treatment, if any, that might be advantageous to the subject” (45 CFR 46.116(a)(4); 21 CFR 50.25(a)(4)) makes little sense for research studies involving no interventions. Because of this requirement, forms often contain illogical statements that can confuse participants. Some IRBs and the Office for Human Research Protections (OHRP) require a statement in the consent form, for example, that the advantageous alternative procedure or treatment is to not participate in the research. Similarly,
including a statement that refusal to participate will involve no penalty or loss of benefits (45 CFR 46.116(a)(8); 21 CFR 5025(a)(8)) raises concerns in some survey research in which there are no benefits to be lost; in such cases, participants may become unduly alarmed (McQuillan et al. 2000).
Although any of these basic elements of consent can be altered or waived, the current criteria for doing so are often difficult to meet. One of the criteria for alteration or waiver is that the research could not practicably be carried out without the waiver or alteration (45 CFR 46.116(d)(3)). Because IRBs often interpret “could not practicably be carried out” to mean impossible to carry out, they require the element of disclosure to be included, often in a less than meaningful way (NBAC 1999b). In other cases, investigators and IRBs spend much time documenting alteration or waiver justifications in order to meet the stringent standards set in the criteria, even though those criteria are not relevant within the context of the research.
Moreover, when considering what information should be provided to prospective participants, IRBs and investigators should strive to ascertain what would be important to the prospective participants from the participants’ perspectives. For example, before initiating research on genetic screening for breast cancer, investigators held focus groups with prospective participants to ask them what they were interested in knowing about the proposed research. Rather than being solely interested in the type of information required under the regulations for disclosure, prospective participants were interested in knowing about options to reduce the risk of developing cancer, the cost of testing, and insurance coverage for the test. This information would not have been included in the informed consent process without input from prospective participants (Bernhardt et al. 1997). Moreover, this information was as relevant to their decisionmaking as the information required under the regulations. Elsewhere, NBAC has recommended that prospective participants can provide valuable input into the type of information other prospective participants might want to learn by involving them in the design of studies and the informed consent process (NBAC 1998; NBAC 1999b; NBAC 2001).
100
One specific type of research worthy of mention is deception research. Methods related to deception are sometimes the only valid ones for addressing an issue or question (e.g., study of prejudice). There has been longstanding debate among psychologists and ethicists about the merits of this research (Nicks et al. 1997). In all imaginable cases, deception research involves interaction with participants and thus the opportunity for an informed consent process. In all cases, the informed consent process must be tailored so that the research study can be carried out. Participants cannot be told the true purpose of the research until after the study is over, or participants can be told some, but not all, purposes of the research until the end of the study. When IRBs approve research studies that involve deception, an informed consent process should be designed that is appropriate to the purpose of the study and that provides adequate protection for the participants. For example, in a study designed to determine whether the stereotypical belief that women are not as proficient as men in mathematics adversely affects women’s test scores, two groups of participants were asked to take the same math test, but were given opposite information about whether the test had been shown to produce gender differences. Some or all of this information was false. The study was not intended to reveal whether the test itself produced gender differences in test scores, but rather whether the threat of stereotyping led to gender difference in test scores. Participants were told the true purpose of the study when the study was completed (Spencer et al. 1999).
Although it is tempting to require a set of basic elements of disclosure to be used during the informed consent process, it is unlikely that any single set of basic elements can be applied feasibly and credibly to all types of research. Whether an investigator has included the eight basic elements of disclosure is often open to interpretation. Further guidance is needed not only to describe required elements for different types of research, but also to describe procedures for ascertaining what information should be included in a particular research study.
In certain circumstances, monitoring the informed consent process could increase participant protection, and monitoring procedures could be used to measure
National Bioethics Advisory Commission
participants’ understanding of the nature of the research and the risks involved. Such circumstances might include research involving significant risk, research enrolling participants who might have difficulty in understanding the risks associated with the study, or research for which the IRB is concerned about whether the informed consent is being carried out according to the stipulations in the approved protocol. In these cases, IRBs should require some type of monitoring of the informed consent process, although it need not perform the monitoring itself. These mechanisms could be either temporary, lasting until the concerns of the IRB are satisfied, or permanent, for the duration of the research study.
Prospective Participants Who Cannot Give Voluntary Informed Consent
The discussion thus far has focused on obtaining informed consent from competent adults.6 When individuals who do not have the capacity or who have lost the capacity to provide voluntary informed consent are enrolled in research, other mechanisms must be used to satisfy the ethical requirement of informed consent. A previous NBAC report has addressed this issue with respect to research involving individuals who have a mental disorder that may affect decisionmaking capacity (NBAC 1998). Other situations in which adults are unable to give voluntary informed consent also should be examined.
Recommendation 5.1: Federal policy should emphasize the process of informed consent rather than the form of its documentation and should ensure that competent participants have given their voluntary informed consent. Guidance should be issued about how to provide appropriate information to prospective research participants, how to promote prospective participants’ comprehension of such information, and how to ensure that participants continue to make informed and voluntary decisions throughout their involvement in the research.
Waiver of Informed Consent
Although NBAC supports the ethical standard of informed consent in research, circumstances arise in which the requirement of seeking informed consent from competent participants may be waived (NBAC 1998;
101
Chapter 5: Ensuring Voluntary Informed Consent and Protecting Privacy and Confidentiality
NBAC 1999a; NBAC 1999b; NBAC 2001).7 One circumstance involves the use of existing identifiable data. As noted in an earlier NBAC report on research involving the use of human biological materials, when the study offers the potential to gain important knowledge, poses little or no risk to participants, and consent would be difficult or impossible to obtain, it is appropriate to waive the informed consent requirement (NBAC 1999b).
The current federal regulations stipulate four criteria that must all be met to waive informed consent:
| 1. |
The research involves no more than minimal risk to the participants. |
|
| 2. |
The waiver will not adversely affect the rights and welfare of the participants. |
|
| 3. |
The research could not practicably be carried out without the waiver. |
|
| 4. |
Whenever appropriate, the participants will be provided with additional pertinent information after participating (45 CFR 46.116(d); 21 CFR 50.23, 50.24) |
|
| [ |
FDA regulations only provide for waiver of informed consent in limited emergency situations]). |
|
At the time these requirements were written, one set of criteria was wanted that would address waivers and alterations for both biomedical and behavioral research. Although the third and fourth criteria appear to apply primarily to deception research (Dresser 1981) and are useful in an IRB’s deliberation about altering the informed consent process in deception research, they are not readily applied to other types of research in which an alteration is requested or even when a waiver of the entire informed consent process is requested. As mentioned earlier, one of the most poorly understood terms by IRBs and investigators is practicably.8 Only in a few research studies would it be impossible to obtain informed consent; however, in many studies the financial cost and time required to obtain informed consent would be prohibitive and a potentially poor use of limited research resources.
Indeed, four recommendations in NBAC’s report on the use of human biological materials address the interpretation of these waiver criteria when conducting research with stored biological material (NBAC 1999b). These recommendations, displayed in Exhibit 5.3, were intended to add clarity to the regulations, not to change them.
Exhibit 5.3: Previous NBAC
Recommendations Regarding Waiver of Informed Consent
102
As a matter of policy, two options are available. The first, which NBAC recommended in previous reports, is to specify the clarifications necessary to existing regulations for granting waivers (NBAC 1999b; NBAC 2001). The second is to revise the regulations, given a broader understanding of the ethical standards of informed consent. With this second approach, criteria for altering the consent process would no longer be needed if Recommendation 5.1 of this report were adopted. However, criteria for waiving the informed consent process altogether would still be required.
Waiver of the informed consent process is justifiable in research studies when there is no interaction between investigators and participants, such as in studies using existing identifiable data (e.g., studies of records). In a previous report, NBAC discussed risks associated with research involving the use of existing data (NBAC 1999b), noting that in such studies, risks will usually not be physical and instead are likely to arise from the acquisition, use, or dissemination of information from the research study and thus are likely to involve threats to privacy and breaches in confidentiality. As discussed in the following section, many steps can be taken to protect both privacy and confidentiality, and with such protections in place, IRBs may waive the requirement for informed consent. The criteria for waiving informed consent in such instances should be revised.
Recommendation 5.2: Federal policy should permit Institutional Review Boards in certain, limited situations (e.g., some studies using existing identifiable data or some observational studies) to waive informed consent requirements if all of the following criteria are met:
a) all components of the study involve minimal risk or any component involving more than minimal risk must also offer the prospect of direct benefit to participants; b) the waiver is not otherwise prohibited by state, federal, or international law; c) there is an adequate plan to protect the confidentiality of the data; d) there is an adequate plan for contacting participants with information derived from the research, should the need arise; and
National Bioethics Advisory Commission
e) in analyzing risks and potential benefits, the Institutional Review Board specifically determines that the benefits from the knowledge to be gained from the research study outweigh any dignitary harm associated with not seeking informed consent.
The first criterion is intended to limit exposure to riskier research when informed consent is not obtained, except in circumstances in which a component involving more than minimal risk also offers the prospect of direct benefit. The second criterion is intended to protect participants’ rights existing under federal, state, or international statute. Protecting welfare is removed from the text of the criterion because welfare is protected through criterion a, c, or e in Recommendation 5.2. The third and fourth criteria are recommended on the assumption that strong confidentiality protections can reduce violations of privacy (see discussion below). The fifth criterion is recommended based on the discussion above regarding the justification of research through risk/potential benefit analysis. A criterion related to practicability is not included, because it would not add to the protection of participants if the other criteria are met.
Documentation of Informed Consent
A similar sort of flexibility is needed for the documentation of informed consent. Current regulations require, with limited exception, that informed consent be documented using a written consent form approved by the IRB and signed by the participant (45 CFR 46.117(a); 21 CFR 40.27(a), 56.109(c), (d)). As NBAC noted previously, this requirement could pose inappropriate barriers to conducting useful and ethically sound research that otherwise conforms to regulatory requirements related to informed consent (NBAC 2001), an observation that is made elsewhere as well (Singer 1978).9 A signed, written consent form may be problematic in at least two ways: it must be signed, and it is in writing. In some cultures, both in the United States and in other countries, requesting a signature raises issues of trust and might suggest inappropriate gravity regarding the research. In addition, a written form has little utility with illiterate populations. In certain types of research, such as survey research, signed, written consent forms are not considered part of
103
Chapter 5: Ensuring Voluntary Informed Consent and Protecting Privacy and Confidentiality
the standard methodology. For example, telephone surveys (such as those used by telemarketers) and face-to-face interviews are often encountered as part of daily living, and individuals either freely participate or refuse, for example, by hanging up the telephone. For these reasons, NBAC has distinguished between the substantive ethical standard of informed consent and the procedures by which consent is obtained and has recommended that investigators should not deviate from the substantive ethical standards, although the procedures by which informed consent is obtained could vary if approved by the IRB (NBAC 2001).
Although the federal regulations may have been attempting to reflect a legal standard for documentation of informed consent, NBAC is aware of no case law in which a signed, written consent form is required.10 To fulfill the substantive ethical standard of informed consent, depending upon the sort of research being proposed, it may be more appropriate to use other forms of documentation, such as audiotape, videotape, witnesses, or telephone calls to participants to verify informed consent and participation in the research study.
Recommendation 5.3: Federal policy should require investigators to document that they have obtained voluntary informed consent, but should be flexible with respect to the form of such documentation. Especially when individuals can easily refuse or discontinue participation, or when signed forms might threaten confidentiality, Institutional Review Boards should permit investigators to use other means of verifying that informed consent has been obtained.
Privacy and Confidentiality
All research, whether clinical, epidemiological, health services, or social science, involves issues related to protecting privacy and confidentiality. In fact, in some research the invasion of privacy or breaches in confidentiality are the only research-related risks to which participants might be exposed (e.g., some anthropological, psychological, and historical research studies; records review studies; and secondary analysis of existing data). Privacy and confidentiality, however, tend to be poorly
understood concepts, often mistakenly used interchangeably, and either ignored or inappropriately handled by investigators and IRBs.11 Privacy refers to persons and to their interests in controlling access of others to themselves (Boruch and Cecil 1979). In the research context, privacy refers to how investigators collect or access identifiable data from participants. Protection of privacy naturally follows as an application of the ethical principle of respect for persons, because in respecting the personhood and autonomy of individuals, investigators must be sensitive in the ways in which they interact with participants who are deciding whether to reveal or withhold information. Americans value their privacy and are concerned about the circumstances under which they share information about themselves and, once shared, how the information will be handled (IOM 2000).
Confidentiality, on the other hand, refers to identifiable data and relates to agreements between the participant and the investigator about how the participant’s data will be handled and to whom it will be disclosed. It is an extension of the concept of privacy.12 Participants are concerned about unauthorized or inadvertent disclosures of their data, and although participants and the public are generally supportive of research, they often want to be assured that they are in control of access to their information and that potential breaches in confidentiality are minimized.13 Current federal regulations incompletely address privacy and confidentiality issues in research. IRBs may only approve a research study when the research study is judged to have adequate provisions to protect the privacy of participants and to maintain the confidentiality of data (45 CFR 46.111(a)(7); 21 CFR 56.111(a)(7)). Federal regulations do not provide any additional requirements, stipulate acceptable protection provisions, or define the terms.
Some federal departments have established additional protections relating to privacy and confidentiality that are applicable to research. The Department of Education complies with the Family Education Rights and Privacy Act of 1974, which is designed to protect student records from disclosure without consent from parents or students over 18 years of age. In addition, the Protection of Pupil
104
Rights Amendment gives parents the right to consent for their children to participate in sensitive research. The Department of Justice provides additional regulatory protections for prisoners (28 CFR 512) that give prisoners control over their data, require at least one prisoner and a majority who are not prison personnel to be members of the IRB reviewing the research, and prohibit prison administrators from accessing research data.
Additional confidentiality protections are provided in the National Center for Educational Statistics Confidentiality Statute and the Public Health Service Act for the Centers for Disease Control and Prevention’s (CDC’s) assurance of confidentiality. These statutes limit the use of research data to those purposes for which the participants consent and prohibit the disclosure of identifiable data to anyone without the consent of participants.
The Health Insurance Portability and Accountability Act of 199614 required that legislation regarding standards for the privacy of individually identifiable health information be enacted by August 21, 1999, or DHHS would be required to issue regulations. Because Congress did not pass such legislation, on December 20, 2000, DHHS announced regulations entitled “Standards for Privacy of Individually Identifiable Health Information,” which became effective April 14, 2001. Although these regulations provide some confidentiality protections for individually identifiable health information, their application in the research context is limited because the regulations apply only to health plans, health care clear-inghouses, and health care providers. The regulations outline conditions under which individually identifiable health information held by any of the three entities may be disclosed without the individual’s authorization to investigators. Such disclosures may be made if an IRB or “privacy board” (as defined in the rule) approves a waiver of authorization based on certain criteria, if the information is sought solely for review preparatory to research, or if the research is on a decedent’s information. Notwithstanding these additional federal protections of privacy and confidentiality, there is little federal guidance for IRBs and investigators regarding the protection of privacy and confidentiality.
National Bioethics Advisory Commission
Privacy
Respecting privacy involves more than seeking informed consent from participants to access their data. Obtaining informed consent serves a vital role in providing participants with control over whether they will allow investigators to have access to them and to data about them. Privacy is influenced by the culture and individual circumstances of participants, the nature and context of the research, and the particular social and political environment in which the research and use of the data will occur. Individuals have different views on what is considered private. For example, telling an investigator about one’s sexual practices is acceptable to some participants, but highly offensive to others. It may be even more difficult to respect a participant’s privacy when he or she is different from the investigator in terms of age, ethnicity, locale, socioeconomic status, or gender.15 Privacy refers to the ways and circumstances in which investigators access information about participants. Consent processes can protect the aspect of privacy that deals with seeking permission to access data, but other research procedures are needed to protect the aspect of privacy related to the circumstances surrounding the actual accessing of that data. Privacy can be respected by using procedures such as written questionnaires rather than face-to-face interviews, by using techniques in which responses are signaled on the telephone touch pad rather than verbally during telephone interviews, or by using private rooms for disrobing for clinical examinations. To respect privacy is to let participants control the access of others to themselves, to provide conditions in which the investigator’s inquiries are welcome, and to provide an opportunity for participants to decline or restrict access.16 To breach privacy is to violate participants’ space or to intrude where one is not welcome or not trusted. In fact, the mere act of contacting people about participating in a research study may be a violation of their privacy, particularly when the prospective participants are identified as having a stigmatizing condition (e.g., HIV/AIDS, drug addiction). Research in which management gives consent to conduct research on employees, or deception research, in which participants are led to behave in a way that they would not wish to be seen, are examples of invasions of
105
Chapter 5: Ensuring Voluntary Informed Consent and Protecting Privacy and Confidentiality
privacy.17 Although there are few publicized cases of violations of privacy, the invasion of privacy may cause any of the following harms: psychological (e.g., worry, irritation, fear, embarrassment, self-doubt), legal (e.g., arrest), social (e.g., stigmatization), or economic (e.g., blackmail or unemployment).18 Because privacy concerns vary by type and by context of research, as well as by the culture and individual circumstances of participants, investigators should be well informed and mindful of participants’ cultural norms. Investigators also need to be aware of the various research procedures and methods that can be used to respect privacy. In addition, a clear, comprehensive regulatory definition of privacy along with guidance for protecting privacy in various types of research is needed.19 NBAC proposes the following as a regulatory definition of privacy: Privacy interests are persons’ interests in controlling access of others to themselves and to information about them. Privacy and confidentiality have long been concerns of social scientists and oral historians, and many professional societies for these disciplines have developed ethics codes and guidance describing specific privacy and confidentiality issues and protections for their disciplines (e.g., psychology, anthropology, and oral history).20 In developing new guidance, policymakers should consult existing codes and guidance.
Guidance can be instrumental in helping investigators and IRBs raise concerns and issues about privacy, however it cannot be expected to provide all of the solutions. It is incumbent on investigators and IRBs, therefore, to tailor research procedures to protect privacy as appropriate for each research study. In addition, IRBs and investigators must be familiar with a variety of procedures and methods that prevent violations of privacy.
Confidentiality
Applied to research, confidentiality, like privacy, is a complex, multifaceted issue. Privacy is an agreement between the investigator and participant regarding disclosures of the participant’s identifiable data and how that data will be handled. Confidentiality involves informing the participant about mandatory reporting requirements (e.g., reports of child abuse), plans for sharing identifiable data with other investigators or interested parties
(e.g., FDA), and the extent to which confidentiality can be protected by law—that is, investigators could be subpoenaed to release identifiable data. In addition, state law must be taken into account in describing confidentiality protection. IRBs and investigators are expected to be knowledgeable about applicable state laws involving privacy and confidentiality, which vary in their coverage of types of data and disclosure requirements.21 Confidentiality also involves informing participants about how the data will be managed. Inherent in this disclosure is review by the IRB of the actual procedures used to ensure protection of confidentiality, which include those used in handling and transmitting data, eliminating linkages of data and identifiers, storing raw data (e.g., questionnaires, records, abstract forms) and data sets, and planning for long-term storage and use, including sharing the data with other investigators.
Research procedures unrelated to data management but generally related to research might compromise confidentiality. For example, reimbursing participants by check or reporting results back to participants could provide a link between the research and the participants that would not otherwise exist. In longitudinal studies, calling or mailing participants might identify them with a research study. IRBs and investigators should review all of the proposed research procedures for possible compromises to confidentiality.
Making stronger legal protections available to all investigators could enhance confidentiality protections. In examining such options and designing such protections, mandatory federal or state reporting requirements (e.g., mandatory child abuse disclosures) should be considered.
In addition to the federal statutes mentioned earlier, another mechanism available to protect confidentiality is the Certificate of Confidentiality. Under the Public Health Service Act, DHHS agencies may issue Certificates of Confidentiality to investigators authorizing them to withhold identifiable data about research participants that may be summoned under federal, state, or local civil, judicial, administrative, legislative, or other proceedings. DHHS regards the certificate’s protection as superceding state law, and some case law supports this position.22 However, Certificates of Confidentiality do not provide the same degree of protection as that provided under
106
CDC’s and the Department of Education’s National Center for Educational Statistics Confidentiality Statutes, because although certificates protect against compulsory disclosures by the investigator, they do not prohibit investigators from making disclosures unauthorized by the participant.
In addition, certificates, which are specific to a research study and not to an investigator or institution, are issued sparingly and only when it is determined that the data are sensitive and that issuance will increase study participation. Stronger confidentiality protections are needed for all research involving sensitive, identifiable data. At a minimum, Certificates of Confidentiality should be issued on request to investigators when it is determined that the research involves sensitive, identifiable data and no other legal protections apply. Much stronger protection of confidentiality would be afforded if a mechanism such as a Certificate of Confidentiality also prohibited investigators from making unauthorized disclosures, however, such additional protection should be imposed cautiously because it may entail serious costs, as investigators would not be able to share data with others without the participant’s permission. Like privacy, confidentiality concerns vary by research type and context, and no one set of procedures to protect confidentiality can be developed that would cover all types of research contexts. IRBs and investigators must tailor confidentiality protections to the specific circumstances and methods used in each specific research study. A recent Institute of Medicine (IOM) report describes procedures investigators can easily adopt to reduce the risks associated with breaches of confidentiality (IOM 2000). This report suggests that IRBs consider recommending or requiring such procedures when appropriate and it encourages IRBs and investigators to consider using strong confidentiality protections to reduce some of the violations associated with privacy. A clear, comprehensive definition of confidentiality along with guidance for protecting confidentiality in various types of research is needed. NBAC offers the following definition of confidentiality: Confidentiality is the protection of identifiable data through agreements between participants and investigators about who may have access to the data and how the data will be managed in order to control access.
National Bioethics Advisory Commission
Recommendation 5.4: Federal policy should be developed and mechanisms should be provided to enable investigators and institutions to reduce threats to privacy and breaches of confidentiality. The feasibility of additional mechanisms should be examined to strengthen confidentiality protections in research studies.
Summary
Once an IRB has determined that risks of a given protocol are reasonable in relation to potential benefits, no person should participate in that research without his or her voluntary informed consent (or that of an appropriate representative). However, for some studies that pose minimal risk, it might be possible to waive this requirement. The process of informed consent involves investigators making appropriate disclosures about their research and participants developing a good understanding of the information provided and their choices regarding participation in a study. Participating in the process of informed consent is one of the best ways investigators can demonstrate their concern and respect for those they wish to enroll in a study.
In this report, NBAC reinforces previous recommendations from earlier reports by recommending that the process of providing information and ensuring comprehension should be emphasized, rather than the documentation of the decision to give consent, and that both the information and the way it is conveyed—while meeting full disclosure requirements—should be tailored to meet the needs of the participants in a particular research context. NBAC also recommends that documentation requirements should be adapted for varying research settings and that the criteria for deciding when informed consent is not necessary should be clarified to better ensure that participants are protected. Finally, because there are no clear policies on protecting privacy interests and confidentiality in the research context, the informed consent process alone is sometimes insufficient for protecting the rights and welfare of participants. Federal policy should be clarified and strengthened in this area to assist investigators in ensuring that the interests of participants are protected.
107
Chapter 5: Ensuring Voluntary Informed Consent and Protecting Privacy and Confidentiality
Notes
1 See Vanderpool, H., “Unfulfilled Promise: How the Belmont Report Can Amend the Code of Federal Regulations Title 45 Part 46—Protection of Human Subjects.” This background paper was prepared for NBAC and is available in Volume II of this report.
16 Ibid.
17 Ibid.
18 See Goldman, J., “Privacy and Confidentiality in Health Research.” This background paper was prepared for NBAC and is available in Volume II of this report.
| 2 Ibid. | 19 In DHHS’ response to NBAC’s report, Research Involving |
| 3 Ibid. | Human Biological Materials: Ethical Issues and Policy Guidance, it |
| was noted that NBAC’s recommendations regarding identifiability | |
| 4 Ibid. | and privacy might be inconsistent with proposed medical record |
| rules promulgated by DHHS. | |
| 5 NBAC Town Meetings: February 9, 2000, Houston, Texas; | |
| April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, | 20 Anthropology: http://www.aaanet.org/committees/ethics/ |
| Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, | ethcode.htm; Psychology: http://www.apa.org/ethics/code.html; |
| Portland, Oregon. | Oral History: http://www.dickinson.edu/organizations/oha/ |
| evaluationguidelines.html. Each last accessed September 4, 2000. | |
| 6 Informed consent requirements pertaining to research involving | |
| children are not addressed in the Common Rule. However, DHHS | 21 See Schwartz, J., “Oversight of Human Subject Research: The |
| regulations include specific requirements relating to research | Role of the States.” This background paper was prepared for NBAC |
| involving children (45 CFR 46 Subpart D). DHHS regulations | and is available in Volume II of this report. |
| require that “when in the judgment of the IRB” children are capa- | |
| ble of providing assent (their affirmative agreement to participate), | 22 People v. Newman (32 N.Y.2d 379, 298 N.E.2d 651, |
| they are asked to do so. In addition, at least one parent or guardian | 345 N.Y.2d 502, 1973). |
| and in some instances both parents or guardians are required to | |
| give permission for the child to participate in the research. NBAC | |
| has not addressed ethical issues pertaining to children participating | References |
| in research. Attention should be given to the ethical standards and | |
| regulatory requirements for adequately protecting children. | Advisory Committee on Human Radiation Experiments (ACHRE). |
| 1995. Final Report. Washington, D.C.: U.S. Government Printing | |
| 7 It should be noted that special provisions exist for waiving | Office. |
| consent in the context of emergency research. | |
| Beauchamp, T.L., and J.F. Childress. 1994. Principles of Biomedical | |
| 8 Ibid. | Ethics, 4th ed. New York: Oxford University Press. |
| 9 Ibid. | Bernhardt, B.A., G. Geller, M. Strauss, K.J. Helzsouer, M. Stefanek, |
| 10 See Schwartz, J., “Oversight of Human Subject Research: The | P.M. Wilcox, and N.A. Holtzman. 1997. “Toward a Model |
| Role of the States.” This background paper was prepared for NBAC | Informed Consent Process for BRCA1 Testing: A Qualitative |
| and is available in Volume II of this report. | Assessment of Women’s Attitudes.” Journal of Genetic Counseling |
| 6(2):207–222. |
11 See Goldman, J., “Privacy and Confidentiality in Health Research.” This background paper was prepared for NBAC and is available in Volume II of this report.
12 See Sieber, J., “Privacy and Confidentiality: As Related to
Human Research in Social and Behavioral Science.” This background paper was prepared for NBAC and is available in Volume II of this report.
13 See Goldman, J., “Privacy and Confidentiality in Health Research.” This background paper was prepared for NBAC and is available in Volume II of this report.
14 Pub. Law 191, 104th Congress.
15 See Sieber, J., “Privacy and Confidentiality: As Related to Human Research in Social and Behavioral Science.” This background paper was prepared for NBAC and is available in Volume II of this report.
Blenkinsop, S. 1997. “Whatever Happened to Plain English? The Gobbledygook Smokescreen that Baffles Research Subjects.”
In Volunteers in Research and Testing, eds. B. Close, R. Combes, A. Hubbard, and J. Illingworth, 89–98. Bristol, PA: Taylor and Francis.
Boruch, R.F., and J.S. Cecil. 1979. Assuring the Confidentiality of Social Research Data. Philadelphia: University of Pennsylvania Press.
Dresser, R.S. 1981. “Deception Research and the HHS Final Regulations.” IRB: A Review of Human Subject Research 3(4):3–4.
Faden, R.R., and T.L. Beauchamp. 1986. A History and Theory of Informed Consent. New York: Oxford University Press.
Hochhauser, M. 1997. “Can Patients Understand Their Consent Forms?” Bender’s Health Care Monthly April:8–10.
108
Institute of Medicine (IOM). 2000. Protecting Data Privacy in Health Services Research. Washington, D.C.: National Academy Press.
McQuillan, G., M.A. Speers, and V. Burt. 2000. “Applying the Federal Regulations for Protecting Human Subjects in Survey Research: The NHANES Experience.” Unpublished manuscript. NBAC archive.
National Bioethics Advisory Commission (NBAC). 1998.
Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity. 2 vols. Rockville, MD: U.S. Government Printing Office.
———. 1999a. Ethical Issues in Human Stem Cell Research. 3 vols. Rockville, MD: U.S. Government Printing Office.
———. 1999b. Research Involving Human Biological Materials: Ethical Issues and Policy Guidance. 2 vols. Rockville, MD: U.S. Government Printing Office.
———. 2001. Ethical and Policy Issues in International Research: Clinical Trials in Developing Countries. 2 vols. Bethesda, MD: U.S. Government Printing Office.
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission). 1979.
Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research. Washington, D.C.: U.S. Government Printing Office.
National Bioethics Advisory Commission
Nicks, S.D., J.H. Korn, and T. Mainieri. 1997. “The Rise and Fall of Deception in Social Psychology and Personality Research.”
Ethics and Behavior 7(1):69–77.
Singer, E. 1978. “Informed Consent: Consequences for Response Rate and Response Quality in Social Surveys.” American Sociological Review 43(2):144–162.
Spencer S.J., C.M. Steele, and D.M. Quinn. 1999. “Stereotype Threat and Women’s Math Performance.” Journal of Experimental Social Psychology 35:4–28.
Verheggen, F., R. Jonkers, and G. Kok. 1996. “Patients’ Perceptions on Informed Consent and the Quality of Information Disclosure in Clinical Trials.” Patient Education and Counseling 29(2):137–153.
Waggoner, W.C., and D.M. Mayo. 1995. “Who Understands? A Survey of 25 Words or Phrases Commonly Used in Proposed Clinical Research Consent Forms.” IRB: A Review of Human Subjects 17(1):6–9.
Young, D.R., D.T. Hooker, and F.E. Freeberg. 1990. “Informed Consent Documents: Increasing Comprehension by Reducing Reading Level.” IRB: A Review of Human Subjects Research 12(3):1–5.
109
Chapter Six 6
Conduct of Research—Selected Issues for Local Institutions
Introduction
This chapter addresses three issues regarding the roles and responsibilities of local institutions and their Institutional Review Boards (IRBs) in protecting research participants:
Continual review and monitoring of research in progress is a critical part of the oversight system. Regular, continual review is necessary to ensure that emerging data or evidence have not altered the risks/potential benefits assessment so that risks are no longer reasonable. In addition, mechanisms are needed to monitor adverse events, unanticipated problems, and changes to a protocol. IRBs could increase their effectiveness in meeting these responsibilities with better guidance and some restructuring of the review and monitoring process.
One of the greatest burdens on IRBs and investigators today is review of multi-site studies. Requiring multiple institutions to review the same protocol is unnecessarily taxing and provides no real added value to protection of participants. Multi-site review poses problems in the initial stages of review as well as in the continual review and monitoring stages, especially in the evaluation of adverse events in clinical research.
A comprehensive system of oversight of human research should include a mechanism to compensate participants for medical and rehabilitative costs resulting
from research-related injuries. Participants who volunteer to be in a research study and are harmed as a direct result should be cared for and compensated. This obligation fulfills the principle of justice.
Monitoring of Ongoing Research
Oversight of research should not end once the study begins. Reports have consistently pointed out that research must be monitored once in progress to minimize harms to research participants (OIG 1998a; OIG 1998c; GAO 1996). This section addresses issues related to monitoring the progress of research, including conducting continuing review, reporting protocol changes and unanticipated problems, and ensuring participants’ safety.
Continuing Review
One means of monitoring ongoing research is the continuing review process. As research progresses, investigators might learn more about the risks and potential benefits of an experimental intervention or how other new research findings might affect judgments about risks and potential benefits. Such changes may warrant modifications to the informed consent process, for example, to include the risk of a newly reported side effect. Other developments might also require modifications to protocol design, or in the extreme case, stopping a research study. A continuing review process generally focuses on updated information provided by investigators, including the status of participant enrollment, summary of changes to the protocol, relevant new reports in the literature, unanticipated problems, and plans for changes to the protocol.
111
Chapter 6: Conduct of Research—Selected Issues for Local Institutions
Continuing review has been a major problem for IRBs for some time. A 1975 study of 61 institutions conducted for the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission), found that roughly half of IRBs seldom or never reviewed interim reports from investigators (Cooke and Tannenbaum 1978, I-44). The National Commission went on to recommend, at a minimum, annual continuing review for research studies involving more than minimal risk or vulnerable populations (National Commission 1978, 16). The President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission) concluded that “many IRBs do not understand what is expected in the way of ‘continuing review’…[and] the problems manifested in these studies clearly need attention” (President’s Commission 1981, 47). More than 15 years later, the Office of Inspector General (OIG) of the Department of Health and Human Services (DHHS) found that IRBs “conduct minimal continuing review of approved research” and that the reviews are “hurried and superficial” (OIG 1998a; OIG 1998c).
As noted in the OIG reports, continuing review suffers in part because of excessive IRB workloads and insufficient regulatory guidance (OIG 1998a; OIG 1998c). Regulations currently require that “an IRB shall conduct continuing review…at intervals appropriate to the degree of risk, but not less than once per year” (45 CFR 46.109(e); 21 CFR 56.109(f)). However, the regulations do not specify the purpose or content of that review. Continuing review should not be a repetition of the initial review, because applying all of the requirements of initial review to continuing review is rarely necessary. Rather, IRBs should be looking for specific new developments that may affect participant protections. Because the federal regulations are incomplete in describing what should be considered in continuing review, it is understandable that IRBs do not always conduct appropriate review. Thus, additional guidance is needed.1 Moreover, the requirement of continuing review itself is overly broad, as the frequency and need for continuing review vary depending on the nature of research. For the purpose of continuing review, IRBs should focus their
attention primarily on research initially determined to involve more than minimal risk. In research involving high or unknown risks, the first few trials of a new intervention may substantially affect what is known about the risks and potential benefits of that intervention. On the other hand, the ethics issues and participant protections necessary in minimal risk research are unlikely to be affected by developments from within or outside the research, in, for example, research involving the use of existing data or in research that is in the data analysis phase when there is no additional contact with participants. Continuing review of such research should not be required because it is unlikely to provide any additional protection to research participants and would merely increase IRB burden. However, because minimal risk research does involve some risk, IRBs may choose to require continuing review when they have concerns. In these cases, other types of monitoring would be more appropriate, such as assessing investigator compliance with the approved protocol or requiring reporting of protocol changes and unanticipated problems. Although such efforts might fail to detect some protocol problems, the resource requirement inherent in conducting continuing reviews for all protocols and the distraction of the IRB’s attention from riskier research do not justify devoting a disproportionate amount of resources to continuing review. Clarifying the nature of the continuing review requirements would allow IRBs to better focus their efforts on reviewing riskier research and thereby increase protection for participants where it is most needed.
Reporting Protocol Changes and Unanticipated Problems
In addition to the periodic re-evaluation of risks and potential benefits as part of continuing review, IRBs conduct as-needed reviews when investigators request an amendment to approved protocols or in the event of unanticipated problems with a research study. Indeed, current regulations require institutions to create written procedures for “ensuring prompt reporting to the IRB of proposed changes in a research activity, and for ensuring that such changes in approved research, during the period for which IRB approval has already been given,
112
may not be initiated without IRB review and approval except when necessary to eliminate apparent immediate hazards to the subject” (45 CFR 46.103(b)(4)(iii); 21 CFR 56.108(a)(3)(4)). Institutions are also required to ensure reporting to the IRB “any unanticipated problems involving risks to subjects or…any suspension or termination of IRB approval” (45 CFR 46.103(b)(5); 21 CFR 56.108(b)(1)(3)).
Both requirements are ethically relevant; however, they could be more useful to IRBs with additional guidance, as guidance is slim regarding what types of protocol amendments must be reported to an IRB. Currently, IRBs spend time reviewing amendments that do not include material changes to the protocol, while changes that may affect the rights and welfare of participants at times go unreported or unaddressed. For example, IRBs often find themselves reviewing amendments requesting a change of mailing address or telephone number contacts, while at the same time, investigators sometimes alter recruitment criteria or make substantive changes in consent forms without IRB approval. Guidance is needed regarding the types of changes that must be reported to IRBs as well as those that do not need to be reported, the types of amendments eligible for review using procedures other than a full IRB review (see Chapter 2), and the types of amendments that must be reviewed by the full IRB, so that IRBs can focus on what is important—ethically significant changes to research protocols.
Similar issues arise with regard to the requirement that unanticipated problems be reported to the IRB and to federal agencies, such as when consent forms or questionnaires containing identifiable information are lost. Interpreting the requirements for reporting unanticipated problems to federal agencies has been a significant problem for IRBs and their institutions.2 IRBs and investigators need additional guidance regarding the types of unanticipated problems that must be reported and how they should be addressed.
Monitoring the Safety of Participants
Reporting and reviewing adverse events are particularly important in clinical research. Although regulations and guidance are available for IRBs, investigators, and sponsors to follow in reporting and evaluating adverse
National Bioethics Advisory Commission
events, they have not been revised to reflect changes in the way research is conducted (e.g., movement toward more multi-site clinical trials). Moreover, other entities not considered in federal regulations (45 CFR 46 Subpart A), such as Data and Safety Monitoring Boards (DSMBs), are beginning to play an increasingly important role in safety monitoring (DeMets et al. 1999; Gordon et al. 1998) and are potentially well situated for this role because they review data from all participating sites and have access to unblinded data. Just how such safety monitoring is consonant with current requirements for continuing review, reporting of unanticipated problems, and reporting of adverse events should be clarified. Until recently, communication among federal agencies requiring reporting of adverse events has been lacking to the extent that relevant agencies, IRBs, and investigators are not all adequately informed (Prentice and Gordon 1997).3 However, new efforts are under way to improve communication and to harmonize the reporting requirements among federal agencies (DHHS 2000).4
Reporting of Adverse Events
As one of the requirements for approval of research, IRBs must ensure that as “…appropriate, the research plan makes adequate provision for monitoring the data collected to ensure the safety of subjects” (45 CFR 46.111(a)(6); 21 CFR 56.111(a)(6)). Although this requirement was designed with clinical research in mind, it provides little guidance about what is required of IRBs or investigators conducting clinical research. The National Institutes of Health (NIH) recently issued guidance on data and safety monitoring in Phase I and II trials,5 and because much clinical research involves the use of an investigational drug, device, or biologic product, the Food and Drug Administration (FDA) regulations also apply and are much more specific than the Common Rule. (Citations, language, and annotation of the FDA’s reporting regulations appear in Appendix H.)
FDA Requirements for Adverse Event Reporting
FDA regulations generally refer to five phases in the development and marketing of products, which include three phases of product development and two distinct post-market approval phases. All three stages of drug and biologic product development are classified as research
113
Chapter 6: Conduct of Research—Selected Issues for Local Institutions
(21 CFR 312.21). Phase I includes safety studies of the product’s initial use in humans, in which a small number of participants (20 to 80) receive the drug to verify its safety. Phase II includes early controlled clinical trials to obtain preliminary effectiveness data and information on short-term side effects, with a larger number of participants (sometimes several hundred). Phase III includes expanded controlled and uncontrolled studies to gather information about the effectiveness and safety of products and usually involve several hundred to several thousand participants. After Phase III studies, FDA grants or denies market approval. All adverse events must be reported during the three phases of product development, and some mandatory post-approval reporting requirements must also be met, particularly for medical devices. Although these three phases comprise the research portion of product development, information continues to be gathered after market approval. Medical device development follows a similar process, although it is not defined as three distinct phases in the regulations (21 CFR 812.25, 812.35).
Drugs and biologic products have two distinct post-market approval phases, while for devices, there is only one. FDA may require sponsors to conduct Phase IV studies after drug approval to obtain further information about risks, potential benefits, and optimal use of a drug (21 CFR 312.85). However, only some Phase IV studies might be research, and there is no equivalent Phase IV reporting for medical devices. Accumulating information on the public’s experience with the approved drug or other FDA-regulated product could be considered a fifth phase (21 CFR 314.80, 314.81, 814.82, 814.34). (FDA refers to this phase as post-marketing reporting.) Phase V primarily includes voluntary reporting to FDA on product experience, but some mandatory reporting also occurs regarding the effectiveness of new medical devices. After device approval, user facilities are required by federal regulations to report medical device-related deaths to FDA and the device manufacturer and medical device-related serious injuries to device manufacturers (21 CFR Part 803), but other reporting of adverse events with approved products is voluntary.
FDA reporting requirements pertain to investigators and sponsors, and the requirements differ for drugs and
biologic products and devices. The various types of adverse events are defined in regulations along with required timeframes for reporting. (See Exhibit 6.1.) For example, FDA regulations contain definitions of the following terms: life-threatening adverse drug experience, serious adverse drug experience, unexpected adverse drug experience, associated with the use of the drug, and unanticipated adverse device effect (21 CFR 312.32(a); 21 CFR 812.2(s)).
Investigators are required to report to the sponsor “any adverse effect that may reasonably be regarded as caused by, or probably caused by, the drug” being studied (21 CFR 312.64(b)) or to the sponsor and the investigator’s IRB any unanticipated adverse device effect (21 CFR 812.150(a)(1)). Serious adverse drug effects are to be reported by sponsors to FDA (21 CFR 312.32(c)(1)(i)(A) and (c)(1)(ii)), and unanticipated adverse device effects are to be reported by sponsors to FDA and all reviewing IRBs (21 CFR 812.150(b)(1)).
Review of Adverse Events
Reporting of adverse events is only the first step in monitoring safety; evaluation of reports and distribution of findings to all relevant parties are also critical. Under
45 CFR 46 Subpart A, IRBs are required to evaluate adverse events, and FDA regulations contain requirements for evaluation of reports and reporting back findings (21 CFR 312.56(c)-(d); 21 CFR 812.46(b)(1)-(2)). Multi-site clinical trials pose special challenges for IRBs because a single IRB is unable to review and evaluate adverse event reports for these trials. Even if local IRBs were to receive all adverse event reports from all institutions with complete information in a timely fashion, and even if each IRB, through its membership or the use of consultants, had the expertise to analyze these events, it would be inefficient to have hundreds of local IRBs duplicatively perform this meticulous and time-consuming task (Prentice and Gordon 1997), when one complete and reliable analysis should be sufficient. Thus, the burden on local IRBs could be greatly reduced and participant protections improved if a mechanism were put in place to handle all required safety monitoring at the level of the sponsor, or the lead organization managing the research study, rather than the local IRB.
More important, local IRBs that receive an adverse event report cannot determine whether the event is
114
National Bioethics Advisory Commission
Exhibit 6.1: FDA Requirements for Adverse Event Reporting
The FDA regulations include several definitions to assist in identifying what constitutes an adverse event that triggers each of the various reporting requirements. These definitions include the following:
For drugs and biologic products:
Associated with the use of the drug. There is a reasonable possibility that the experience may have been caused by the drug.
Disability. A substantial disruption of a person’s ability to conduct normal life functions.
Life-threatening adverse drug experience. Any adverse drug experience that places the patient or subject, in the view of the investigator, at immediate risk of death from the reaction as it occurred, i.e., it does not include a reaction that, had it occurred in a more severe form, might have caused death.
Serious adverse drug experience. Any adverse drug experience occurring at any dose that results in any of the following outcomes: death, a life-threatening adverse drug experience, inpatient hospitalization or prolongation of existing hospitalization, a persistent or significant disability/incapacity, or a congenital anomaly/birth defect. Important medical events that may not result in death, be life-threatening, or require hospitalization may be considered a serious adverse drug experience when, based on appropriate medical judgment, they may jeopardize the patient or subject and may require medical or surgical intervention to prevent one of the outcomes listed in this definition. Examples of such medical events include allergic bronchospasm requiring intensive treatment in an emergency room or at home, blood dyscrasias or convulsions that do not result in inpatient hospitalization, or the development of drug dependency or drug abuse.
Unexpected adverse drug experience. Any adverse drug experience, the specificity or severity of which is not consistent with the current investigator brochure; or, if an investigator brochure is not required or available, the specificity or severity of which is not consistent with the risk information described in the general investigational plan or elsewhere in the current application, as amended. For example, under this definition, hepatic necrosis would be unexpected (by virtue of greater severity) if the investigator brochure only referred to elevated hepatic enzymes or hepatitis. Similarly, cerebral thromboembolism and cerebral vasculitis would be unexpected (by virtue of greater specificity) if the investigator brochure only listed cerebral vascular accidents. Unexpected, as used in this definition, refers to an adverse drug experience that has not been previously observed (e.g., included in the investigator brochure) or anticipated based on the pharmacological properties of the pharmaceutical product (21 CFR 312.32(a)).
For devices:
Unanticipated adverse device effect. Any serious adverse effect on health or safety or any life-threatening problem or death caused by or associated with a device, if that effect, problem, or death was not previously identified in nature, severity, or degree of incidence in the investigational plan or application (including a supplementary plan or application), or any other unanticipated serious problem associated with a device that relates to the rights, safety, or welfare of subjects (21 CFR 812.2(s)).
frequent or rare, whether it is caused by the research as opposed to the underlying illness or standard treatment, or whether the adverse event is more common in the intervention group than in the control groups. Local IRBs lack access to the essential data needed to evaluate adverse event reports and are thus not only are wasting time attempting to analyze them but are also unable to make use of the data.
DSMBs, as monitoring bodies independent from investigators conducting the research, are one such mechanism growing in use. DSMBs monitor the safety and effectiveness of the experimental intervention and recommend stopping a trial if significant benefits or risks have developed or if the trial is unlikely to be concluded successfully (DeMets et al. 1999; Gordon et al. 1998). Membership qualifications, composition, functions, and
115
Chapter 6: Conduct of Research—Selected Issues for Local Institutions
oversight of DSMBs generally differ from those of IRBs. NIH has issued several policies regarding data and safety monitoring and now requires the use of DSMBs for multi-site clinical trials involving interventions that entail potential risk to the participants,6,7 and NIH institutes and centers have flexibility in implementing the requirement for DSMBs—that is, they may conduct or sponsor the monitoring activities or delegate such responsibility to a grantee or contractor.
NIH policy also requires that adverse events be reported to IRBs. This reporting includes communication between the IRB and DSMB when one is used and requires that investigators submit any reports from the DSMB to the IRB. Most recently, NIH has issued a policy requiring a monitoring plan as part of all Phase I and Phase II clinical trials.8 Unlike the FDA regulations, NIH policy is not specific regarding what constitutes an adverse event, in what timeframe it must be reported, and to whom. Further, unlike FDA regulations, DHHS regulations (45 CFR 46) are silent regarding the reporting of adverse events, except to require that institutions provide written procedures for reporting any unanticipated problems involving risks to participants.
Difficulties in Following Reporting Requirements
Because of the sheer volume and quality of reports and the complexity of the regulatory requirements, assessing reports of adverse events is a major burden for IRBs and investigators (Prentice and Gordon 1997). In every town meeting conducted by the National Bioethics Advisory Commission (NBAC), IRB members said that they were inundated with adverse event report forms and they often had little understanding of the context in which to evaluate the event in a multi-site trial.9 Investigators reported frustration with their lack of understanding of what constituted an adverse event, required reporting times, and to whom adverse events should be reported, and they reported confusion about knowing when reporting is mandatory or voluntary and which reporting requirements apply to the FDA’s five phases of product development and use. Investigators also complained about the requirement to report separately to sponsors, NIH, and their IRBs. These findings echo those of the OIG (OIG 1998a; OIG 1998c).
Because protecting the rights and welfare of participants should be the top priority of IRBs and investigators, having a monitoring system that is effective, easy to use, efficient, and responsive to serious events should be the goal of all who participate in the review process. Several deficiencies are noted in the current system, including complex, confusing, and fragmented regulations; regulations that may not be fully enforced; and lack of effective communication among the relevant parties. Although the FDA regulations for reporting adverse events are detailed, they are complex, fragmented, and often confusing, and investigators suggest that they cannot understand the regulations as they are written.10 For example, there appears to be a range of views about what constitutes an adverse event and what should be reported, from reporting practically all physical problems (e.g., spiked temperature) to suggestions that adverse events are “trade secrets” that should not be reported.11 In addition, the relationship between holding information as confidential and reporting adverse events requires clarification. There are many potential reasons companies might consider such information confidential—for example, new indications regarding existing products, the nature of new drugs under study, the specific populations under study, or a proprietary study design—and handling the reporting and evaluation of adverse events when some, if not all, information about the trial is confidential can be difficult. Adverse events that are related to investigational drugs must be reported to sponsors by investigators (21 CFR 312.64(b)), who must, in turn, report serious adverse events to FDA and all participating investigators. Reporting requirements are different for investigational medical devices, however. For devices, investigators must report adverse events to sponsors and their reviewing IRBs (21 CFR 812.150(a)(1)). Sponsors, in turn, must report significant new information to all reviewing IRBs, participating investigators, and FDA (21 CFR 812.150(b)(1)). Problems potentially arise when sharing information about adverse events may involve disclosing confidential information about the study. However, protecting research participants should take priority over protecting the financial interests of sponsors.
116
One set of simplified regulations for safety monitoring is needed, and regulations and guidance should be written so that investigators and sponsors understand what constitutes an adverse event, what type of event must be reported within what time period, and to whom an event should be reported. In addition, regulations and guidance should be clear regarding whose responsibility it is to analyze and evaluate adverse event reports and should describe the required communication and coordination channels among IRBs and safety monitoring entities, such as DSMBs, investigators, sponsors, and federal agencies.
NBAC could not determine the extent to which evaluation of adverse events and the reporting of resulting findings are a problem. Although investigators and IRBs attending NBAC’s town meetings suggested that they were either not receiving reports or not receiving them in a timely fashion from sponsors,12 NBAC also heard from witnesses that whenever adverse event reports are serious or unexpected, sponsors of drug trials go to great lengths to investigate them, including interviewing investigators and conducting site visits.13 Nevertheless, a more transparent and effective system is needed for reporting, evaluating, and reporting back results of adverse events.
Recommendation 6.1: Federal policy should describe how sponsors, institutions, and investigators should monitor ongoing research.
Recommendation 6.2: Federal policy should describe clearly the requirements for continuing Institutional Review Board review of ongoing research. Continuing review should not be required for research studies involving minimal risk, research involving the use of existing data, or research that is in the data analysis phase when there is no additional contact with participants. When continuing review is not required, other mechanisms should be in place for ensuring compliance of investigators and for reporting protocol changes or unanticipated problems encountered in the research.
Recommendation 6.3: Federal policy should clarify when changes in research design or context require review and new approval by an Institutional Review Board.
National Bioethics Advisory Commission
Recommendation 6.4: The federal government should create a uniform system for reporting and evaluating adverse events occurring in research, especially in multi-site research. The reporting and evaluation responsibilities of investigators, sponsors, Institutional Review Boards, Data Safety Monitoring Boards, and federal agencies should be clear and efficient. The primary concern of the reporting system should be to protect current and prospective research participants.
Review of Cooperative Research Studies
The Range of Cooperative Research
One of the major challenges facing IRBs is the review of cooperative research or multi-site studies, or research involving multiple institutions. Although most collaborative studies occur at multiple institutions, investigators from different institutions might collaborate in conducting research at a single site. Research of all types can be conducted cooperatively and can involve hundreds of institutions. Each institution might perform experimental interventions (e.g., clinical trials) or simply provide investigators access to data (e.g., epidemiological studies). In clinical research, for example, institutions might all perform the same or different functions (e.g., one institution collects tissue samples, another analyzes them) and may be geographically proximate or on different continents.
Some cooperative research involves investigators or institutions that conduct little or no research and are too small to establish their own IRBs, such as a single community physician, a small private practice unaffiliated with a larger institution, or a small college. In some cases, it may be possible to rely on an IRB at a neighboring institution, which could provide some degree of local review because of its familiarity with the community from which the participants come, although it would not necessarily be familiar with the investigators or the circumstances under which the research will be conducted. Some institutions do not take advantage of this option because of concerns regarding potential institutional liability.
117
Chapter 6: Conduct of Research—Selected Issues for Local Institutions
Current Requirements and Problems
Each institution engaged in cooperative research must comply with the federal regulations (45 CFR 46.114). In practical terms, this means that each institution must have its IRB review the research protocol or make other arrangements for review, which must be approved by the department or agency head. For example, an institution that does not have its own IRB could use an IRB from another institution or an independent IRB to review its research (45 CFR 46.114). This requirement is imposed on IRBs that must follow the Common Rule (45 CFR 46 Subpart A), but is not on IRBs that must comply only with the FDA regulations, which do not require that all institutions or individuals engaged in the research have their IRBs review the research protocol. Instead, FDA requires only one IRB to review the protocol (21 CFR 56.103(a)). As discussed in Chapter 1, a research study may be subject to review under the Common Rule if it is conducted or funded by a federal department or agency bound to the Common Rule or if it is conducted at one of the institutions that has voluntarily agreed to apply the requirements of the Common Rule to all research that is conducted as part of a Multiple Project Assurance (MPA).
The federal requirements regarding collaborative studies are problematic for several reasons. Investigators and sponsors are frustrated by having to submit protocols to multiple IRBs, particularly because changes requested by any one board establish a new round of discussions and negotiations among all the collaborating IRBs, a repetitive process that causes time delays and increased costs and that is perceived as providing little or no additional protection to the research participants.14 IRBs are frustrated because they are spending scarce resources on reviewing the same research protocol that, in some cases, is being reviewed by hundreds of other IRBs, even when overall design and methods can only be changed with great difficulty. Moreover, investigators, sponsors, and IRBs all believe that multiple reviews of the same protocol usually do not increase protection of research participants.15 Moreover, in large multi-site research studies, local IRBs often have little or no substantive authority over protocol design, short of disapproving local participation
in the protocol. Although local IRBs can make changes to consent forms, often they find sponsors unreceptive to questions or proposed changes to study design.16 In part, this reluctance may reflect the need for protocol standardization across institutions, as procedures at each site must be sufficiently similar to allow data aggregation. Sponsors pressed for time also may be reluctant to make changes to the protocol in response to a single, local IRB, because each modification to the protocol requires resub-mission to each of the IRBs that had already approved the protocol. Moreover, private sponsors of research are not required to follow the IRB review requirements in the Common Rule; rather they are only required to follow the FDA regulations, which do not require each institution participating in the research to provide an IRB review of the research. Academic institutions with MPAs, on the other hand, are almost always required to follow two sets of regulations in multi-site research involving FDA-regulated investigational products: one set that requires them to review the research unless other arrangements are made (45 CFR 46) and the other that does not require every institution to review the research (20 CFR 50). The result is that the institutions are held responsible for IRB review even when they have little direct control over research design.
Arguments in Favor of Local IRB Review
In the United States, independent review of research involving human participants primarily occurs at the local level. The development of local review grew out of the peer review process used to evaluate scientific merit and the NIH requirement that grantee institutions take responsibility for the ethical conduct of human research. It is a model of review that reflected the nature of research at the time—single research studies conducted by one investigator from a single institution.17 At the time of inception of review committees, local institutional review was seen as offering distinct advantages, and in its early evaluation of the IRB system, the National Commission supported the use of “local review committees…located in institutions where research is conducted,” suggesting that such committees have four advantages:
118
1. Local committees are familiar with the “actual conditions surrounding the conduct of research.”
2. Local committees “can work closely with investigators” to ensure that participants are protected and investigators are treated fairly.
3. Local committees can serve as a resource and assist in educating investigators and the public.
4. Local committees can communicate with federal officials and among themselves about protection and ethical issues (National Commission 1978, 1–2).
In recent years, the first of these advantages has been given particular prominence and was largely supported and promulgated by the Office for Protection from Research Risks (OPRR). As stated by Gary Ellis, former director of OPRR:
We embrace the local IRB at the research site as the cornerstone of the American system of protection of human subjects….It is local review by individuals who are in the best position to know the research at the site, the resources at the institution, the capabilities and the reputations of the investigators and staff, the prevailing attitudes and ethics of the community and most importantly, the likely subject population (Ellis 1994, 31–32).
Features of the local community and participant population may be relevant, for example, in determining whether the selection of participants is equitable and whether consent forms and materials are likely to be understandable. Institutional factors, such as the availability of emergency care or patient record keeping, may be relevant in determining whether risks are minimized and confidentiality is assured.
There are additional advantages to using local IRBs. For example, Steven Peckman, director of an academic human participant protection program, argues that not only do local IRBs have knowledge about the local setting, they serve other important functions: “The local IRB demystifies the review process and creates an environment of collegiality.”18 Further, local IRBs interact with investigators, with each party able to discuss concerns about the research study. Peckman also argues that local IRBs hear the concerns of prospective research participants either by having them sit as IRB members or by seeking their input in other ways. Local IRBs also provide
National Bioethics Advisory Commission
a place for investigators and participants to address complaints.
Why IRB Review Need Not Always Be Local
Although there are advantages to local review—that is, situating IRBs in the institutions that are conducting the research and that are geographically close to the research site—they should not be overstated. For example, factual information about an institution, such as facilities and policies, is usually easy to determine by asking an investigator or institutional official. Moreover, in a small institution, more subtle institutional information, such as that regarding an investigator’s work experience, may be available to an institutional IRB, but in large institutions, this might not be the case.
Similar caveats apply to knowledge of a local community. Geographically remote IRBs can learn the basic demographic characteristics of a community from afar. Moreover, information about the local (geographic) community is often of questionable relevance. In few cases do geographic communities have sufficiently uniform and distinct beliefs relevant to research to judge a protocol as ethical in one locale but not in another. On the other hand, in the town meetings NBAC conducted, IRBs frequently reported turning down protocols that had been approved by other IRBs,19 although, in such cases the declining IRB rarely thought that it was a local factor that rendered the protocol inappropriate and instead turned it down because it found some aspect of study design, such as fees paid to participants, inappropriate. Although the absence of data is not equivalent to the absence of a problem, no data are available regarding specific research studies that after some negotiation became acceptable in one community but not in another.
Moreover, institutions that enroll participants from a defined geographic community might not enroll participants from a single cultural community. In a paper supporting local review, Peckman notes the wide range of linguistic and cultural groups in Los Angeles, illustrating that even within one metropolitan area, an IRB might encounter numerous diverse groups.20 When an institution serves a wide range of communities, it is unclear how the beliefs of any one group should inform IRB decisions. There are, of course, a few cases in which geographically cohesive groups have readily identifiable beliefs
119
Chapter 6: Conduct of Research—Selected Issues for Local Institutions
relevant to the interests of research participants (e.g., some American Indian communities); but these cases appear to be the exception rather than the rule (Norton and Manson 1996; Sharp and Foster 2000).
One ethical requirement for which sensitivity to local cultures has clear relevance is the review of the consent process, especially consent forms. Institutions that serve diverse communities must be attentive to the information needs of participants from various cultural backgrounds, even as local variability in, for example, language and educational attainment, influences IRB review of protocols. However, the use of such community-wide generalizations must not overshadow the importance of individual variability. Similarly, consent forms should differ between an institution enrolling primarily college-educated participants and one enrolling participants who generally have less than a high school education. Thus, although there may be ways in which knowledge of local factors affects local IRB decisions about protocols, the importance of such knowledge should not be overstated, and community differences should not be used as an explanation for variable decisionmaking by IRBs when the differences have no substantive bearing on the review.
As discussed earlier, there are other advantages of using local IRBs, which can sometimes more easily engage local investigators, for example, by routinely inviting them to meetings at which their protocols are discussed. However, the advantages of local IRB review, in terms of both factual knowledge and local access, may not be as significant as they initially appear to be. For example, for an institution that infrequently conducts research involving human participants, relying on a remote IRB with frequent meetings rather than a local IRB that meets infrequently may actually improve communication between investigators and the board, and for multi-site research, local investigators often have little direct control over issues of protocol design, which must be standardized across sites. For them, the only decision is whether to participate. In fact, with large multi-site studies, a system of exclusively local IRB review undermines precisely the kind of interaction between investigator and IRB that was intended when the IRB system was created. If the investigators who designed the study are not affiliated with any institution conducting the research, it may be
that there is no IRB interaction with the investigators who actually design and run the study, such as the staff of an industry sponsor of research. In the case of such studies, a single, central IRB would foster significantly more relevant IRB-investigator interaction.
Alternatives to Local IRB Review
Although many acknowledge the arguments that favor local IRB review, they also recognize that local review is not always necessary or appropriate. Even the National Commission, which strongly supported a system of local IRBs, recognized that in some cases, research studies did not require review by an IRB located in or near the institution where the research would be conducted. For small institutions, other arrangements, such as use of another institution’s IRB or several institutions forming a joint IRB, were acceptable (National Commission 1978). For multi-site research studies, the National Commission stated that:
Review by one IRB (generally at the entity most substantially involved with the research) should satisfy statutory and regulatory requirements. Other entities that are involved with the research may also require review by their IRBs, however. In such instances, IRBs should give priority to consideration of protocols that are receiving multiple review, in order to reduce the extended time period that such review may entail (National Commission 1978, 8).
These exceptions suggest that the National Commission did not view local IRB review as an absolute requirement.
Although IRBs must have knowledge of the local community, there are no regulatory requirements that preclude review by IRBs that are not organizationally part of the institution(s) conducting research and/or are not geographically close to the research site. What is required is that the IRB has sufficient knowledge of the local research context—in terms of the relevant institution(s), the relevant investigators, and the relevant com-munity(ies)—to conduct an effective review (45 CFR 46.107, 46.111(a)(3)-(4), (7), (b), 46.116).21 As mentioned above, since 1981 FDA has allowed nonlocal review of research (review by an IRB geographically remote from the
120
research site and/or independent of the institution conducting the research), as long as the IRB obtains sufficient knowledge of the local research context (21 CFR 56.107, 56.111(a)(3), 56.111(a)(7), 56.111(b); FDA 1998, 19–20).
In recent years, guidance from the Office for Human Research Protections (OHRP)/OPRR also has moved in this direction, emphasizing the types of local knowledge that may be required for different types of studies.22 OHRP allows “institutional sites that are geographically close enough to comfortably contribute membership to a common IRB” to create such a shared, or common, IRB.23 In addition, in 1995, OPRR began approving assurances in which an institution designates an independent IRB (OIG 1998b). Recently, OHRP has approved a pilot program using a central IRB for review of certain National Cancer Institute-funded cooperative cancer trials and a cooperative review arrangement by a group of geographically disparate institutions.24 OHRP has also joined FDA in accepting IRBs that routinely meet by teleconference, facilitating the work of IRBs whose members are truly representative of various geographic areas.25
Models of IRB Review in Other Countries
Although the United States has generally conducted ethics reviews using committees located within institutions, several countries have established systems of regional review committees that are not affiliated with a particular institution (e.g., Denmark). In addition to using regional review committees, these systems generally have a central or national review committee that provides guidance to the regional committees and resolves disagreements. For example, Denmark has a national research ethics committee that holds higher authority than its regional research ethics committees.26 Whether a regional review committee system functions better or worse than the individual institutional review system is unclear. The major advantages of regional review systems are their independence and ability to review expeditiously multi-site studies, and at least in one instance, a regional system seems to be more efficient and effective.27 In Denmark, the regional review system is established and supported by the government with virtually no influence from the local institutions conducting the research. Local institutions have no
National Bioethics Advisory Commission
choice over the committee that reviews their research and have no power to appoint members to that committee. Thus, the independence of the review system is attributed both to the location of the review committee outside the institutions conducting the research and to the selection process for review committee members. This means, in a sense, that in a system that uses institutionally based review committees (e.g., IRBs), greater independence could be achieved by selecting members who are not affiliated with the institution.28 The United Kingdom makes use of local review committees for most research, but has created an additional layer of regional committees for multi-site research. Once a multi-center protocol has been approved by a regional committee, all local review committees are to review expeditiously the protocol by the executive subcommittee, considering only matters that may affect local acceptability, such as the qualifications of the investigators, the suitability of the site and prospective participants, and the language and information in the consent form. Even though problems have been noted with this system (Al-Shahi and Warlow 1999; Lux et al. 2000), critics suggest the regional system is better than the solely local system used previously in the United Kingdom (Alberti 2000; Tully et al. 2000).
In Denmark, review of multi-site studies is handled by assigning their review to the lead investigator’s review committee, which is then responsible for seeking input from the other participating investigators’ review committees. Søren Holm has reported that multi-site studies are generally reviewed and approved in a relatively short period (fewer than 60 days).29 The advantages to this review process for multi-site studies are that the lead investigator has contact with only one regional review committee, changes to the protocol are negotiated with only one investigator, and there is only one approved protocol from which all investigators conduct the study.
On the other hand, a regional system of review may have at least two disadvantages, especially in a large country. Regional review committees may lack knowledge about the institutions in which the research will be conducted and the investigators who carry it out. This is not an uncommon problem in large institutions in which the IRBs may lack certain types of knowledge about the
121
Chapter 6: Conduct of Research—Selected Issues for Local Institutions
investigators or the context of the research. Another disadvantage is that the regional review committee may not be able to adequately monitor the research study once it is approved. However, even if regional review systems are more effective than local institutional review systems, it is unclear that a model of regional review, such as a system of government-run regional review committees and a single, national committee with authority to manage appeals, could be adopted effectively in the United States, which differs from other countries in terms of population size and geographic area and the number of research studies that are conducted annually.30
Role of Local IRBs and Institutions
Discussions about IRB review of multi-site research studies often confuse local institutional control of research with local institutional review. Local institutions must be able to maintain some oversight over the research their investigators conduct, and each institution must decide whether it wants to participate in a multi-site research study and must maintain its authority to decline to participate, even if another IRB has approved the research. As long as an accredited IRB reviews and approves the research protocol, multiple IRB reviews of the same research protocol are not always necessary to ensure the protection of research participants. For research studies conducted solely by one institution, it often makes sense for that institution’s IRB to conduct the review. But for cooperative research, IRB review by all institutions participating in the research should be the exception.
A number of arrangements between a local institution and the reviewing IRB could be possible. The reviewing, or lead, IRB might be, for example, the IRB of the institution where the research study was developed, an IRB at a participating institution with particular expertise in the areas of research, or an independent IRB. It is essential that the terms of the arrangement are clearly defined in advance with respect to the roles and responsibilities to be assumed by each party. It must be clear who will have responsibility for providing ongoing educational programs for investigators and staff, who will be responsible for conducting appropriate verification activities, to whom participants’ complaints and concerns should be
addressed, and how local knowledge will be brought to bear on IRB review. For example, if an institution has its own IRB and programs in education and verification, the institution might retain those responsibilities. Members of the local IRB could provide local knowledge to the external IRB during its review or by reviewing the decisions of the lead IRB as part of local control. Alternatively, the local IRB might arrange to have the option of tailoring the consent process and documentation to the needs of the local institution and participants. In either case, the lead IRB assumes responsibility only for review, while the local institution retains most of the other responsibilities. Institutional IRBs might be willing to take on the primary responsibility of review voluntarily or for a fee.
If, on the other hand, the local institution has had little experience with research and does not have its own IRB, it may be appropriate for the reviewing IRB to assume more responsibilities, such as those for providing education and verification. The IRB also might be required to gather information about the local institution and community, as independent IRBs do now. Creative pilot projects are needed to identify those cooperative models that work best. In the cooperative model, the lead IRB must assume a number of responsibilities beyond the review of the protocol. Institutional and independent lead IRBs may wish to charge for such services.
It is clear that innovative and creative alternative mechanisms and processes for reviewing protocols in multi-site research are needed. To allow for such projects and to support a change in the current system toward a more flexible review system, federal policy should be clear about the functions that must be performed, but less restrictive about who performs each function. Institutions have research responsibilities, such as providing education and monitoring investigator compliance, that go beyond conducting IRB reviews, and reliance on a nonlocal IRB must not allow those other activities to go unfulfilled. The same considerations apply to studies conducted at a single site.
Recommendation 6.5: For multi-site research, federal policy should permit central or lead Institutional Review Board review, provided that participants’ rights and welfare are rigorously protected.
122
Compensation for Research-Related Injuries
A comprehensive system of oversight of human research should include a mechanism to compensate participants for medical and rehabilitative costs resulting from research-related injuries. The inclusion of this mechanism has long been justified on ethical grounds. First, research participants are entitled to be left no worse off than they would have been had they not participated in the research.31 Second, without compensation, those with limited access to health care services may not be able to afford treatment and rehabilitation for research-related injuries (Kolata and Eichenwald 1999; Vasgird et al. 2000). Third, even if consent forms and the disclosures within them are comprehensive, completely unforeseen iatrogenic harms might occur in research (President’s Commission 1982, 58). Fourth, research participation is often an act of social beneficence in which people agree to volunteer in order to further larger societal goals. Thus, injuries should not be summarily dismissed merely because the individuals were provided information in the consent process and agreed to accept the risk of certain harms (President’s Commission 1982, 59–60).
NBAC supports those arguments favoring compensation for research-related injuries, and other international ethics and research bodies support the principle of compensating research injuries in various ways as well. A previous report (NBAC 2001) identified 15 international research ethics policies with statements that address compensation for research-related injuries, including the Council of Europe (Council of Europe 1997), the Council for International Organizations of Medical Sciences (CIOMS 1991; CIOMS 1993), the International Conference on Harmonization (ICH 1996), and the Joint United Nations Programme on HIV/AIDS (UNAIDS 2000). In addition, a number of national guidelines and policies also support this principle, including Australia (NHMRC 1999), Brazil (NHC 1996), China (Committee on Research Involving Human Subjects 1998), France,32 India (Indian Council of Medical Research 2000), the Netherlands,33 New Zealand (Health Research Council of New Zealand 1997), South Africa (Medical Research Council of South Africa 1993), Uganda (National
National Bioethics Advisory Commission
Consensus Conference on Bioethics and Health Research in Uganda 1997), and the United Kingdom (MRC 1998).
In addition to the ethical arguments for a research-related compensation program, a practical argument can be made in the context of the previous discussion on the review of cooperative research. The parties, including the institution(s), the investigator(s), the sponsor(s), and the product manufacturer (if not the sponsor and the injury involves a product), that benefit directly from research in which participants suffer injury are primary candidates for bearing liability for compensation. However, because research involves risk, which can manifest itself as an actual harm, research participants can suffer injury even if all parties perform to the highest standard. As a result, liability for research-related injuries is one of the concerns institutions often raise when considering whether to rely on an IRB other than their own to review a multi-site research study. Therefore, developing systems for review of cooperative research will depend in part on addressing issues related to institutional liability for harms, as institutions hesitate to relinquish control of local review if they retain the same degree of liability. Thus, centralizing IRB review would be more acceptable to such institutions if liability were also centralized in some way.
Current Regulatory Requirements
Current federal regulations do not impose requirements for compensating participants injured by research (45 CFR 46 Subpart A; 21 CFR 50, 56).34 Regulations require only that the informed consent process for research involving more than minimal risk contains “an explanation as to whether any compensation and an explanation as to whether any medical treatments are available if injury occurs and, if so, what they consist of, or where further information may be obtained” (45 CFR 46.116(a); 21 CFR 56.116(a)). However, the regulations also prohibit exculpatory language in the informed consent process—that is, language that makes the participant “waive or appear to waive any…legal rights, or releases or appears to release the investigator, the sponsor, the institution or its agents from liability for negligence” (45 CFR 46.116; 21 CFR 50.20). Thus, in cases in which no compensation will be made available, investigators and IRBs
123
Chapter 6: Conduct of Research—Selected Issues for Local Institutions
must craft language that fits both of these requirements. OHRP guidance suggests the use of language such as the following: “This hospital makes no commitment to provide free medical care or payment for any unfavorable outcomes resulting from participation in this research. Medical services will be offered at the usual charge.”35
Previous Recommendations for a Compensation Program
Because the regulations do not require compensation to research participants who are injured in the course of research, and because few institutions offer such compensation, the only option an injured participant has to obtain compensation for research-related injuries is through legal action. For over 30 years, various parties have discussed alternatives to this option (Bergen 1967; Ladimer 1963), and many national panels and advisory groups in the United States have recommended the establishment of a compensation program (ACHRE 1995; President’s Commission 1982; DHEW 1977; Tuskegee Syphilis Study Ad Hoc Advisory Panel 1973). The Tuskegee Syphilis Study Ad Hoc Advisory Panel, in its 1973 Final Report, noted that:
No policy for the compensation of research subjects harmed as a consequence of their participation in research has been formulated, despite the fact that no matter how careful investigators may be, unavoidable injury to a few is the price society must pay for the privilege of engaging in research which ultimately benefits the many. Remitting injured subjects to the uncertainties of the law court is not a solution (Tuskegee Syphilis Study Ad Hoc Advisory Panel 1973, 23).
The panel recommended the development of a “no-fault” clinical research insurance plan, in which receipt of compensation does not depend on the establishment of wrongdoing or negligence.36 The same year, the Department of Health, Education, and Welfare’s (DHEW, now DHHS) Medical Malpractice Commission recommended to the Secretary that federal research grants include funding for institutions to purchase insurance or to self-insure in order to provide compensation for participants injured in research. DHEW formed the
Secretary’s Task Force on the Compensation of Injured Research Subjects in 1974, and in 1977 the task force recommended that participants injured in Public Health Service-sponsored research be compensated to the extent that injuries exceeded those reasonably associated with their disease and treatment (DHEW 1977). The National Commission endorsed the task force recommendations, but it could not prepare a report on this topic before it expired. The DHEW Ethics Advisory Board next took up the issue; however, this board was terminated in 1980. Finally, in 1982, the President’s Commission issued a report on the subject entitled Compensating for Research Injuries, in which it recommended that an experiment be conducted that would have entailed providing federal support to a small number of institutions to cover the costs associated with providing compensation on a non-fault basis to injured research participants (President’s Commission 1982). Results from the experiment were intended for use in a report to DHHS about the need for a full-fledged compensation program.
However, the experiment was never conducted, and the issue of compensation was not addressed until the Advisory Committee on Human Radiation Experiments (ACHRE) recommended in its 1995 Final Report that the federal government should consider a system of compensation for injuries to research participants resulting from participation in federally funded research. ACHRE supported the approach recommended by the President’s Commission, noting that “so that years from now others do not have to revisit and struggle with this issue, the federal government must take steps now to address the issue of compensation for injured research subjects” (ACHRE 1995, 827). The federal government’s response to the ACHRE recommendation reflected a concern regarding the extent of research-related injuries:
In the absence of a finding that a significant number of modern research subjects are unfairly denied compensation, the Administration is not prepared to propose a system outside the existing network of Federal and state liability and insurance systems. The Administration does, however, view the debate over the extent and effectiveness of our current human subject protections to encompass this issue. The
124
Administration would be open to considering any recommendations from NBAC or legislation from Congress that seek to address this issue. The desire to spread the cost of research injury is a reason to consider a compensation scheme (Human Radiation Interagency Working Group 1997, 17).
The Current Situation
The availability, circumstances, and procedures for compensating injured research participants have remained unchanged from those observed by the Tuskegee Advisory Panel. Unless a participant is privately insured or is injured at one of the few research sites that provide treatment or compensation (Marwick 1998), he or she must take legal action through filing a lawsuit and showing that one or more wrongful acts (e.g., battery) or a breach of duty was committed (e.g., negligence in performance or failure to perform contractual obligations) by someone involved in the research and that the wrongful act(s) caused the injury. Because research participants also can be injured by their underlying conditions or by the research itself, even if all parties perform their tasks with the greatest care, it is often difficult to determine whether a specific injury arises directly from a research intervention. Further, even where warranted and successful, such lawsuits are costly and slow and thus are of limited value at the time the injury is incurred.
Nevertheless, the threat of legal action often forces institutions to adopt procedures to protect themselves. For example, consent forms often disclose every foreseeable risk, and institutions require comprehensive and multiple IRB reviews of multi-site studies in order to ensure that the associated risks and potential benefits are acceptable to the various sites as well as to the research participants.
In addition, the threat of legal action often leads to difficult negotiations between research institutions and sponsors regarding the type and amount of compensation that can be offered or what is to be stated in the consent form.37 In some cases, sponsors wish to offer more treatment and compensation than do institutions, and in other cases, institutions are prepared to offer more than the sponsors will permit. Further, institutions want to include their own language in the consent form
National Bioethics Advisory Commission
regarding the availability of compensation rather than the sample language proposed by the sponsor. When differences between institutions and sponsors arise, the negotiations are often time consuming and contentious, with institutions finding themselves in a compromised position because they do not want to lose the opportunity to collaborate in the research.
The Need for a Compensation System
No adequate database exists that describes the number of injuries or illnesses that are suffered by research participants, the proportion of these illnesses or injuries that are caused by the research, and the medical treatment and rehabilitation expenses that are subsequently borne by the participants. It may be argued that, regardless of the magnitude of the problem, the costs of research injuries should never be borne by participants. If they are injured by research participation, those who benefit from the research (e.g., institutions and sponsors) bear some obligation to compensate participants who risked and suffered injury on their behalf. However, currently, injured research participants alone bear both the cost of lost health and the expense of medical care, unless they are adequately insured or pursue successful legal action to gain compensation from specific individuals or organizations involved in conducting the research. Although the adoption of a system of universal health care or a national health insurance program in the United States would eliminate some, if not all, medical costs incurred by research participants, under the current system, even though institutions and sponsors are most likely to be required to provide such compensation, ultimately institutions pass the costs on to sponsors, who in turn pass the costs on to society in the form of higher prices or taxes. A national compensation program outside the current fault-based system would mean that research participants would not have to bear the burden of seeking compensation, and at the same time the costs of compensation would not be imposed unfairly on investigators, institutions, or sponsors in cases in which no negligence or wrongdoing might have occurred. Arguably, this would be appropriate, because the public ultimately benefits from the results of research through new and improved products and services.
125
Chapter 6: Conduct of Research—Selected Issues for Local Institutions
Because the costs of research injuries should not be borne by the injured participants and because support for a compensation system should be provided by those most likely to profit or derive other benefits from it, sponsors and institutions should be assigned responsibility for funding such a system. The funding mechanism could be flexible and could include options such as privately purchased liability insurance, self-insurance, or a centrally administered fund supported by premiums paid by sponsors and institutions.
More information is needed about the nature and extent of research-related injuries and uncompensated research injuries. Efforts are needed to obtain information from both public and private sponsors of research as well as to develop effective systems for collecting data regarding the nature and extent of research-related injuries. The Department of Veterans Affairs (VA), for example, has adopted regulations requiring compensation for research-related injuries to individuals who participate in VA-funded research, but the agency does not have a central mechanism for collecting local data on research-related injuries (38 CFR 17.85).
However, any such data collection efforts, whether centralized or not, should use existing sources of data—for example, reports to FDA of serious adverse events. Information regarding the extent of injuries also would be needed as part of the development and evaluation of a compensation program.
The scope and nature of a national research-related injury compensation system should support the system’s goals of providing the following:
Recommendation 6.6: The federal government should study the issue of research-related injuries to determine if there is a need for a compensation program. If needed, the federal government should implement the recommendation of the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (1982) to conduct a pilot study to evaluate possible program mechanisms.
Summary
Local institutions have three major responsibilities in the conduct of research involving human participants. Initial IRB review is a powerful tool for protecting participants because, when used appropriately, it can prevent abuses before the research begins. Oversight, however, should not end when the research begins, and continuing review and monitoring should be intensified as the risk and complexity of the research increase. These processes at all times should emphasize the protection of research participants.
In addition, the review process should facilitate rather than hinder collaborative research among institutions, provided that participants are protected. Local institutional review has many advantages and is appropriate for studies conducted by a single institution. However, when multiple institutions engage in a collaborative study, the local review system can become cumbersome and less effective. Alternative review mechanisms, such as the use of a lead or central IRB, should be encouraged, tested, and evaluated.
Ultimately, however, even with all the available protective mechanisms in place, some participants will be harmed by participating in research, and these participants should be cared for and compensated for this harm. This presents an enormous challenge, because it is often difficult to separate injuries traceable to the research from those that stem from the underlying disease being studied. However, this challenge should not deter movement toward the development of an appropriate compensation program for those who are harmed by participating in research.
126
Notes
1 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
2 Ibid.
3 Ibid.
4 Notice of proposed actions under the NIH Guidelines for Research Involving Recombinant DNA Molecules. 65 Fed. Reg. 77655–77659.
5 NIH Guide for Grants and Contracts, “Further Guidance on a Data and Safety Monitoring for Phase I and Phase II Trials.” Notice OD-00-038. June 5, 2000. Available at http://grants.nih.gov/ grants/guide. Last accessed December 4, 2000.
6 NIH Guide for Grants and Contracts, “Further Guidance on a Data and Safety Monitoring for Phase I and Phase II Trials.” Notice OD-00-038. June 5, 2000; “Guidance on Reporting Adverse Events to Institutional Review Boards for NIH-Supported Multicenter Clinical Trials.” Notice 99-107. June 11, 1999; “NIH Policy for Data and Safety Monitoring.” Notice 98-084. June 10, 1998. All available at http://grants.nih.gov/grants/guide. Last accessed December 4, 2000.
7 NIH Guide for Grants and Contracts, “NIH Policy for Data and Safety Monitoring.” Notice 98-084. June 10, 1998. Available at http://grants.nih.gov/grants/guide. Last accessed December 4, 2000.
8 NIH Guide for Grants and Contracts, “Further Guidance on a Data and Safety Monitoring for Phase I and Phase II Trials.” Notice OD-00-038. June 5, 2000. Available at http://grants.nih.gov/ grants/guide. Last accessed December 4, 2000.
9 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
10 Ibid.
11 Recombinant DNA Advisory Committee Meeting, NIH. 8–10 December, 1999. Bethesda, Maryland.
12 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
13 Personal communication from B. Spilker, PhRMA, to M. Speers, NBAC. October 24, 2000.
14 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
National Bioethics Advisory Commission
15 Ibid.
16 Ibid.
17 See McCarthy, C.R., “Reflections on the Organizational Locus of the Office for Protection from Research Risks.” This background paper was prepared for NBAC and is available in Volume II of this report.
18 See Peckman, S., “Local Institutional Review Boards.” This background paper was prepared for NBAC and is available in Volume II of this report.
19 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
20 See Peckman, S., “Local Institutional Review Boards.” This background paper was prepared for NBAC and is available in Volume II of this report.
21 OHRP Guidance: “IRB Knowledge of Local Research Context.” September 27, 1998 (Updated July 21, 2000). Available at http://ohrp.osophs.dhhs.gov/humansubjects/guidance/local.htm. Last accessed December 4, 2000.
22 Ibid.
23 OHRP Guidance: “General Guidance on the Use of Another Institution’s IRB.” August 9, 1991. Available at http://ohrp.osophs.dhhs.gov/humansubjects/guidance/irb-rely.htm. Last accessed December 4, 2000.
24 Personal communication from J. Goldberg, National Cancer Institute, to NBAC staff. May 23, 2000; Schuster, D.P., Testimony before NBAC. September 12, 2000. Washington, D.C.
25 OHRP Guidance: “IRB Meetings Convened via
Telephone Conference Call.” March 28, 2000. Available at http://ohrp.osophs.dhhs.gov/references/irbtel.pdf. Last accessed December 8, 2000.
26 See Holm, S., “The Danish Research Ethics Committee System—Overview and Critical Assessment.” This background paper was prepared for NBAC and is available in Volume II of this report.
27 Holm, S., Testimony before NBAC. September 12, 2000. Washington, D.C.
28 See Holm, S., “The Danish Research Ethics Committee System—Overview and Critical Assessment.” This background paper was prepared for NBAC and is available in Volume II of this report.
29 Ibid.
30 Ibid.
31 Faden, R., Testimony before NBAC. January 13, 2000. Washington, D.C.
127
Chapter 6: Conduct of Research—Selected Issues for Local Institutions
32 Law 88-1138 Regarding the Protection of Persons Agreeing to Biomedical Research.
33 Law of 26 February 1998, containing regulations with regard to medical-scientific research on humans, Staatblad (Official Law Gazette of the Netherlands) 161.
34 The Department of Veterans Affairs adopted regulations to provide medical treatment for research-related injuries. These regulations are not part of the Common Rule, but are supplemental.
35 OHRP Guidance: “‘Exculpatory Language’ in Informed Consent.” November 15, 1996. Available at http://ohrp.osophs.dggs.gov/ humansubjects/guidance/exculp.htm. Last accessed December 4, 2000.
36 Since the time of the Tuskegee Ad Hoc Advisory Panel’s report, no fault has come to have a very specific meaning in the context of automotive insurance. As the automotive context is not strictly analogous, we will use the term nonfault rather than no fault to refer to compensation that does not depend on establishment of wrongdoing or negligence.
37 NBAC Town Meetings: February 9, 2000, Houston, Texas; April 5, 2000, Pittsburgh, Pennsylvania; May 3, 2000, Orlando, Florida; June 7, 2000, Chicago, Illinois; August 14, 2000, Portland, Oregon.
References
Advisory Committee on Human Radiation Experiments (ACHRE). 1995. Final Report. Washington, D.C.: U.S. Government Printing Office.
Alberti, K.G.G.M. 2000. “Multicentre Research Ethics Committees: Has the Cure Been Worse Than the Disease?” British Medical Journal 320:1157–1158.
Al-Shahi, R., and C.P. Warlow. 1999. “Ethical Review of a Multicentre Study in Scotland: A Weighty Problem.” Journal of the Royal College of Physicians of London 33(6):549–552.
Bergen, R.P. 1967. “Insurance Coverage for Clinical Investigation.”
Journal of the American Medical Association 201:305–306.
Committee on Research Involving Human Subjects. 1998.
Guidelines on Ethical Review of Medical Research. China: Ministry of Public Health.
Cooke, R.A., and A.S. Tannenbaum. 1978. A Survey of Institutional Review Boards and Research Involving Human Subjects. In Appendix to Report and Recommendations: Institutional Review Boards, 1-1–1-310. Washington, D.C.: Department of Health, Education, and Welfare.
Council for International Organizations of Medical Sciences (CIOMS). 1991. International Guidelines for Ethical Review of Epidemiological Studies. Geneva: CIOMS.
———. 1993. International Ethical Guidelines for Biomedical Research Involving Human Subjects. Geneva: CIOMS.
Council of Europe. 1997. Convention for the Protection of Human Rights and Dignity of the Human Being with Regard to the Application of Biology and Medicine. France: Council of Europe.
DeMets, D.L., S.J. Pocock, and D.G. Julian. 1999. “The Agonizing Negative Trend in Monitoring of Clinical Trials.” The Lancet 354(9194):1983–1988.
Department of Health and Human Services (DHHS). 2000.
“Secretary Shalala Bolsters Protections for Human Research Subjects.” [Press Release.] 23 May. Available at http://www.hhs.gov/news/. Last accessed November 6, 2000.
Department of Health, Education, and Welfare (DHEW). Secretary’s Task Force on the Compensation of Injured Research Subjects. 1977. Final Report. Pub. OS-77-003. Bethesda, MD: DHEW.
Ellis, G. 1994. “Perspectives from OPRR.” In Changes and Choices for Institutional Review Boards: The Inclusion of Women and Minorities, and Other Emerging Issues in Research, 31–32. Boston: Public Responsibility in Medicine and Research.
Food and Drug Administration (FDA). 1998. Information Sheets: Guidance for Institutional Review Boards and Clinical Investigators. Available at http://www.fda.gov/oc/ohrt/irbs/default.htm. Last accessed December 8, 2000.
General Accounting Office (GAO). 1996. Scientific Research: Continued Vigilance Critical to Protecting Human Subjects. GAO/HEHS-96-72. Washington, D.C.: GAO.
Gordon, V.M., J. Sugarman, and N. Kass. 1998. “Toward a More Comprehensive Approach to Protecting Human Subjects.” IRB: A Review of Human Subjects Research 20(1):1–5.
Health Research Council of New Zealand. 1997. HRC Guidelines on Ethics in Health Research. Auckland, New Zealand: Health Research Council of New Zealand.
Human Radiation Interagency Working Group. 1997. Building Public Trust: Actions to Respond to the Report of the Advisory Committee on Human Radiation Experiments. Washington, D.C.: U.S. Government Printing Office.
Indian Council of Medical Research. 2000. Ethical Guidelines on Biomedical Research Involving Human Subjects. New Delhi: Indian Council of Medical Research.
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). 1996. ICH Harmonised Tripartite Guideline. Guideline for Good Clinical Practice. Geneva: ICH Secretariat, International Federation for Pharmaceutical Manufacturers Association.
Joint United Nations Programme on HIV/AIDS (UNAIDS). 2000.
Guidance Document: Ethical Considerations in HIV Preventive Vaccine Research. Geneva: UNAIDS.
Kolata, G., and K. Eichenwald. 1999. “Stopgap Medicine: A Special Report for the Uninsured, Drug Trials Are Health Care.” New York Times, 22 June, A-1.
128
Ladimer, I. 1963. “Clinical Research Insurance.” Journal of Chronic Diseases 16:1229, 1233.
Lux, A.L., S.W. Edwards, and J.P. Osborne. 2000. “Responses of Local Research Ethics Committees to a Study with Approval from a Multicentre Research Ethics Committee.” British Medical Journal 320(7243):1182–1183.
Marwick, C. 1998. “Compensation for Injured Research Subjects.”
Journal of the American Medical Association 279(23):1854.
Medical Research Council (MRC). 1998. Guidelines for Good Clinical Practice in Clinical Trials. London: MRC.
Medical Research Council of South Africa. 1993. Guidelines on Ethics for Medical Research. South Africa: Medical Research Council of South Africa.
National Bioethics Advisory Commission (NBAC). 2001. Ethical and Policy Issues in International Research: Clinical Trials in Developing Countries. 2 vols. Bethesda, MD: U.S. Government Printing Office.
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission). 1978.
Report and Recommendations: Institutional Review Boards. Washington, D.C.: U.S. Government Printing Office.
National Consensus Conference on Bioethics and Health Research in Uganda. 1997. Guidelines for the Conduct of Health Research Involving Human Subjects in Uganda. Kampala, Uganda.
National Health and Medical Research Council (NHMRC). 1999.
National Statement on Ethical Conduct in Research Involving Humans. Australia: NHMRC.
National Health Council (NHC). 1996. Resolution No. 196/96 on Research Involving Human Subjects. Brazil: NHC. Addition: 1997. Resolution No. 251. Addition: 1999. Resolution No. 292. Brazil: NHC.
Norton, I.M., and S.M. Manson. 1996. “Research in American Indian and Alaska Native Communities Navigating the Cultural Universe of Values and Process.” Journal of Consulting and Clinical Psychology 64(5):856–860.
National Bioethics Advisory Commission
Office of Inspector General (OIG), DHHS. 1998a. Institutional Review Boards: A Time for Reform. OEI-01-97-00193. Washington, D.C.: DHHS.
———. 1998b. Institutional Review Boards: The Emergence of Independent Boards. OEI-01-97-00192.Washington, D.C.: DHHS.
———. 1998c. Institutional Review Boards: Their Role in Reviewing Approved Research. OEI-01-97-00190. Washington, D.C.: DHHS.
Prentice, E.D., and B. Gordon. 1997. “IRB Review of Adverse Events in Investigational Drug Studies.” IRB: A Review of Human Subjects Research 19(6):1–4.
President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission). 1981. Protecting Human Subjects. Washington, D.C.: U.S. Government Printing Office.
———. 1982. Compensating for Research Injuries: The Ethical and Legal Implications of Programs to Redress Injured Subjects. Washington, D.C.: U.S. Government Printing Office.
Sharp, R.R., and M.W. Foster. 2000. “Involving Study Populations in the Review of Genetic Research.” Journal of Law, Medicine, and Ethics 28(1):41–51.
Tully, J., N. Ninis, R. Booy, and R. Viner. 2000. “The New System of Review by Multicentre Research Ethics Committees: Prospective Study.” British Medical Journal 320:1179–1182.
Tuskegee Syphilis Study Ad Hoc Advisory Panel. 1973. Final Report. Washington, D.C.: DHEW.
Vasgird, D.R., M. Hensleigh, A. Berkman, E. Schachne, and C. Trinh. 2000. “Protecting the Uninsured Human Research Subject.”
Journal of Public Health Management Practice 6(6):37–47.
129
Chapter Seven 7
Implications of This Report
Introduction
This report proposes an oversight system for research involving human participants that addresses the needs, interests, and responsibilities of all parties involved in the research enterprise. Adopting the recommendations in this report will lead to better protection of human participants while also promoting ethically sound research and reducing unnecessary bureaucratic burdens. Achieving these goals will, in turn, support the public’s trust in research, enhance enthusiasm for all research involving human beings, and restore the respect of investigators for the oversight system. Ultimately, the needs of the American people will be better met through research that is both ethically sound and of the highest scientific quality.
Resources
The recommendations made in this report will generate additional costs for institutions, sponsors, and the federal government (through the establishment of a new federal oversight office). The importance of identifying the needed resources to support a robust system of protection of research participants has long been a concern. In 1978, the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission), concerned that Institutional Review Boards (IRBs) could not function without adequate resources, recommended that the oversight system be appropriately funded (National Commission 1978). In 1998, the Department of Health and Human Service (DHHS) Office of Inspector General called attention to the critical shortage of resources, including staff, office
space, and information technology, and recommended that IRBs be given access to adequate resources (OIG 1998). The scarcity of resources for IRBs has been attributed to low levels of support from sponsors and institutions (GAO 1996).
In previous reports, the National Bioethics Advisory Commission (NBAC) recognized that research should not be thwarted because resources are not available to provide the necessary protections for human participants and that compliance would require additional resources (NBAC 1998; NBAC 1999). Sponsors of research, whether public or private, should work together with institutions conducting research to make the necessary resources available.
Limited information is available concerning the human and financial resources that federal agencies, other sponsors, and research institutions dedicate to oversight. In fact, the only information NBAC could obtain in this regard was for the federal agencies. However, it is clear that the current system relies on other sponsors and research institutions also contributing substantial resources to oversight. Thus, the information presented below is an incomplete picture of the current resources used to support the oversight system.
Human and financial resources that directly support human research protection activities are limited at the federal level.1 As shown in Table 7.1, of the 16 federal agencies responding to an NBAC survey, 5 have no dedicated administrative unit or staff. Even within DHHS, four agencies have no dedicated administrative units or staff. Most departments spend only 1 to 2 percent of their human research budget on protection activities. The National Institutes of Health (NIH) spent less
131
Chapter 7: Implications of This Report
Table 7.1: Federal Agency Support for Research Participant Protections for FY 1999
| Budget for | Human | |||||||
| Agency | Dedicated Administrative Unit | Dedicated | Research | |||||
| Human Research | Administrative Unit | Budget | ||||||
| Central Intelligence Agency | Yes, Chief of Human Subjects Research Panel and | |||||||
| Contracting Officer’s Technical Representatives | Classified | Classified | ||||||
| Department of Commerce | National Telecommunications and Information | |||||||
| Administration - No | None reported | $ | 17,600,000 | |||||
| National Institute of Standards and Technology (NIST)- | ||||||||
| personnel in Office of NIST Counsel and elsewhere | $ | 41,024,000 | ||||||
| Census - No | $ | 158,500,000 | ||||||
| Department of Defense | 60 full-time equivalents (FTEs) | $ | 3,500,000 | $ | 37,100,000 | |||
| Department of Education | 1.5 FTEs | $ | 200,000 | $ | 50,000,000 | |||
| Department of Energy | 2 FTEs | $ | 425,000 | $ | 27,000,000 | |||
| Department of Health | ||||||||
| and Human Services | ||||||||
| Administration for Children | ||||||||
| and Families | No | None reported | $ | 30,000,000 | ||||
| Agency for Healthcare | ||||||||
| Research and Quality | No | None reported | $ | 109,411,000 | ||||
| Centers for Disease | ||||||||
| Control and Prevention | 6 FTEs | $ | 651,505 | $ | 172,025,000 | |||
| Food and Drug Administration | ~200 FTEs | ~$37,000,000 | $ | 11,329,000 | ||||
| Health Care Financing | ||||||||
| Administration | 6 FTEs | None reported | $ | 15,000,000 | ||||
| Health Resources and | ||||||||
| Services Administration | No | None reported | $ | 75,908,000 | ||||
| Indian Health Service | 4.7 FTEs | $ | 1,000,000 | $ | 22,400,000 | |||
| National Institutes of Health | 4 FTEs in Office of Human Subjects Research, | $ | 480,000 | $ | 8,580,000,000 | |||
| another 10.73 in Institute’s administrative units, | $ | 334,965 | ||||||
| 19 FTEs in OPRR* | $ | 2,700,000 for OPRR | ||||||
| Substance Abuse and Mental | ||||||||
| Health Services Administration | No | None reported | $ | 338,349,000 | ||||
| Department of Housing | No (Office of Lead Hazard Control conducts | None reported | $ | 11,000,000 | ||||
| and Urban Development | some oversight) | |||||||
| Department of Justice | 2 part-time employees | Not available | $ | 110,705,000 | ||||
| Department of Transportation | No | None reported | Not reported | |||||
| Department of Veterans | 3 units, 3 FTEs** | None reported | ||||||
| Affairs (VA) | for FY 1999 | $ | 175,600,000 | |||||
| National Aeronautics and | ||||||||
| Space Administration | 5.9 FTEs | $ | 625,000 | $ | 20,000,000 | |||
| National Science Foundation | Part-time employee | None reported | $ | 150,360,000 | ||||
| Social Security Administration | No | None reported | $ | 40,000,000 | ||||
| Agency for International | ||||||||
| Development | Cognizant Human Subjects Officers | None reported | $ | 60,000,000 | ||||
| Consumer Product Safety | ||||||||
| Commission | No | None reported | <$100,000 | |||||
| Environmental Protection Agency | No | None reported | $ | 76,000,000 | ||||
*Until June 2000, OPRR was the administrative unit within DHHS that provided assurances for all DHHS agencies as well as other federal departments.
**During FY 2000, VA planned to increase to 8 FTE, and in FY 2001 there will be 12 FTE in Headquarters. There are plans for five field offices to be established in FY 2000.
Source: NBAC, “Federal Agency Survey on Policies and Procedures for the Protection of Human Subjects in Research.” This staff analysis is available in Volume II of this report.
132
than .05 percent of its human research budget directly on protection activities, even when the Office for Protection from Research Risks (OPRR) was administratively part of the agency.
The lack of adequate resources also has been noted in site visits by the Office for Human Research Protections (OHRP), formerly OPRR, and in testimony presented to NBAC. For example, in a site visit by OPRR to Duke University, OPRR found that the university had assigned only 2.5 to 3 full-time staff members to the IRB, even though the university reviewed over 1,700 protocols annually.2 IRB administrators, in particular, who testified to NBAC described the need for resources and strongly encouraged NBAC to consider the issue of resources in its recommendations.3 A report commissioned by NIH, Reducing Regulatory Burden, noted that resources available to IRBs are decreasing despite increases in workload and recommended providing additional federal resources when adding to IRB duties (Mahoney 1999).
Data estimating the costs of institutional protection programs are nearly 20 years old (Brown et al. 1979; Cohen 1982). In 1982, it was estimated that the total cost (administrative plus meeting costs) of IRB review at universities without medical schools was $137 per protocol, $154 at universities with medical schools, and $207 at hospitals. Adjusting these cost estimates for inflation using the Consumer Price Index, the total cost of IRB review at universities without medical schools would currently be estimated at $245 per protocol, $275 per protocol at universities with medical schools, and $369 per protocol at hospitals. Based on these inflation-adjusted figures, the current cost of protecting human research participants at research institutions nationwide is estimated to be $32.12 million. It is difficult, however, to estimate IRBs’ costs at academic institutions, because costs are not itemized separately in grant applications; rather they are part of the costs recovered, at least partly though the indirect cost mechanism (Goldman and Williams 2000).
Some information, however, is available about the prices (which may be higher or lower than costs) being charged for IRB review. One commentator reported that her independent IRB charged $1,000 for an initial review of a protocol and $275 for initial review of a research
National Bioethics Advisory Commission
site.4 Another commentator estimated that the average fee for a protocol review was $1,000.5 The number of academic institutions charging for IRB review of research that is not federally funded has been increasing in the past few years, with fees ranging from $500 to $2,000 for initial review and an average fee of $1,130.6 In addition to the call for additional resources, academic institutions have expressed concerns about the costs of the IRB being billed through the indirect cost mechanism, because an administrative capitation prevents them from requesting additional resources (AAU 2000). Alternatives, such as a direct budget line for protection of research participants, have been suggested (AAU 2000).
Recommendation 7.1: The proposed oversight system should have adequate resources to ensure its effectiveness and ultimate success in protecting research participants and promoting research:
a) Funds should be appropriated to carry out the functions of the proposed federal oversight office as outlined in this report. b) Federal appropriations for research programs should include a separate allocation for oversight activities related to the protection of human participants. c) Institutions should be permitted to request funding for Institutional Review Boards and other oversight activities. d) Federal agencies, other sponsors, and institutions should make additional funds available for oversight activities.
Future Research
This report has raised many questions about ethical issues that could not be answered because of insufficient or nonexistent empirical evidence. Little research has been conducted on the actual conduct of research, although some studies are under way.7 Current thinking about ethical issues in research—including the importance of conducting analyses of risks and potential benefits, obtaining informed consent, protecting privacy and confidentiality, and protecting those who are vulnerable—would greatly benefit from research designed to address
133
Chapter 7: Implications of This Report
these specific issues. For example, approaches for assessing cognitive competency, evaluating what participants want to know about research, and ascertaining best practices for seeking informed consent are deserving of more study. Clearer guidance could be developed from a stronger knowledge base.
In addition, although a good deal of ethical analysis has been conducted on many research issues, some (e.g., ethical standards for informed consent) have received more attention from ethicists than others (e.g., understanding what constitutes undue inducement or exploitation in research). In general, increased interdisciplinary discussion that includes biomedical and social scientists, lawyers, and historians would advance understanding of the ethical conduct of research.
Recommendation 7.2: The federal government, in partnership with academic institutions and professional societies, should facilitate discussion about emerging human research protection issues and develop a research agenda that addresses issues related to research ethics.
The New System as a Dynamic and Responsive Structure
Research is an ever-evolving enterprise, responding to scientific interests, the biological and social condition of humans, the practicalities of resource availability, and competing demands on academic institutions and others involved in sponsoring or conducting research. Thus, the oversight system also must be dynamic and should be enhanced in order to ensure that sound ethical principles and standards are followed, while interpretation and application of those principles and standards accommodate and remain relevant to new discoveries, influences, and ethical challenges. The system should provide both the stable foundation and the flexibility necessary to respond to the changing research enterprise.
The proposed oversight system reflects a rethinking of the entire structure for protecting research participants, taking into account the roles of various parties, including participants, investigators, institutions, IRBs, sponsors, and other entities, such as professional societies, a federal oversight office, state regulators, Congress,
and the public. Articulating the roles of each of these parties serves several purposes. It encourages a range of individuals, organizations, and communities to take more seriously their roles in the protection of research participants and holds them accountable for their actions. Distributing roles and responsibilities among more parties in the oversight system also reduces the burden placed on any one entity, such as IRBs. In articulating roles in various areas, such as monitoring and education, responsibility should be delegated to one or more parties, such as a central IRB, a local IRB, another entity within an institution, a Data and Safety Monitoring Board, or the research sponsor. It is critical that all parties understand their responsibilities and those of others in the system.
Once these roles have been articulated, it becomes clear that the oversight system must be more than a federal structure. Outside parties play a crucial role. Indeed, the system functions in large part because of the goodwill of the thousands of people who aspire to meet their obligations to conduct research ethically. Although attention is often focused on those who do not take their ethical obligations seriously, a majority of those involved in research are committed to ethical behavior. However, the awareness of and commitment to ethically sound research can be strengthened and enriched by educational institutions, credentialing and professional organizations, editors and publishers, and advocacy groups, organizations that have tremendous influence over their constituencies and that are in an excellent position to harness the energy of their members to support an efficient and effective oversight system.
Six distinct functions or responsibilities of a consolidated oversight system are recommended: policy development through regulations and guidance; educational programs aimed at the responsibilities of all parties; ethical review of proposed research; monitoring of ongoing research; enforcement of the requirements; and accountability for all those involved in the review and conduct of research. By focusing attention and resources on these functions, the system will provide the necessary infrastructure for protecting research participants and promoting research. To ignore any one of them is to have a system that functions less than optimally.
134
The functions of the system are, and should be, interconnected, with changes in one function affecting outcomes in another. For example, the creation of a single set of regulations and a single office to promulgate them makes it possible to implement specific regulatory changes without amending the regulations of each of the agencies, as is now required. Creation of a single, independent oversight office would also facilitate issuance of regulatory guidance, because direction from that office would apply to all parties conducting research involving human participants.
The greater flexibility proposed for IRBs in deciding what information must be provided to participants and in determining frequency of continuing review demands a higher level of competency among IRBs. Certification of IRB members and staff and accreditation of institutions can validate such competency. Higher and consistent standards for IRB members will ensure more effective assessments of risks and potential benefits as well as determinations of vulnerability. Similarly, certification of investigators allows for flexibility in implementing the recommended informed consent process. More education should make certification of investigators easier to achieve, because investigators will receive the knowledge and skills necessary for certification as part of their professional training. The recommendations regarding verification, adverse event monitoring, and accreditation of research institutions all make single review of multi-site research more feasible.
Thus, just as the oversight functions are interconnected, so too are the recommendations for improving them, and, therefore, consideration should be given to adopting them more or less in total, for to do otherwise may result in diminished success in implementing many of the recommendations in this report. For example, a crucial component of the proposed system is the creation of a new independent oversight office. If this new office were not created, it would be imperative to find an alternative mechanism that would allow the Common Rule to be modified more easily.
Now is the time for strategic change. With more than 20 years of experience with much of the current regula-
National Bioethics Advisory Commission
tory language and 10 years of experience with the Common Rule structure, all parties understand the strengths and weaknesses of the current oversight system, and it is evident that making minor tactical changes to it will not result in the corrections that are needed. However, the proposed changes should be implemented in a planned, phased-in fashion, to avoid creating major disruptions that could compromise the protection of research participants or the conduct of ethical research.
General Themes in the Recommendations
Three general themes are found in this report. First, the recommendations related to policy generally suggest that there should be fewer regulations and more guidance, which clarifies regulatory responsibilities, suggests ways of fulfilling those responsibilities, and assists institutions in formulating institution-specific policies—improving consistency across institutions. At the same time, guidance permits reasoned deviations from the regulations.
Second, the recommendations generally focus attention on research in which participants need the most protection, striving to make the level of protection commensurate with the level of risk. This theme is strongest in the recommendations regarding continuing review, verification, reporting of adverse events, informed consent, review of minimal risk research, and additional scrutiny for research involving a high level of risk.
Third, the recommendations somewhat increase the scope of regulated research while streamlining the process of regulatory compliance. The recommendations regarding privately funded research and the removal of exemptions close some gaps in the current system and thus increase the amount of research subject to regulation. However, it is also recommended that requirements for initial and continuing review of all minimal risk research be reduced. Thus, although the scope of the oversight system is broadened, changes are proposed that will streamline the system and attenuate the need for additional resources by increasing efficiency, making it safer and more productive.
135
Chapter 7: Implications of This Report
What This Report Means to Those Who Use the System
One unified and comprehensive federal policy embodied in a single set of regulations and guidance should govern all research with human participants. Furthermore, one office should have the authority to promulgate those regulations and issue appropriate guidance (see Recommendations 2.1–2.3). At the same time, there is a continuing role for the various federal departments and agencies that conduct and fund research with human participants—a role focused particularly on education, monitoring, and enforcement (see Chapter 2). Nonfederal accreditation and certification bodies should be created to improve the quality and accountability of institutions and bodies responsible for the review and conduct of research (see Recommendations 3.3 and 3.4). In addition, it is time to encourage and foster the participation of groups often overlooked in the protection of research participants, including professional organizations, journal editors, and patient advocacy groups (see Chapters 2 and 3).
As a whole, these recommendations strengthen oversight by dispersing responsibilities more widely. However, although a new federal oversight office will provide much needed leadership and uniformity in the system, the research community must move to implement self-regulation through nonfederal credentialing organizations. The desired outcome of these reforms would be a system that emphasizes education as a means for improving protections and avoiding the need for large-scale federal enforcement actions.
How This Report Will Affect IRBs, Investigators, and Institutions
Almost all of the recommendations in this report directly or indirectly affect investigators, IRBs, and institutions by increasing their responsibilities. For example, it is recommended that the scope of regulated research be expanded (see Recommendations 2.1 and 2.4); that risky research be more closely monitored (see Recommendations 6.1–6.4); and that additional educational and credentialing requirements be imposed on
those who conduct and review human research (see Recommendations 3.1–3.4). Although these increased responsibilities are significant, numerous changes are proposed that would reduce the regulatory burden on IRBs, investigators, and institutions. Under the recommendations, these groups would be required to follow only one set of rules (see Recommendation 2.3), would have one office providing authoritative interpretation (see Recommendation 2.2), and would be given more guidance on issues not requiring a regulatory response (see Recommendations 4.1–4.3 and 5.1–5.4). Specifically, investigators, IRBs, and institutions would have greater flexibility to:
These changes, as well as the recommendation that the distribution of IRB members with relevant expertise and experience should be commensurate with the types of research reviewed by the IRB (see Recommendation 3.9), should make the oversight system more applicable to research in the social sciences and humanities.
Overall, the recommendations suggest three broad changes for IRBs, investigators, and institutions. First, they shift requirements away from procedure and toward competence. That is, although investigators and IRB members are given greater discretion, they are also required to demonstrate appropriate levels of competence in the protection of research participants. Second, the recommendations encourage more strategic use of IRB review, with IRBs reviewing more protocols, but doing so using mechanisms that would allow them to focus on riskier research. Third, the recommendations propose a more strategic use of monitoring by increasing
136
the focus primarily and more intensely on risky research and by reducing the monitoring requirements for less risky research.
How This Report Will Affect Research Participants
Although the recommendations in this report aim to improve the operation of the oversight system, they are not intended simply to reduce inefficiency and needless regulatory burden. Rather, they seek to effect real change in the protections offered to research participants. Under the proposed system, with one oversight office providing enforcement authority and accountability for the oversight of all research:
National Bioethics Advisory Commission
Summary
This report proposes 30 recommendations for changing the oversight system. These changes are proposed at both the national and local levels and are directed at all who are involved in the research enterprise. The time is right for changes of such magnitude and breadth, because only such across-the-board modifications will reform the oversight system so that it is capable of adequately protecting the rights and welfare of research participants while promoting ethically sound research.
Notes
1 NBAC, “Federal Agency Survey on Policies and Procedures for the Protection of Human Subjects in Research.” This staff analysis is available in Volume II of this report.
2 Telephone conversation between M. Carome, OHRP, and M. Speers, NBAC. November 13, 2000.
3 Keane, M.A., and Nelson, D.K., Testimony before NBAC. July 11, 2000. Bethesda, Maryland.
4 Heath, E., Testimony before NBAC. June 6, 2000. San Francisco, California.
5 Spilker, B., Testimony before NBAC. April 7, 2000. Washington, D.C.
6 Baylor College of Medicine. “Fees for IRB Review.” Available at http://research.bcm.tmc.edu/A_C/a_c.html. Last accessed December 11, 2000.
7 NIH Guide for Grants and Contracts, “Informed Consent in Research Involving Human Participants.” September 27, 1996. Request for Applications RFA-OD-97-001. Available at http://grants.nih.gov/grants/guide. Last accessed December 4, 2000.
137
Chapter 7: Implications of This Report
References
Association of American Universities (AAU). Task Force on Research Accountability. 2000. Report on University Protections of Human Beings Who Are the Subjects of Research. Washington, D.C.: AAU. Available at http://www.aau.edu/HumSubReport06.28.00.pdf. Last accessed May 5, 2001.
Brown, J.H.U., L.S. Schoenfeld, and P.W. Allan. 1979. “The Costs of an Institutional Review Board.” Journal of Medical Education 54(4):294–299.
Cohen, J. 1982. The Costs of IRB Review. In Human Subjects Research: A Handbook for Institutional Review Boards, eds. R.A. Greenwald, M.K. Ryan, and J.E. Mulvihill, 39–47. New York: Plenum Press.
General Accounting Office (GAO). 1996. Scientific Research: Continued Vigilance Critical to Protecting Human Subjects. GAO/HEHS-96-72. Washington, D.C.: GAO.
Goldman, C.A., and T. Williams. 2000. Paying for University Research Facilities and Administration. Washington, D.C.: RAND.
Mahoney, J. 1999. NIH Initiative to Reduce Regulatory Burden: Identification of Issues and Potential Solutions. Bethesda, MD: National Institutes of Health. Available at http://grants.nih.gov/ grants/policy/regulatoryburden/index.htm. Last accessed November 29, 2000.
National Bioethics Advisory Commission (NBAC). 1998.
Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity. 2 vols. Rockville, MD: U.S. Government Printing Office.
———. 1999. Ethical Issues in Human Stem Cell Research. 3 vols. Rockville, MD: U.S. Government Printing Office
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission). 1978.
Report and Recommendations: Institutional Review Boards. Washington, D.C.: U.S. Government Printing Office.
Office of Inspector General (OIG), Department of Health and Human Services (DHHS). 1998. Institutional Review Boards: A Time for Reform. OEI-01-97-00193. Washington, D.C.: DHHS.
138
Appendix A
Acknowledgments
This report benefited from the input of many individuals and groups. The Commission asked the following individuals to review portions of the draft report or to assist Commission staff in data collection and analysis. The contributions made by these individuals improved the quality and outcome of the report and are greatly appreciated:
139
Appendix B
May 1999
Letter from Harold T. Shapiro to President William J. Clinton and President Clinton’s Response
141

6100 Executive Blvd Suite 5B01 Rockville, MD 20892-7508
Telephone 301-402-4242
Facsimile 301-480-6900
Website www.bioethics.gov
Harold T. Shapiro, Ph.D.
Chair
N AT I O N A L B I O E T H I C S A D V I S O R Y C O M M I S S I O N
The President The White House Washington, DC 20005
Dear Mr. President:
May 4, 1999
Patricia Backlar
Arturo Brito, M.D.
Alexander Morgan Capron, LL.B. Eric J. Cassell, M.D., M.A.C.P. R. Alta Charo, J.D.
James F. Childress, Ph.D.
David R. Cox, M.D., Ph.D.
Rhetaugh Graves Dumas, Ph.D., R.N. Laurie M. Flynn Carol W. Greider, Ph.D.
Steven H. Holtzman Bette O. Kramer Bernard Lo, M.D.
Lawrence H. Miike, M.D., J.D. Thomas H. Murray, Ph.D. Diane Scott-Jones, Ph.D.
Eric M. Meslin, Ph.D.
Executive Director
Consistent with your October 3,1995, Executive Order 12975, the National Bioethics Advisory Commission has focused a good deal of its efforts over the last three years on issues surrounding the protection of human research subjects. I know of your interest, as well as that of the Congress, which has rightfully inquired about the adequacy of existing protections. To supplement the reports we have already submitted, I take this opportunity to provide you with a summary of our concerns and preliminary findings, some of which will, in our judgment, require further actions by the Federal Government. The attached memo provides additional details, but our key concerns are the following:
We will submit a more comprehensive report to you in the coming months, which will provide specific recommendations for changes in the federal system for protecting human subjects. Of course, as with all NBAC studies, we will continue to ensure that the public, the research community, and the agencies of the Federal Government have an opportunity to participate in the report’s preparation.
Sincerely,
Harold T. Shapiro Chair
National Bioethics Advisory Commission
NBAC Summary of Preliminary Findings:
Adequacy of Federal Protections for Human Subjects in Research
Extending Federal Protections for Human Research Subjects to All Americans
In 1997, President Clinton stated that “science must respect the dignity of every American. We must never allow our citizens to be unwitting guinea pigs in scientific experiments….” That same month, the National Bioethics Advisory Commission (NBAC) resolved, as a matter of ethical principle, that no person should be enrolled in research without the twin protections of informed consent and independent review of the research. NBAC notes with concern that this goal remains unmet.
In particular, the protections of the Federal Policy for the Protection of Human Subjects, also known as the Common Rule, do not extend to all Americans; the Common Rule applies only to subjects in research regulated by the Food and Drug Administration (FDA) or to subjects in research sponsored by some Federal departments and agencies.i Among the Common Rule’s most important protections are the requirements for informed consent by research subjects and for independent review of the research by a local Institutional Review Board (IRB). Despite the fact that many research institutions voluntarily apply the Common Rule—even to their privately financed research—there are other significant sectors of privately funded research that remain ungoverned either by State or Federal law.
NBAC finds that the absence of Federal jurisdiction over much privately funded research means that the U.S. government cannot know how many Americans currently are subjects in experiments, cannot influence how they have been recruited, cannot ensure that research subjects know and understand the risks they are undertaking, and cannot ascertain whether they have been harmed.
Not only does this prevent the Federal Government from protecting Americans enrolling in research, but it affects the Federal Government’s ability to craft policies governing emerging technologies. While preparing its 1997 report Cloning Human Beings, for example, NBAC noted that the Common Rule’s lack of jurisdiction over privately funded research made it impossible to rely on IRBs as the primary mechanism for protecting human subjects against inappropriate uses of those technologies.
Implementation of the Common Rule
Beginning in 1996, Federal departments and agencies responded to NBAC’s request for information pursuant to Executive Order 12975. NBAC is pleased to report that agencies have responded to the Executive Order not only by reporting on their current protections, but by evaluating those protections and taking steps to strengthen them. Based on the agency reports and actions and on its own investigations and contracted studies, NBAC concludes that the Common Rule has significantly reduced, but not eliminated, the possibility for harm to human subjects. As a result, NBAC also concludes that there is a need for significant improvement, both to enforce Federal protections and to make their implementation less burdensome for Federal agencies and researchers.
Research regulated by the FDA or sponsored by one of the Federal departments or independent agencies that have adopted the Common Rule requires prior approval and continuing oversight by an IRB. NBAC has found that all the Federal departments and agencies that sponsor substantial amounts of biomedical research with human subjects have implemented these requirements. On the other hand, several departments and agencies that sponsor behavioral and other nonbiomedical research have not fully implemented the provisions of the Common Rule, despite the fact that
145
Appendix B: Letters of Request and Response
such research may pose serious nonphysical risks, such as loss of insurance or employment, discrimination, incarceration, and invasion of privacy. Although various Federal regulations do provide protections for certain vulnerable populations, these are not incorporated in the Common Rule. In addition, NBAC notes that the Common Rule does not require any special protections for especially vulnerable populations, such as children.
NBAC has identified occasions when nonbiomedical research that posed more than minimal risk was conducted on certain vulnerable populations; in some of these instances, the research was supported or conducted by one of the agencies that has not adopted additional protections, such as those found in Subparts B, C, or D of the Department of Health and Human Services regulations.
Federal departments and agencies do face obstacles in fully implementing the Common Rule. NBAC notes that many agencies find the Common Rule confusing or its provisions too burdensome in light of the type or amount of research they sponsor. Although some agencies have been taking steps to bring themselves into compliance with the Common Rule, nearly all of them agree that increased protection of human subjects cannot be achieved without additional staffing and highly visible statements of commitment from the leadership of their respective departments. Some also have suggested that a central authority governing human subjects research could help to interpret the Common Rule’s requirements, create the oversight structures needed for its implementation, and advise on ethically complex protocols.
NBAC also finds that centralized leadership is needed to achieve consistent interpretation of key statutory and regulatory requirements. Lack of a single authority also means that improvement in human subjects protections, such as those specific to vulnerable populations, requires that every affected department independently adopt new regulations. This is inevitably slower and more inefficient than adoption by a central authority.
For example, in its 1998 report Research Involving Persons with Mental Disorders That May Affect Decisionmaking
Capacity, NBAC observed that some affected agencies were hard-pressed to reconcile their agency mission of fostering much-needed research into the causes and cures for mental illness with the shared Federal commitment to paying scrupulous attention to the interests of vulnerable human subjects. In addition, the absence of a single, authoritative Federal office to oversee human subjects protections will make it difficult to ensure that all affected departments will issue regulations implementing NBAC’s recommendations; indeed, similar recommendations were made 20 years ago by another national bioethics commission regarding the same population, but they were never adopted.
Ensuring Adequate and Accountable Local Oversight
The decentralized local system is sorely strained by inadequate staffing and education of IRBs; by the explosion in research activity; by emerging ethical issues arising from ethical issues raised by epidemiological and public health research; by the trend toward collaborative, multi-centered research; and by an absence of comprehensive public accountability.
NBAC’s work highlights many of these problems and offers some solutions. Its upcoming report on research involving human biological materials, for example, suggests some solutions to the difficult problem of applying current Federal protections to epidemiological research on stored tissue, while its project on international research norms is revealing the dilemmas posed by collaborative research across national boundaries. Its report, Research Involving Persons With Mental Disorders That May Affect Decisionmaking Capacity, emphasized the need to maintain the public’s trust in the integrity of the scientific endeavor. To that end, NBAC suggested “IRBs can effectively use the mechanisms of audit (both internal and external) and disclosure to improve accountability and inspire public confidence in their oversight activities.”
146
National Bioethics Advisory Commission
Conclusion
NBAC finds that the current Federal regulations have served to prevent most recurrences of the gross abuses associated with biomedical research in the earlier part of this century. Nonetheless, some abuses still occur, and the system is in need of significant revision in order to provide clear, efficient, and authoritative guidance to Federal departments and agencies and to ensure that local oversight is effective and accountable to the public. This is essential to improving protections for human research subjects. It is also a necessary first step toward extending these protections to those Americans not yet protected by any State or Federal standards for human subjects in research.
i As of 1997, 16 departments and independent agencies had formally adopted the Common Rule as signatories. In addition, one other independent agency—the Central Intelligence Agency (CIA)—had adopted the Rule in accordance with Executive Order 12333:
Departments
U.S. Department of Agriculture Department of Commerce Department of Defense Department of Education Department of Energy
Department of Health and Human Services Department of Housing and Urban Development Department of Justice Department of Transportation Department of Veterans Affairs
Independent Agencies
International Development Cooperation Agency (Agency for International Development) Consumer Product Safety Commission Environmental Protection Agency National Aeronautics and Space Administration National Science Foundation Social Security Administration
Central Intelligence Agency *
Office of Science and Technology Policy **
* As of 1997, the CIA was not a formal signatory to the Common Rule but had adopted the Rule in accordance with Executive Order 12333. At the time of this report, the CIA is in the process of becoming a formal signatory.
** The Office of Science and Technology Policy is a signatory to the Common Rule, even though it does not itself conduct or support research directly. It has “accepted” the Common Rule, but does not have its own Code of Federal Regulations.
147

Appendix C
The Current Oversight System: History and Description
History
By the early 1900s, concern about the involvement of human participants in research was part of the American political agenda, and since that time it has remained a topic of discussion among policymakers, most notably as medical research expanded in the 1940s and 1950s, although discussions during that period were rarely converted to policy (ACHRE 1995, 83–129). In the late 1940s, however, formal statements of what was required to ensure ethically sound research began to emerge. These early declarations came largely in response to atrocities committed by Nazi investigators, mostly physicians, who were tried before the Nuremberg Military Tribunal.1 A key task of the prosecutors in this case was to distinguish between the Nazi experiments and U.S. wartime research, both of which used human beings. With the trial looming, and prompted by its liaison to the Nuremberg prosecutors, the American Medical Association adopted its first code of research ethics (AMA 1946), which influenced the American judges’ own set of standards for ethical research (ACHRE 1995, 133–135; Moreno 1999, 75). In an unusual move, as part of their decision the judges presented ten “basic principles” for research using human participants, now known as the Nuremberg Code.2,3 However, adherence to such ethical standards was not automatically assured by the development of formal codes of ethics. For example, many saw the Nuremberg Code as an appropriate response to Nazi war crimes but unnecessary for American medicine (Rothman 1991,
62–63). Additionally, the Nuremberg Code is difficult to implement because, among things, its first principle, “the voluntary consent of the human subjects is absolutely
essential,” would appear to allow for no exceptions. This absolute requirement may reflect the code’s origins in deliberations about research with healthy rather than sick participants and with medical research as opposed to other types of research. Moreover, despite its principled stringency, the Nuremberg Code embodies a deep trust of the research community and gives little weight to the inherent conflicts of interest that might be involved. Investigators alone would be responsible for obtaining informed consent and deciding whether their research met the code’s principles.
Although the Nuremberg Code received little attention in the immediate aftermath of the trial, it was not entirely ignored. In 1953, U.S. Secretary of the Department of Defense (DOD), Charles Wilson, issued a Top Secret memorandum establishing policy for research related to atomic, biological, and chemical warfare (Wilson 1953). The policy incorporated the principles of the Nuremberg Code and two additional protections—a prohibition on research involving prisoners of war and a requirement that the Secretary of the appropriate military service approve research studies.
Later that year the National Institutes of Health (NIH) Clinical Center opened and established a policy requiring independent review of research and participants’ written consent, at least for research involving patient volunteers and/or “unusual hazard” (NIH 1953). In 1954, these protections of independent review and written informed consent were extended to all NIH intramural research involving “normal volunteers.”4 These bold statements of policy did not lead immediately to dramatic shifts in actual research practices. The DOD policy remained largely Top Secret and was not
151
Appendix C: The Current Oversight System: History and Description
disseminated to investigators (ACHRE 1995, 108), and at the NIH Clinical Center, procedures for independent review were rarely invoked (Rothman 1991, 56).
Moreover, both the DOD and NIH policies were limited by confusion over the definition of research. It is not clear whether the Nuremberg Code was meant to apply to research with patients as participants or only to research with healthy individuals. As a result, until 1961, the NIH Clinical Center policy did not require independent review for most research with patients.5 In part, policymakers were concerned about intruding into the doctor-patient relationship. More generally, though, until the late 1960s, concern about human protections seemed unnecessary to many observers. The development of penicillin, the conquest of polio, and the emergence of new medical devices and procedures—all apparently unmarked by inappropriate conduct—bolstered the public prestige of and trust in the biomedical research community.6 In 1962, fetal abnormalities linked to maternal use of thalidomide shook public confidence in the regulation of drug-related research. In response, Congress passed the Kefauver-Harris amendments to the Federal Food, Drug, and Cosmetic Act, requiring the Food and Drug Administration (FDA) to evaluate new drugs for efficacy in addition to safety.7 The amendments also specifically required the informed consent of participants in the testing of investigational drugs, but this requirement was weakened by vague language outlining permissible exceptions. Nonetheless, the Kefauver-Harris amendments gave FDA broad authority to regulate drug-related research.
Other events soon influenced NIH policy. A 1962 NIH-commissioned study found that few institutions had their own policies for the protection of research participants (ACHRE 1995, 174; Faden and Beauchamp 1986, 158). The potential for problems was borne out when, in early 1964, newspapers began to describe an NIH-funded study at the Brooklyn Jewish Chronic Disease Hospital in which investigators had injected cancerous cells into elderly patients. The investigators claimed to have obtained informed consent, but many of the patients were incapacitated or did not speak English, and those able to give consent were not told that the cells to
be injected were cancerous (Faden and Beauchamp 1986, 161; Jonsen 1998, 143).
In response (and against the recommendations of a committee appointed to examine the issue), NIH Director James Shannon argued that the institutes should take more responsibility for research ethics (Faden and Beauchamp 1986, 208). At Shannon’s request, the Public Health Service (PHS) issued a policy for extramural projects (PHS 1966). The initiative was important, but the policy was primarily procedural. It required independent review of research by a committee of the investigator’s “institutional associates,” but offered little guidance about the content of that review (PHS 1966, 475). The policy did not originally specify the composition of the review committee, but an accompanying memo stated that a group of people from different disciplines, familiar with the investigator but “free to assess his judgment without placing in jeopardy their own goals,” would be required (Stewart 1966, 474). Importantly, it marked a shift away from sole “reliance on the judgment and virtue of the individual researcher” in the assessment of the ethics of research.8 At NIH, the PHS policy was implemented through the Institutional Relations Branch of the Division of Research Grants. The branch addressed this policy in the way it handled other matters of institutional relations, by negotiating “assurances of compliance” with the PHS policy from each institution receiving funding. For enforcement, the branch could withhold, or, more likely, threaten to withhold funds.9 The NIH Institutional Relations Branch later became the NIH Office for Protection from Research Risks (OPRR) and assumed a lead role in the protection of research participants within the Department of Health and Human Services (DHHS). In June 2000, OPRR was moved out of NIH to the Office of the Secretary, DHHS, becoming the Office for Human Research Protections (OHRP).10 However, in 1966, the PHS “Policy for Clinical Investigations with Human Subjects” applied only to extramural research, and the Division of Research Grants dealt only with NIH grantees. Also in 1966, the NIH Clinical Center strengthened its separate system of independent review (Faden and Beauchamp 1986, 209). At the same time, FDA issued guidance to clarify the
152
requirements for and exceptions from informed consent as specified in the Kefauver-Harris amendments (Faden and Beauchamp 1986, 204).
While NIH and FDA were revising their policies for the protection of human subjects, Henry Beecher published a damning indictment of contemporary research practice in the United States (Beecher 1966). He presented 22 examples of “unethical or questionably ethical studies” published in major medical journals. The article was influential largely because it showed that abuses were occurring in research conducted by mainstream investigators (Rothman 1991, 75). A review of published studies conducted in Great Britain revealed similar abuses (Pappworth 1962).
Several years after Beecher’s article, one of the studies he had discussed became a subject of particular controversy—an investigation of hepatitis involving the injection of a mild strain of the virus into children at the time of admission to the Willowbrook State School for the Retarded in New York. Parental consent was obtained, but the consent form may have been misleading, and desperate parents may have been unduly influenced by the availability of preferential and later exclusively research-related admissions to the institution. One notable feature of the case was that the Willowbrook research had been reviewed and approved by the Armed Forces Epidemiological Board, which funded the research, as well as a local review committee for human experimentation (ACHRE 1995, 178; Faden and Beauchamp 1986, 163–164).
The debates about the Willowbrook State School experiments became part of a more general concern that local institutional review was not adequately protecting individuals who participated in research (Faden and Beauchamp 1986, 211). In 1971, five years after the PHS policy was put into place, the then Department of Health Education and Welfare (DHEW) developed more detailed guidance and justification for review committees in the form of the “Yellow Book” (DHEW 1971).
In the summer of 1972, the New York Times published details of the Tuskegee Syphilis Study, sponsored by PHS since the early 1930s (Heller 1972). Although a formal protocol never existed, the study intended to trace the natural history of syphilis in poor African American
National Bioethics Advisory Commission
males living in Macon County, Alabama. Participants were not told of the purpose of the study; in fact, they were misled into believing that they were being treated for syphilis. Throughout this project, research-related procedures, such as lumbar punctures, were described as “special free treatments.” Investigators continued the study even after penicillin became widely available and prescribed for the treatment of syphilis. In exchange for participation, the men received some unrelated health care, free meals, and transportation, and later in the study, to encourage autopsy, a $50 burial stipend (Jones 1981).
Following media attention about the study, a PHS Advisory Panel was formed to examine whether it was justified at its inception or after penicillin became generally available, to recommend whether the study should be continued, and to determine whether existing human research protection policies were adequate. The study was found to be ethically unjustified; it was halted, and surviving participants were offered treatment. In addition, the Advisory Panel determined that existing procedures for protecting research participants were not adequate. They recommended that “Congress should establish a permanent body with the authority to regulate at least all Federally-supported research involving human subjects” (Tuskegee Syphilis Study Ad Hoc Advisory Panel 1973, 23).
In 1973, the Senate Labor and Public Welfare Committee began a series of hearings on human experimentation (Jonsen 1998, 92–98). Interest was intensified by controversy over NIH-funded research involving aborted fetuses (Cohn 1973). A number of bills were circulated, including one that would have created a National Human Experimentation Board.11 However, it became clear that there was not enough support for such a body, and a compromise was reached. The proposal for a national oversight board was dropped in exchange for an agreement that DHEW would issue regulations governing research with human subjects (ACHRE 1995, 181).12 DHEW quickly developed regulations based on existing “Yellow Book” policies. The regulations were issued by DHEW in May of 1974,13 and the National Research Act was signed in July of that year.14 The National Research Act also established the National Commission for the Protection of Human
153
Appendix C: The Current Oversight System: History and Description
Subjects of Biomedical and Behavioral Research (National Commission) to provide ethical and policy analysis related to conducting human research. The National Commission produced ten reports and is best known for the Belmont Report (National Commission 1979) (see Appendix D), which identified three fundamental ethical principles applicable to research with human participants—respect for persons, beneficence, and justice—which translated respectively into provisions for informed consent, assessment of risk and potential benefits, and selection of participants.
The Belmont Report was important primarily because it linked desired ethical standards and practices and existing concerns to fundamental ethical principles. DHEW regulations already contained specific provisions for obtaining and documenting informed consent and guidance on assessing risk and benefit. The report recommended that additional attention be given to the equitable selection of participants. In response to the Belmont Report, DHHS and FDA simultaneously revised their regulations. Thus, the two sets of regulations were unified in significant ways. In January 1981, the DHHS Secretary signed the revised regulations (45 CFR 46; 21 CFR 50, 56).15 The revisions, however, did not incorporate many of the National Commission’s recommendations. The regulations still placed their greatest emphasis on obtaining and documenting voluntary informed consent, while giving minimal attention to the assessment of risk and potential benefit or the selection of participants. Moreover, the regulations were not supplemented with the substantive ethical rationale that the Belmont Report provided, leaving the regulations less detailed and potentially more difficult to interpret. By 1981, the recommendations in the National Commission’s report on fetal research were codified as a separate Subpart of the DHHS regulations (Subpart B),16 as had the recommendations from their report on research with prisoners (Subpart C).17 However, their recommendations regarding children and those institutionalized as mentally infirmed were not implemented.
In 1981, the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission) was
established. In its first of two biennial reports on research, the President’s Commission examined the general structure of research protections (President’s Commission 1981). The second biennial report (President’s Commission 1983) focused on their implementation. (See Exhibit C.1.) Agency responses to the President’s Commission recommendations were mixed. In 1983, DHHS promulgated regulations governing research with children (Subpart D).18 OPRR took steps to improve interpretation and implementation of the regulations, such as sponsoring regional workshops, issuing regulatory guidance in the form of OPRR Reports, and chairing the Human Subjects Research Subcommittee of the Committee on Science, National Science and Technology Council, Office of the President.
Perhaps the most promising area of agency action was in the standardization of regulations across agencies and departments. The White House convened an interagency ad hoc committee to develop what would become the “Common Rule” (the Federal Policy for the Protection of Human Subjects [Federal Policy]), a set of identical regulations codified by various agencies. Initially, the group worked under the Federal Coordinators Council for Science, Engineering, and Technology. The standardization process was slow. In addition, in a time of severe budget constraints, few were willing to issue new regulations and establish new offices, and many departments requested extra protections or specific exceptions from any proposed rule.19 Nevertheless, in 1991, the regulations known as the Common Rule were simultaneously published in the Federal Register by 15 departments and agencies.20 The Common Rule expanded the scope of regulated research and provided some standardization across departments, with DHHS, primarily through OPRR, playing a key role in its development.
Description of the Federal Regulatory Structure
This history has resulted in a complex and sometimes overlapping, but also incomplete, structure for the protection of human research participants. Each codification
154
National Bioethics Advisory Commission
Exhibit C.1: Recommendations of the President’s Commission’s First Biennial Report—
Protecting Human Subjects (1981)
n “Private” research organizations receiving direct congressional appropriations should be required to follow federal regulations for the protection of human subjects (President’s Commission 1981).
Recommendations of the President’s Commission’s Second Biennial Report—
Implementing Human Research Regulations (1983)
of the Common Rule by a department or agency is equivalent to 45 CFR 46.101–46.124 (Subpart A), the DHHS codification. The DHHS codification is cited throughout this report when referring to the 15 sets of regulations promulgated by federal departments that comprise the Common Rule.21
The Office of Science and Technology Policy (OSTP) did not codify the Common Rule, even though it signed the Federal Policy, because it does not conduct or sponsor research. The Common Rule also regulates research conducted or sponsored by two other agencies that are not signatories to the Common Rule but that are bound
155
Appendix C: The Current Oversight System: History and Description
to DHHS regulations and therefore to the Common Rule either through public law (the Social Security Administration),22 or Executive Order (the Central Intelligence Agency).23 Thus, the Common Rule has 15 codifications and 16 signatories, and it covers 18 federal agencies —17 by force of law. Its structure is depicted in Figure C.1.
The Common Rule applies to all research involving human participants “conducted, supported or otherwise subject to regulation by any federal department or agency which takes appropriate administrative action to make this policy applicable to such research.” Thus, it specifically allows agencies with regulatory authority (notably, FDA) to apply the Common Rule to regulated research (45 CFR 46.101(a)). However, DHHS does not require FDA to apply the Common Rule to the research FDA regulates. FDA also has its own regulatory authority over research involving food and color additives, investigational drugs for human use, medical devices for human use, biological products for human use being developed for marketing, and electronic products that emit radiation (e.g., microwave ovens, laser printers, sun lamps, diagnostic x-ray equipment). FDA also regulates research intended to support a change in the labeling of marketed products. To this regulated research, FDA applies its own set of regulations (21 CFR 50, 56) that are generally, but not entirely the same as the Common Rule (45 CFR 46 Subpart A). However, FDA is bound to DHHS regulations when it conducts its own research.
Recent Events
Since the creation of the Common Rule, government agencies, Congress, academic scholars, and the private sector have called attention to problems with the current oversight system and have recommended or taken steps to improve it.
Government Activities
In 1994, the Advisory Committee on Human Radiation Experiments (ACHRE) was created to investigate reports of federally sponsored human research involving radioactive materials conducted between 1944 and 1972 and to assess the current state of protections for research participants. Concerning the latter charge they found, “evidence of serious deficiencies in some parts of the current system” (ACHRE 1995, 797). Specifically, ACHRE was concerned with variability in the quality of IRBs, persistent confusion among human participants regarding whether they were involved in research or therapy, and insufficient attention to the implications of diminished decisionmaking capacity in the consent process. Several of the committee’s recommendations addressed the protection of the rights and welfare of human research participants in the future and are directly related to the present report. (Relevant recommendations are reproduced in their entirety in Exhibit C.2.)
Congressional Interest
In general, Congress has been a generous and enthusiastic supporter of biomedical research, but at the same time some members of Congress have expressed concerns about the adequacy of oversight of human research.24 In recent years, several congressional committees have held hearings on various aspects of protection, including overall DHHS oversight of human research, oversight of gene therapy trials,25 privacy in health research,26 the administration of investigational drugs to members of the armed forces,27 and the use of placebo-controls and washout periods in clinical trials.28 Legislation to strengthen the system of protection has been introduced, although none has been enacted. In addition, the Children’s Research Act of 200029 included provisions to extend protections for children participating in research.
156
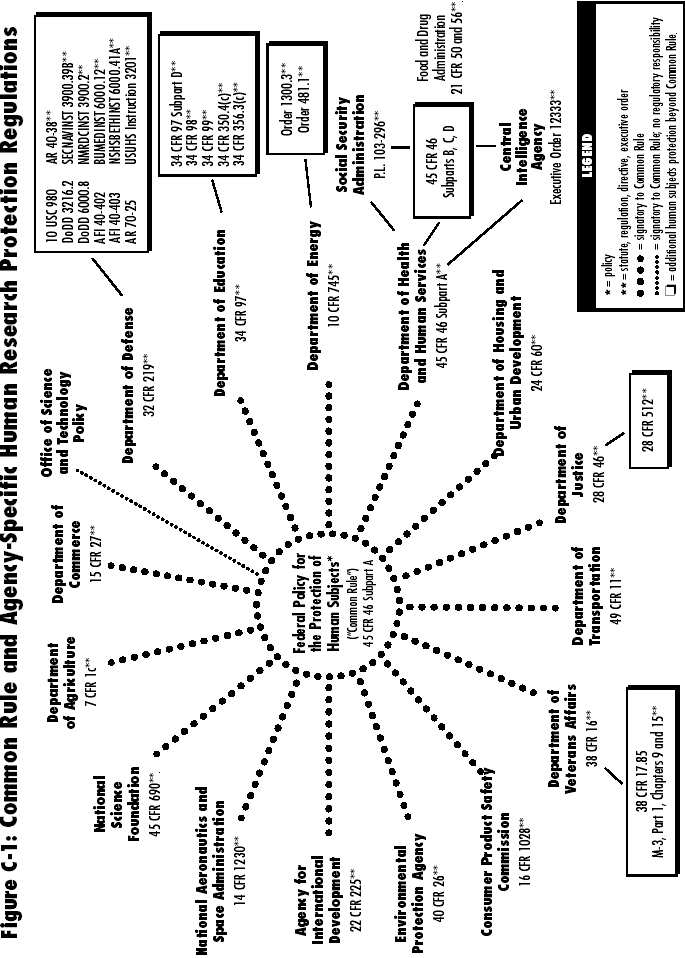
Appendix C: The Current Oversight System: History and Description
Exhibit C.2: ACHRE Recommendations Relevant to This Report
| n |
Recommendation 9: The Advisory Committee recommends to the Human Radiation Interagency Working Group [the cabinet-level group to which ACHRE reported] that efforts be undertaken on a national scale to ensure the centrality of ethics in the conduct of scientists whose research involves human subjects. |
| n |
Recommendation 10: The Advisory Committee recommends to the Human Radiation Interagency Working Group that the IRB component of the federal system for the protection of human subjects be changed in at least the five critical areas described below. 1. Mechanisms for ensuring that IRBs appropriately allocate their time so they can adequately review studies that pose more than minimal risk to human subjects. This may include the creation of alternative mechanisms for review and approval of minimal-risk studies. 2. Mechanisms for ensuring that the information provided to potential subjects 1) clearly distinguishes research from treatment, 2) realistically portrays the likelihood that subjects may benefit medically from their participation and the nature of the potential benefits, and 3) clearly explains the potential for discomfort and pain that may accompany participation in the research. 3. Mechanisms for ensuring that the information provided to potential subjects clearly identifies the federal agency or agencies sponsoring or supporting the research project in whole or in part and all purposes for which the research is being conducted or supported. 4. Mechanisms for ensuring that the information provided to potential subjects clearly identifies the financial implications of deciding to consent to or refuse participation in research. 5. Recognition that if IRBs are to adequately protect the interests of human subjects, they must have the responsibility to determine that the science is of a quality to warrant the imposition of risk or inconvenience on human subjects and, in the case of research that purports to offer a prospect of medical benefit to subjects, to determine that participating in the research affords patient-subjects at least as good an opportunity of securing this medical benefit as would be available to them without participating in research. |
| n |
Recommendation 11: The Advisory Committee recommends to the Human Radiation Interagency Working Group that a mechanism be established to provide for the continuing interpretation and application of ethics rules and principles for the conduct of human subject research in an open and public forum. This mechanism is not provided for in the Common Rule. |
| n |
Recommendation 13: The Advisory Committee recommends that the Human Radiation Interagency Working Group take steps to improve three elements of the current federal system for the protection of the rights and interests of human subjects—oversight, sanctions, and scope: 1) oversight mechanisms to examine outcomes and performance, 2) appropriateness of sanctions for violations of human subjects protections, and 3) extension of human subjects protections to nonfederally funded research (ACHRE 1995, 816–826). |
Notes
1 United States v. Karl Brandt et al. (“The Medical Case”), Trials of War Criminals Before the Nuernberg Military Tribunals Under Control Council Law 10. Nuernberg, October 1946–April 1949. Volumes I–II. Washington, D.C.: U.S. Government Printing Office.
2 United States v. Karl Brandt, et al. (“The Medical Case”), Trials of War Criminals Before the Nuernberg Military Tribunals Under Control Council Law 10. Nuernberg, October 1946–April 1949. Volumes I–II. Washington, D.C.: U.S. Government Printing Office. The Nuremberg Code has been widely reproduced. See, e.g., Shuster 1997.
3 During World War II, unethical medical experiments were also performed by the Japanese, primarily against Chinese prisoners. However, these incidents played less of a role in the development of U.S. policy, partly because the researchers were not prosecuted. Some have alleged that the U.S. government traded immunity for investigators’ data (Harris 1994).
4 See Fletcher, J., “Location of the Office for Protection from Research Risks Within the National Institutes of Health: Problems of Status and Independent Authority.” This background paper was prepared for NBAC and is available in Volume II of this report.
5 Ibid.
158
6 See Moreno, J., “Protectionism in Research Involving Human Subjects.” This background paper was prepared for NBAC and is available in Volume II of this report.
7 Pub. Law 781, 87th Congress.
8 See Moreno, J., “Protectionism in Research Involving Human Subjects.” This background paper was prepared for NBAC and is available in Volume II of this report.
9 See McCarthy, C.R., “Reflections on the Organizational Locus of the Office for Protection from Research Risks.” This background paper was prepared for NBAC and is available in Volume II of this report.
10 65 Fed. Reg. 37136.
11 S. 934, 93rd Congress.
12 See McCarthy, C.R., “Reflections on the Organizational Locus of the Office for Protection from Research Risks.” This background paper was prepared for NBAC and is available in Volume II of this report.
13 39 Fed. Reg. 18914–18920. 14 Pub. Law 348, 93rd Congress. 15 46 Fed. Reg. 8366; 46 Fed. Reg. 8942. 16 40 Fed. Reg. 33526.
17 43 Fed. Reg. 53655.
18 48 Fed. Reg. 9814.
19 Porter, J., Testimony before NBAC. November 23, 1997. Bethesda, Maryland; see McCarthy, C.R., “Reflections on the Organizational Locus of the Office for Protection from Research Risks.” This background paper was prepared for NBAC and is available in Volume II of this report.
20 56 Fed. Reg. 28002–28023.
21 The 15 signatories to the Common Rule are the Department of Agriculture, the Department of Commerce, the Department of Defense, the Department of Education, the Department of Energy, the Department of Health and Human Services, the Department of Housing and Urban Development, the Department of Justice, the Department of Transportation, the Department of Veterans Affairs, the Agency for International Development, the Consumer Product Safety Commission, the Environmental Protection Agency, the National Aeronautics and Space Administration, and the National Science Foundation.
22 Pub. Law 296, 103rd Congress.
23 Executive Order. “United States Intelligence Activities, Executive Order 12333.” 46 Fed. Reg. 59941.
24 U.S. Congress. House. Committee on Government Reform. Subcommittee on Criminal Justice, Drug Policy and Human Resources. Human Subject Research Protections. 106th Congress, 2nd sess., 3 May 2000; U.S. Congress. House. Committee on
National Bioethics Advisory Commission
Government Reform. Subcommittee on Criminal Justice, Drug Policy and Human Resources. Protecting Human Clinical Research Patients. 105th Congress, 2nd sess., 11 June 1998; U.S. Congress. House. Committee on Government Reform. Subcommittee on Criminal Justice, Drug Policy and Human Resources. Oversight of HHS: Bioethics and the Adequacy of Informed Consent. 105th Congress, 1st sess., 8 May 1997.
25 U.S. Congress. Senate. Committee on Health, Education, Labor, and Pensions. Subcommittee on Public Health. Gene Therapy: What Is the Federal Response for Patient Safety? 106th Congress, 2nd sess., 25 May 2000; U.S. Congress. Senate. Committee on Health, Education, Labor, and Pensions. Subcommittee on Public Health.
Gene Therapy: Is There Oversight for Patient Safety? 106th Congress, 2nd sess., 2 February 2000.
26 For example, U.S. Congress. House. Committee on Commerce. Subcommittee on Health and Environment. Medical Records Confidentiality in the Modern Delivery of Health Care. 106th Congress, 1st Session, 27 May 1999; U.S. Congress. Senate. Committee on Health, Education, Labor, and Pensions. Privacy Under a Microscope: Balancing the Needs of Research and Confidentiality. 106th Congress, 1st sess., 24 February 1999.
27 U.S. Congress. House. Committee on Government Reform. Subcommittee on National Security, Veterans Affairs, and International Relations. Force Protection: Improving Safeguards for Administration of Investigational New Drugs to Members of the Armed Forces. 106th Congress, 1st sess., 9 November 1999.
28 U.S. Congress. House. Committee on Veterans Affairs.
Subcommittee on Oversight and Investigations and Subcommittee on Health. Suspension of Medical Research at West Los Angeles and Sepulveda VA Medical Facilities and Informed Consent and Patient Safety in VA Medical Research. 106th Congress, 1st sess., 21 April 1999; U.S. Congress. House. Committee on Government Reform.
Clinical Trial Subjects: Adequate FDA Protection? 105th Congress, 2nd sess., 22 April 1998.
29 Pub. Law 310, 106th Congress.
References
Advisory Committee on Human Radiation Experiments (ACHRE). 1995. Final Report. Washington, D.C.: U.S. Government Printing Office.
American Medical Association (AMA). Judicial Council. 1946. “Supplementary Report of the Judicial Council of the American Medical Association.” Journal of the American Medical Association
132:1090.
Beecher, H.K. 1966. “Ethics and Clinical Research.” New England
Journal of Medicine 274(24):1354–1360.
Cohn, V. 1973. “Live-Fetus Research Debated.” Washington Post, 10 April, A-1.
159
Appendix C: The Current Oversight System: History and Description
Department of Health, Education, and Welfare (DHEW). 1971. The Institutional Guide to DHEW Policy on Protection of Human Subjects. Washington, D.C.: U.S. Government Printing Office.
Faden, R.R., and T.L. Beauchamp. 1986. A History and Theory of Informed Consent. New York: Oxford University Press.
Harris, S.H. 1994. Factories of Death: Japanese Biological Warfare, 1932–1945, and the American Cover-Up. New York: Routledge.
Heller, J. 1972. “Syphilis Victims in U.S. Study Went Untreated for 40 Years.” New York Times, 26 July, A-1.
Jones, J.H. 1981. Bad Blood: The Tuskegee Syphilis Experiment.
New York: The Free Press.
Jonsen, A.R. 1998. The Birth of Bioethics. New York: Oxford University Press.
Moreno, J.D. 1999. Undue Risk: Secret State Experiments on Humans. New York: W.H. Freeman.
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission). 1979.
Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research. Washington, D.C.: U.S. Government Printing Office.
National Institutes of Health (NIH). 1953. Group Consideration of Clinical Research Procedures Deviating from Accepted Medical Practice or Involving Unusual Hazard. In Final Report, Supplemental Vol. I, 321–324. Washington, D.C.: U.S.
Government Printing Office.
Pappworth, M.H. 1962. “Human Guinea Pigs: A Warning.”
Twentieth Century Autumn:66–75.
President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission). 1981. Protecting Human Subjects. First Biennial Report on the Adequacy and Uniformity of Federal Rules and Policies, and of their Implementation, for the Protection of Human Subjects.
Washington, D.C.: U.S. Government Printing Office.
———. 1983. Implementing Human Research Regulations. Second Biennial Report on the Adequacy and Uniformity of Federal Rules and Policies, and of their Implementation, for the Protection of Human Subjects. Washington, D.C.: U.S. Government Printing Office.
Public Health Service (PHS). 1966. Clinical Investigations Using Human Subjects. In Final Report, Supplemental Vol. I, 475–476. Washington, D.C.: U.S. Government Printing Office.
Rothman, D.J. 1991. Strangers at the Bedside: A History of How Law and Bioethics Transformed Medical Decision Making. New York: Basic Books.
Shuster, E. 1997. “Fifty Years Later: The Significance of the Nuremberg Code.” New England Journal of Medicine
337(20):1436–1440.
Stewart, W.H. 1966. Clinical Research Investigations Using Human Subjects. In Final Report, Supplemental Vol. I, 473–474.
Washington, D.C.: U.S. Government Printing Office.
Tuskegee Syphilis Study Ad Hoc Advisory Panel. 1973. Final Report. Washington, D.C.: DHEW.
Wilson, C. 1953. Memorandum for the Secretary of the Army, Secretary of the Navy, Secretary of the Air Force. In Final Report, Supplemental Vol. I, 308–310. Washington, D.C.: U.S.
Government Printing Office.
160
Appendix D
The Belmont Report
Ethical Principles and Guidelines for the Protection of Human Subjects of Research
The National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research
April 18, 1979
Ethical Principles and Guidelines for Research Involving Human Subjects
Scientific research has produced substantial social benefits. It has also posed some troubling ethical questions. Public attention was drawn to these questions by reported abuses of human subjects in biomedical experiments, especially during the Second World War. During the Nuremberg War Crime Trials, the Nuremberg code was drafted as a set of standards for judging physicians and scientists who had conducted biomedical experiments on concentration camp prisoners. This code became the prototype of many later codes intended to assure that research involving human subjects would be carried out in an ethical manner.
The codes consist of rules, some general, others specific, that guide the investigators or the reviewers of research in their work. Such rules often are inadequate to cover complex situations; at times they come into conflict, and they are frequently difficult to interpret or apply. Broader ethical principles will provide a basis on which specific rules may be formulated, criticized and interpreted.
Three principles, or general prescriptive judgments, that are relevant to research involving human subjects are identified in this statement. Other principles may also be relevant. These three are comprehensive, however, and are stated at a level of generalization that should assist scientists, subjects, reviewers and interested citizens to understand the ethical issues inherent in research involving human subjects. These principles cannot always be applied so as to resolve beyond dispute particular ethical problems. The objective is to provide an analytical framework that will guide the resolution of ethical problems arising from research involving human subjects.
This statement consists of a distinction between research and practice, a discussion of the three basic ethical principles, and remarks about the application of these principles.
A. Boundaries Between Practice and Research
It is important to distinguish between biomedical and behavioral research, on the one hand, and the practice of accepted therapy on the other, in order to know what activities ought to undergo review for the protection of human subjects of research. The distinction between research and practice is blurred partly because both often occur together (as in research designed to evaluate a therapy) and partly because notable departures from standard practice are often called “experimental” when the terms “experimental” and “research” are not carefully defined.
For the most part, the term “practice” refers to interventions that are designed solely to enhance the well being of an individual patient or client and that have a reasonable expectation of success. The purpose of medical or behavioral practice is to provide diagnosis, preventive treatment or therapy to particular individuals. By contrast, the term “research” designates an activity designed to test an hypothesis, permit conclusions to be drawn, and thereby to develop or contribute to general-izable knowledge (expressed, for example, in theories, principles, and statements of relationships). Research is usually described in a formal protocol that sets forth an objective and a set of procedures designed to reach that objective.
161
Appendix D: The Belmont Report
When a clinician departs in a significant way from standard or accepted practice, the innovation does not, in and of itself, constitute research. The fact that a procedure is “experimental,” in the sense of new, untested or different, does not automatically place it in the category of research. Radically new procedures of this description should, however, be made the object of formal research at an early stage in order to determine whether they are safe and effective. Thus, it is the responsibility of medical practice committees, for example, to insist that a major innovation be incorporated into a formal research project.
Research and practice may be carried on together when research is designed to evaluate the safety and efficacy of a therapy. This need not cause any confusion regarding whether or not the activity requires review; the general rule is that if there is any element of research in an activity, that activity should undergo review for the protection of human subjects.
B. Basic Ethical Principles
The expression “basic ethical principles” refers to those general judgments that serve as a basic justification for the many particular ethical prescriptions and evaluations of human actions. Three basic principles, among those generally accepted in our cultural tradition, are particularly relevant to the ethic of research involving human subjects: the principles of respect for persons, beneficence and justice.
1. Respect for Persons. Respect for persons incorporates at least two ethical convictions; first, that individuals should be treated as autonomous agents, and second, that persons with diminished autonomy are entitled to protection. The principle of respect for persons thus divides into two separate moral requirements: the requirement to acknowledge autonomy and the requirement to protect those with diminished autonomy.
An autonomous person is an individual capable of deliberation about personal goals and of acting under the direction of such deliberation. To respect autonomy is to give weight to autonomous persons’ considered opinions and choices while refraining from obstructing their actions unless they are clearly detrimental to others. To show lack of respect for an autonomous agent is to
repudiate that person’s considered judgments, to deny an individual the freedom to act on those considered judgments, or to withhold information necessary to make a considered judgment, when there are no compelling reasons to do so.
However, not every human being is capable of self-determination. The capacity for self-determination matures during an individual’s life, and some individuals lose this capacity wholly or in part because of illness, mental disability, or circumstances that severely restrict liberty. Respect for the immature and the incapacitated may require protecting them as they mature or while they are incapacitated.
Some persons are in need of extensive protection, even to the point of excluding them from activities which may harm them; other persons require little protection beyond making sure they undertake activities freely and with awareness of possible adverse consequences. The extent of protection afforded should depend upon the risk of harm and the likelihood of benefit. The judgment that any individual lacks autonomy should be periodically reevaluated and will vary in different situations.
In most cases of research involving human subjects, respect for persons demands that subjects enter into the research voluntarily and with adequate information. In some situations, however, application of the principle is not obvious. The involvement of prisoners as subjects of research provides an instructive example. On the one hand, it would seem that the principle of respect for persons requires that prisoners not be deprived of the opportunity to volunteer for research. On the other hand, under prison conditions they may be subtly coerced or unduly influenced to engage in research activities for which they would not otherwise volunteer. Respect for persons would then dictate that prisoners be protected. Whether to allow prisoners to “volunteer” or to “protect” them presents a dilemma. Respecting persons, in most hard cases, is often a matter of balancing competing claims urged by the principle of respect itself.
2. Beneficence. Persons are treated in an ethical manner not only by respecting their decisions and protecting them from harm, but also by making efforts to secure their well being. Such treatment falls under the principle of beneficence. The term “beneficence” is often understood to
162
cover acts of kindness or charity that go beyond strict obligation. In this document, beneficence is understood in a stronger sense. as an obligation. Two general rules have been formulated as complementary expressions of beneficent actions in this sense: (1) do not harm and (2) maximize possible benefits and minimize possible harms.
The Hippocratic maxim “do no harm” has long been a fundamental principle of medical ethics. Claude Bernard extended it to the realm of research, saying that one should not injure one person regardless of the benefits that might come to others. However, even avoiding harm requires learning what is harmful; and, in the process of obtaining this information, persons may be exposed to risk of harm. Further, the Hippocratic Oath requires physicians to benefit their patients “according to their best judgment.” Learning what will in fact benefit may require exposing persons to risk. The problem posed by these imperatives is to decide when it is justifiable to seek certain benefits despite the risks involved, and when the benefits should be foregone because of the risks.
The obligations of beneficence affect both individual investigators and society at large, because they extend both to particular research projects and to the entire enterprise of research. In the case of particular projects, investigators and members of their institutions are obliged to give forethought to the maximization of benefits and the reduction of risk that might occur from the research investigation. In the case of scientific research in general, members of the larger society are obliged to give forethought the longer term benefits and risks that may result from the improvement of knowledge and from the development of novel medical. psychotherapeutic. and social procedures.
The principle of beneficence often occupies a well-defined justifying role in many areas of research involving human subjects. An example is found in research involving children. Effective ways of treating childhood diseases and fostering healthy development are benefits that serve to justify research involving children - even when individual research subjects are not direct beneficiaries. Research also makes is possible to avoid the harm that may result from the application of previously
National Bioethics Advisory Commission
accepted routine practices that on closer investigation turn out to be dangerous. But the role of the principle of beneficence is not always so unambiguous. A difficult ethical problem remains, for example, about research that presents more than minimal risk without immediate prospect of direct benefit to the children involved. Some have argued that such research is inadmissible, while others have pointed out that this limit would rule out much research promising great benefit to children in the future. Here again, as with all hard cases, the different claims covered by the principle of beneficence may come into conflict and force difficult choices.
3. Justice. Who ought to receive the benefits of research and bear its burdens? This is a question of justice, in the sense of “fairness in distribution” or “what is deserved.” An injustice occurs when some benefit to which a person is entitled is denied without good reason or when some burden is imposed unduly. Another way of conceiving the principle of justice is that equals ought to be treated equally. However, this statement requires explication. Who is equal and who is unequal? What considerations justify departure from equal distribution? Almost all commentators allow that distinctions based on experience, age, deprivation, competence, merit and position do sometimes constitute criteria justifying differential treatment for certain purposes. It is necessary, then, to explain in what respects people should be treated equally. There are several widely accepted formulations of just ways to distribute burdens and benefits. Each formulation mentions some relevant property on the basis of which burdens and benefits should be distributed. These formulations are (1) to each person an equal share, (2) to each person according to individual need, (3) to each person according to individual effort,
| (4) |
to each person according to societal contribution, and |
| (5) |
to each person according to merit. Questions of justice have long been associated with |
social practices such as punishment, taxation and political representation. Until recently these questions have not generally been associated with scientific research. However, they are foreshadowed even in the earliest reflections on the ethics of research involving human subjects. For example, during the 19th and early 20th centuries the burdens of serving as research subjects fell
163
Appendix D: The Belmont Report
largely upon poor ward patients, while the benefits of improved medical care flowed primarily to private patients. Subsequently, the exploitation of unwilling prisoners as research subjects in Nazi concentration camps was condemned as a particularly flagrant injustice. In this country, in the 1940’s, the Tuskegee syphilis study used disadvantaged, rural black men to study the untreated course of a disease that is by no means confined to that population. These subjects were deprived of demonstrably effective treatment in order not to interrupt the project, long after such treatment became generally available.
Against this historical background, it can be seen how conceptions of justice are relevant to research involving human subjects. For example, the selection of research subjects needs to be scrutinized in order to determine whether some classes (e.g., welfare patients, particular racial and ethnic minorities, or persons confined to institutions) are being systematically selected simply because of their easy availability, their compromised position, or their manipulability, rather than for reasons directly related to the problem being studied. Finally, whenever research supported by public funds leads to the development of therapeutic devices and procedures, justice demands both that these not provide advantages only to those who can afford them and that such research should not unduly involve persons from groups unlikely to be among the beneficiaries of subsequent applications of the research.
C. Applications
Applications of the general principles to the conflict of research leads to consideration of the following requirements: informed consent, risk/benefit assessment, and the selection of subjects of research.
1. Informed Consent. Respect for persons requires that subjects, to the degree that they are capable, be given the opportunity to choose what shall or shall not happen to them. This opportunity is provided when adequate standards for informed consent are satisfied. While the importance of informed consent is unquestioned, controversy prevails over the nature and possibility of an informed consent. Nonetheless, there is widespread
agreement that the consent process can be analyzed as containing three elements: information, comprehension and voluntariness.
Information. Most codes of research establish specific items for disclosure intended to assure that subjects are given sufficient information. These items generally include: the research procedure, their purposes, risks and anticipated benefits, alternative procedures (where therapy is involved), and a statement offering the subject the opportunity to ask questions and to withdraw at any time from the research. Additional items have been proposed, including how subjects are selected, the person responsible for the research, etc.
However, a simple listing of items does not answer the question of what the standard should be for judging how much and what sort of information should be provided. One standard frequently invoked in medical practice, namely the information commonly provided by practitioners in the field or in the locale, is inadequate since research takes place precisely when a common understanding does not exist. Another standard, currently popular in malpractice law, requires the practitioner to reveal the information that reasonable persons would wish to know in order to make a decision regarding their care. This, too, seems insufficient since the research subject, being in essence a volunteer, may wish to know considerably more about risks gratuitously undertaken than do patients who deliver themselves into the hand of a clinician for needed care. It may be that a standard of “the reasonable volunteer” should be proposed: the extent and nature of information should be such that persons, knowing that the procedure is neither necessary for their care nor perhaps fully understood, can decide whether they wish to participate in the furthering of knowledge. Even when some direct benefit to them is anticipated, the subjects should understand clearly the range of risk and the voluntary nature of participation.
A special problem of consent arises where informing subjects of some pertinent aspect of the research is likely to impair the validity of the research. In many cases, it is sufficient to indicate to subjects that they are being invited to participate in research of which some features will not be revealed until the research is concluded. In all cases of research involving incomplete disclosure, such
164
research is justified only if it is clear that (1) incomplete disclosure is truly necessary to accomplish the goals of the research, (2) there are no undisclosed risks to subjects that are more than minimal, and (3) there is an adequate plan for debriefing subjects, when appropriate, and for dissemination of research results to them. Information about risks should never be withheld for the purpose of eliciting the cooperation of subjects, and truthful answers should always be given to direct questions about the research. Care should be taken to distinguish cases in which disclosure would destroy or invalidate the research from cases in which disclosure would simply inconvenience the investigator.
Comprehension. The manner and context in which information is conveyed is as important as the information itself. For example, presenting information in a disorganized and rapid fashion, allowing too little time for consideration or curtailing opportunities for questioning, all may adversely affect a subject’s ability to make an informed choice.
Because the subject’s ability to understand is a function of intelligence, rationality, maturity and language, it is necessary to adapt the preservation of the information to the subject’s capabilities. Investigators are responsible for ascertaining that the subject has comprehended the information. While there is always an obligation to ascertain that the information about risk to subjects is complete and adequately comprehended, when the risks are more serious, that obligation increases. On occasion, it may be suitable to give some oral or written tests of comprehension.
Special provision may need to be made when comprehension is severely limited—for example, by conditions of immaturity or mental disability. each class of subjects that one might consider as incompetent (e.g., infants and young children, mentally disabled patients, the terminally ill and the comatose) should be considered on its own terms. Even for these persons, however, respect requires giving them the opportunity to choose to the extent they are able, whether or not to participate in research. The objections of these subjects to involvement should be honored, unless the research entails pro-providing them a therapy unavailable elsewhere. Respect for persons also requires seeking the permission of other
National Bioethics Advisory Commission
parties in order to protect the subjects from harm. Such persons are thus respected both by acknowledging their own wishes and by the use of third parties to protect them from harm.
The third parties chosen should be those who are most likely to understand the incompetent subject’s situation and to act in that person’s best interest. The person authorized to act on behalf of the subject should be given an opportunity to observe the research as it proceeds in order to be able to withdraw the subject from the research, if such action appears in the subject’s best interest.
Voluntariness. An agreement to participate in research constitutes a valid consent only if voluntarily given. This element of informed consent requires conditions free of coercion and undue influence. Coercion occurs when an overt threat of harm is intentionally presented by one person to another in order to obtain compliance. Undue influence, by contrast, occurs through an offer of an excessive, unwarranted, inappropriate or improper reward or other overture in order to obtain compliance. Also, inducements that would ordinarily be acceptable may become undue influences if the subject is especially vulnerable.
Unjustifiable pressures usually occur when persons in positions of authority or commanding influence—especially where possible sanctions are involved—urge a course of action for a subject. A continuum of such influencing factors exists, however, and it is impossible to state precisely where justifiable persuasion ends and undue influence begins. But undue influence would include actions such as manipulating a person’s choice through the controlling influence of a close relative and threatening to withdraw health services to which an individual would otherwise be entitled.
2. Assessment of Risks and Benefits. The assessment of risks and benefits requires a careful arrayal of relevant data, including, in some cases, alternative ways of obtaining the benefits sought in the research. Thus, the assessment presents both an opportunity and a responsibility to gather systematic and comprehensive information about proposed research. For the investigator, it is a means to examine whether the proposed research is properly designed. For a review committee, it is a method for
165
Appendix D: The Belmont Report
determining whether the risks that will be presented to subjects are justified. For prospective subjects, the assessment will assist the determination whether or not to participate.
The Nature and Scope of Risks and Benefits. The requirement that research be justified on the basis of a favorable risk/benefit assessment bears a close relation to the principle of beneficence, just as the moral requirement that informed consent be obtained is derived primarily from the principle of respect for persons.
The term “risk” refers to a possibility that harm may occur. However, when expressions such as “small risk” or “high risk” are used, they usually refer (often ambiguously) both to the chance (probability) of experiencing a harm and the severity (magnitude) of the envisioned harm.
The term “benefit” is used in the research context to refer to something of positive value related to health or welfare. Unlike “risk,” “benefit” is not a term that expresses probabilities. Risk is properly contrasted to probability of benefits, and benefits are properly contrasted with harms rather than risks of harm. Accordingly, so-called risk/benefit assessments are concerned with the probabilities and magnitudes of possible harms and anticipated benefits. Many kinds of possible harms and benefits need to be taken into account. There are, for example, risks of psychological harm, physical harm, legal harm, social harm and economic harm and the corresponding benefits. While the most likely types of harms to research subjects are those of psychological or physical pain or injury, other possible kinds should not be overlooked.
Risks and benefits of research may affect the individual subjects, the families of the individual subjects, and society at large (or special groups of subjects in society). Previous codes and Federal regulations have required that risks to subjects be outweighed by the sum of both the anticipated benefit to the subject, if any, and the anticipated benefit to society in the form of knowledge to be gained from the research. In balancing these different elements, the risks and benefits affecting the immediate research subject will normally carry special weight. On the other hand, interests other than those of the subject may on some occasions be sufficient by themselves to
justify the risks involved in the research, so long as the subjects’ rights have been protected. Beneficence thus requires that we protect against risk of harm to subjects and also that we be concerned about the loss of the substantial benefits that might be gained from research.
The Systematic Assessment of Risks and Benefits. It is commonly said that benefits and risks must be “balanced” and shown to be “in a favorable ratio.” The metaphorical character of these terms draws attention to the difficulty of making precise judgments. Only on rare occasions will quantitative techniques be available for the scrutiny of research protocols. However, the idea of systematic, nonarbitrary analysis of risks and benefits should be emulated insofar as possible. This ideal requires those making decisions about the justifiability of research to be thorough in the accumulation and assessment of information about all aspects of the research, and to consider alternatives systematically. This procedure renders the assessment of research more rigorous and precise, while making communication between review board members and investigators less subject to misinterpretation, misinformation and conflicting judgments. Thus, there should first be a determination of the validity of the presuppositions of the research; then the nature, probability and magnitude of risk should be distinguished with as much clarity as possible. The method of ascertaining risks should be explicit, especially where there is no alternative to the use of such vague categories as small or slight risk. It should also be determined whether an investigator’s estimates of the probability of harm or benefits are reasonable, as judged by known facts or other available studies.
Finally, assessment of the justifiability of research should reflect at least the following considerations: (i) Brutal or inhumane treatment of human subjects is never morally justified. (ii) Risks should be reduced to those necessary to achieve the research objective. It should be determined whether it is in fact necessary to use human subjects at all. Risk can perhaps never be entirely eliminated, but it can often be reduced by careful attention to alternative procedures. (iii) When research involves significant risk of serious impairment, review committees should be extraordinarily insistent on the justification of the risk (looking usually to the
166
likelihood of benefit to the subject - or, in some rare cases, to the manifest voluntariness of the participation). (iv) When vulnerable populations are involved in research, the appropriateness of involving them should itself be demonstrated. A number of variables go into such judgments, including the nature and degree of risk, the condition of the particular population involved, and the nature and level of the anticipated benefits. (v) Relevant risks and benefits must be thoroughly arrayed in documents and procedures used in the informed consent process.
3. Selection of Subjects. Just as the principle of respect for persons finds expression in the requirements for consent, and the principle of beneficence in risk/benefit assessment, the principle of justice gives rise to moral requirements that there be fair procedures and outcomes in the selection of research subjects.
Justice is relevant to the selection of subjects of research at two levels: the social and the individual. Individual justice in the selection of subjects would require that researchers exhibit fairness: thus, they should not offer potentially beneficial research only to some patients who are in their favor or select only “undesirable” persons for risky research. Social justice requires that distinction be drawn between classes of subjects that ought, and ought not, to participate in any particular kind of research, based on the ability of members of that class to bear burdens and on the appropriateness of placing further burdens on already burdened persons. Thus, it can be considered a matter of social justice that there is an order of preference in the selection of classes of subjects (e.g., adults before children) and that some classes of potential subjects (e.g., the institutionalized mentally infirm or prisoners) may be involved as research subjects, if at all, only on certain conditions.
Injustice may appear in the selection of subjects. even if individual subjects are selected fairly by investigators
National Bioethics Advisory Commission
and treated fairly in the course of research. Thus injustice arises from social. racial, sexual and cultural biases institutionalized in society. Thus, even if individual researchers are treating their research subjects fairly, and even if IRBs are taking care to assure that subjects are selected fairly within a particular institution. unjust social patterns may nevertheless appear in the overall distribution of the burdens and benefits of research. Although individual institutions or investigators may not be able to resolve a problem that is pervasive in their social setting. They can consider distributive justice in selecting research subjects.
Some populations, especially institutionalized ones, are already burdened in many ways by their infirmities and environments. When research is proposed that involves risks and does not include a therapeutic component, other less burdened classes of persons should be called upon first to accept these risks of research, except where the research is directly related to the specific conditions of the class involved. Also, even though public funds for research may often flow in the same directions as public funds for health care, it seems unfair that populations dependent on public health care constitute a pool of preferred research subjects if more advantaged populations are likely to be the recipients of the benefits.
One special instance of injustice results from the involvement of vulnerable subjects. Certain groups, such as racial minorities, the economically disadvantaged, the very sick, and the institutionalized may continually be sought as research subjects, owing to their ready availability in settings where research is conducted. Given their dependent status and their frequently compromised capacity for free consent, they should be protected against the danger of being involved in research solely for administrative convenience, or because they are easy to manipulate as a result of their illness or socioeconomic condition.
167
Appendix E
Code of Federal Regulations, Title 45 Part 46
Subpart A: Federal Policy for the Protection of Human Subjects (Basic DHHS Policy for Protection of Human Research Subjects)
Source: 56 FR 28003, June 18, 1991.
§ 46.101 To what does this policy apply?
| (a) |
Except as provided in paragraph (b) of this section, this policy applies to all research involving human subjects conducted, supported or otherwise subject to regulation by any Federal Department or Agency which takes appropriate administrative action to make the policy applicable to such research. This includes research conducted by Federal civilian employees or military personnel, except that each Department or Agency head may adopt such procedural modifications as may be appropriate from an administrative standpoint. It also includes research conducted, supported, or otherwise subject to regulation by the Federal Government outside the United States. |
|
| (1) |
Research that is conducted or supported by a Federal Department or Agency, whether or not it is regulated as defined in §46.102(e), must comply with all sections of this policy. |
|
| (2) |
Research that is neither conducted nor supported by a Federal Department or Agency but is subject to regulation as defined in §46.102(e) must be reviewed and approved, in compliance with § 46.101, §46.102, and §46.107 through §46.117 of this policy, by an Institutional Review Board (IRB) that operates in accordance with the pertinent requirements of this policy. |
|
| (b) |
Unless otherwise required by Department or Agency heads, research activities in which the only involve- |
|
ment of human subjects will be in one or more of the following categories are exempt from this policy:1
| (1) |
Research conducted in established or commonly accepted educational settings, involving normal educational practices, such as (i) research on regular and special education instructional strategies, or (ii) research on the effectiveness of or the comparison among instructional techniques, curricula, or classroom management methods. |
|
| (2) |
Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures or observation of public behavior, unless: |
|
| (i) |
information obtained is recorded in such a manner that human subjects can be identified, directly or through identifiers linked to the subjects; and (ii) any disclosure of the human subjects’ responses outside the research could reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects’ financial standing, employability, or reputation. |
|
| (3) |
Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures, or observation of public behavior that is not exempt under paragraph (b)(2) of this section, if: |
|
| (i) |
the human subjects are elected or appointed public officials or candidates for public office; or (ii) Federal statute(s) require(s) without exception that the confidentiality of the personally identifiable information will be maintained throughout the research and thereafter. |
|
| (4) |
Research involving the collection or study of existing data, documents, records, pathological |
|
169
Appendix E: Code of Federal Regulations, Title 45 Part 46
specimens, or diagnostic specimens, if these sources are publicly available or if the information is recorded by the investigator in such a manner that subjects cannot be identified, directly or through identifiers linked to the subjects.
| (5) |
Research and demonstration projects which are conducted by or subject to the approval of Department or Agency heads, and which are designed to study, evaluate, or otherwise examine: |
||
| (i) |
Public benefit or service programs; (ii) procedures for obtaining benefits or services under those programs; (iii) possible changes in or alternatives to those programs or procedures; or (iv) possible changes in methods or levels of payment for benefits or services under those programs. |
||
| (6) |
Taste and food quality evaluation and consumer acceptance studies, (i) if wholesome foods without additives are consumed or (ii) if a food is consumed that contains a food ingredient at or below the level and for a use found to be safe, or agricultural chemical or environmental contaminant at or below the level found to be safe, by the Food and Drug Administration or approved by the Environmental Protection Agency or the Food Safety and Inspection Service of the U.S. Department of Agriculture. |
||
| (c) |
Department or Agency heads retain final judgment as to whether a particular activity is covered by this policy. |
||
| (d) |
Department or Agency heads may require that specific research activities or classes of research activities conducted, supported, or otherwise subject to regulation by the Department or Agency but not otherwise covered by this policy, comply with some or all of the requirements of this policy. |
||
| (e) |
Compliance with this policy requires compliance with pertinent Federal laws or regulations which provide additional protections for human subjects. |
||
| (f) |
This policy does not affect any State or local laws or regulations which may otherwise be applicable and which provide additional protections for human subjects. |
||
| (g) |
This policy does not affect any foreign laws or regulations which may otherwise be applicable and |
||
which provide additional protections to human subjects of research.
| (h) |
When research covered by this policy takes place in foreign countries, procedures normally followed in the foreign countries to protect human subjects may differ from those set forth in this policy. [An example is a foreign institution which complies with guidelines consistent with the World Medical Assembly Declaration (Declaration of Helsinki amended 1989) issued either by sovereign states or by an organization whose function for the protection of human research subjects is internationally recognized.] In these circumstances, if a Department or Agency head determines that the procedures prescribed by the institution afford protections that are at least equivalent to those provided in this policy, the Department or Agency head may approve the substitution of the foreign procedures in lieu of the procedural requirements provided in this policy. Except when otherwise required by statute, Executive Order, or the Department or Agency head, notices of these actions as they occur will be published in the Federal Register or will be otherwise published as provided in Department or Agency procedures. |
| (i) |
Unless otherwise required by law, Department or Agency heads may waive the applicability of some or all of the provisions of this policy to specific research activities or classes or research activities otherwise covered by this policy. Except when otherwise required by statute or Executive Order, the Department or Agency head shall forward advance notices of these actions to the Office for Protection from Research Risks, National Institutes of Health, Department of Health and Human Services (DHHS), and shall also publish them in the Federal Register or in such other manner as provided in Department or Agency procedures.1 |
1 Institutions with DHHS-approved assurances on file will abide by provisions of Title 45 CFR Part 46 Subparts A-D. Some of the other departments and agencies have incorporated all provisions of Title 45 CFR Part 46 into their policies and procedures as well. However, the exemptions at 45 CFR 46.101(b) do not apply to research involving prisoners, fetuses, pregnant women, or human in vitro fertilization, Subparts B and C. The exemption at 45 CFR 46.101(b)(2), for research involving survey or interview procedures or observation of public behavior, does not apply to research with children, Subpart D, except for research involving observations of public behavior when the investigator(s) do not participate in the activities being observed.
170
§ 46.102 Definitions.
| (a) |
Department or Agency head means the head of any Federal Department or Agency and any other officer or employee of any Department or Agency to whom authority has been delegated. |
|
| (b) |
Institution means any public or private entity or Agency (including Federal, State, and other agencies). |
|
| (c) |
Legally authorized representative means an individual or judicial or other body authorized under applicable law to consent on behalf of a prospective subject to the subject’s participation in the procedure(s) involved in the research. |
|
| (d) |
Research means a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge. Activities which meet this definition constitute research for purposes of this policy, whether or not they are conducted or supported under a program which is considered research for other purposes. For example, some demonstration and service programs may include research activities. |
|
| (e) |
Research subject to regulation, and similar terms are intended to encompass those research activities for which a Federal Department or Agency has specific responsibility for regulating as a research activity, (for example, Investigational New Drug requirements administered by the Food and Drug Administration). It does not include research activities which are incidentally regulated by a Federal Department or Agency solely as part of the Department’s or Agency’s broader responsibility to regulate certain types of activities whether research or non-research in nature (for example, Wage and Hour requirements administered by the Department of Labor). |
|
| (f) |
Human subject means a living individual about whom an investigator (whether professional or student) conducting research obtains |
|
| (1) |
data through intervention or interaction with the individual, or |
|
| (2) |
identifiable private information. |
|
Intervention includes both physical procedures by which data are gathered (for example, venipuncture) and manipulations of the subject or the subject’s environment that are performed for research purposes.
National Bioethics Advisory Commission
Interaction includes communication or interpersonal contact between investigator and subject. Private information includes information about behavior that occurs in a context in which an individual can reasonably expect that no observation or recording is taking place, and information which has been provided for specific purposes by an individual and which the individual can reasonably expect will not be made public (for example, a medical record). Private information must be individually identifiable (i.e., the identity of the subject is or may readily be ascertained by the investigator or associated with the information) in order for obtaining the information to constitute research involving human subjects.
| (g) |
IRB means an Institutional Review Board established in accord with and for the purposes expressed in this policy. |
| (h) |
IRB approval means the determination of the IRB that the research has been reviewed and may be conducted at an institution within the constraints set forth by the IRB and by other institutional and Federal requirements. |
| (i) |
Minimal risk means that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests. |
| (j) |
Certification means the official notification by the institution to the supporting Department or Agency, in accordance with the requirements of this policy, that a research project or activity involving human subjects has been reviewed and approved by an IRB in accordance with an approved assurance. |
§46.103 Assuring compliance with this policy —research conducted or supported by any Federal Department or Agency.
| (a) |
Each institution engaged in research which is covered by this policy and which is conducted or supported by a Federal Department or Agency shall provide written assurance satisfactory to the Department or Agency head that it will comply with the requirements set forth in this policy. In lieu of requiring submission of an assurance, individual Department or Agency heads shall accept the |
171
Appendix E: Code of Federal Regulations, Title 45 Part 46
existence of a current assurance, appropriate for the research in question, on file with the Office for Protection from Research Risks, National Institutes Health, DHHS, and approved for Federal wide use by that office. When the existence of a DHHS-approved assurance is accepted in lieu of requiring submission of an assurance, reports (except certification) required by this policy to be made to Department and Agency heads shall also be made to the Office for Protection from Research Risks, National Institutes of Health, DHHS.
| (b) |
Departments and agencies will conduct or support research covered by this policy only if the institution has an assurance approved as provided in this section, and only if the institution has certified to the Department or Agency head that the research has been reviewed and approved by an IRB provided for in the assurance, and will be subject to continuing review by the IRB. Assurances applicable to federally supported or conducted research shall at a minimum include: |
|
| (1) |
A statement of principles governing the institution in the discharge of its responsibilities for protecting the rights and welfare of human subjects of research conducted at or sponsored by the institution, regardless of whether the research is subject to Federal regulation. This may include an appropriate existing code, declaration, or statement of ethical principles, or a statement formulated by the institution itself. This requirement does not preempt provisions of this policy applicable to Department- or Agency-supported or regulated research and need not be applicable to any research exempted or waived under §46.101 (b) or (i). |
|
| (2) |
Designation of one or more IRBs established in accordance with the requirements of this policy, and for which provisions are made for meeting space and sufficient staff to support the IRB’s review and recordkeeping duties. |
|
| (3) |
A list of IRB members identified by name; earned degrees; representative capacity; indications of experience such as board certifications, licenses, etc., sufficient to describe each member’s chief anticipated contributions to IRB deliberations; and any employment or other relationship between each member and the |
|
institution; for example: full-time employee, part-time employee, member of governing panel or board, stockholder, paid or unpaid consultant. Changes in IRB membership shall be reported to the Department or Agency head, unless in accord with §46.103(a) of this policy, the existence of a DHHS-approved assurance is accepted. In this case, change in IRB membership shall be reported to the Office for Protection from Research Risks, National Institutes of Health, DHHS.
| (4) |
Written procedures which the IRB will follow |
||
| (i) |
for conducting its initial and continuing review of research and for reporting its findings and actions to the investigator and the institution; (ii) for determining which projects require review more often than annually and which projects need verification from sources other than the investigators that no material changes have occurred since previous IRB review; and (iii) for ensuring prompt reporting to the IRB of proposed changes in a research activity, and for ensuring that such changes in approved research, during the period for which IRB approval has already been given, may not be initiated without IRB review and approval except when necessary to eliminate apparent immediate hazards to the subject. |
||
| (5) |
Written procedures for ensuring prompt reporting to the IRB, appropriate institutional officials, and the Department or Agency head of (i) any unanticipated problems involving risks to subjects or others or any serious or continuing non-compliance with this policy or the requirements or determinations of the IRB; and (ii) any suspension or termination of IRB approval. |
||
| (c) |
The assurance shall be executed by an individual authorized to act for the institution and to assume on behalf of the institution the obligations imposed by this policy and shall be filed in such form and manner as the Department or Agency head prescribes. |
||
| (d) |
The Department or Agency head will evaluate all assurances submitted in accordance with this policy through such officers and employees of the Department or Agency and such experts or consultants engaged for this purpose as the Department or |
||
172
Agency head determines to be appropriate. The Department or Agency head’s evaluation will take into consideration the adequacy of the proposed IRB in light of the anticipated scope of the institution’s research activities and the types of subject populations likely to be involved, the appropriateness of the proposed initial and continuing review procedures in light of the probable risks, and the size and complexity of the institution.
| (e) |
On the basis of this evaluation, the Department or Agency head may approve or disapprove the assurance, or enter into negotiations to develop an approvable one. The Department or Agency head may limit the period during which any particular approved assurance or class of approved assurances shall remain effective or otherwise condition or restrict approval. |
|
| (f) |
Certification is required when the research is supported by a Federal Department or Agency and not otherwise exempted or waived under §46.101 (b) or |
|
| (i). |
An institution with an approved assurance shall certify that each application or proposal for research covered by the assurance and by §46.103 of this policy has been reviewed and approved by the IRB. Such certification must be submitted with the application or proposal or by such later date as may be prescribed by the Department or Agency to which the application or proposal is submitted. Under no condition shall research covered by §46.103 of the policy be supported prior to receipt of the certification that the research has been reviewed and approved by the IRB. Institutions without an approved assurance covering the research shall certify within 30 days after receipt of a request for such a certification from the Department or Agency, that the application or proposal has been approved by the IRB. If the certification is not submitted within these time limits, the application or proposal may be returned to the institution. (Approved by the Office of Management and Budget under Control Number 9999-0020.) |
|
§§ 46.104—46.106 [Reserved]
§ 46.107 IRB membership.
| (a) |
Each IRB shall have at least five members, with varying backgrounds to promote complete and adequate |
National Bioethics Advisory Commission
review of research activities commonly conducted by the institution. The IRB shall be sufficiently qualified through the experience and expertise of its members, and the diversity of the members, including consideration of race, gender, and cultural backgrounds and sensitivity to such issues as community attitudes, to promote respect for its advice and counsel in safeguarding the rights and welfare of human subjects. In addition to possessing the professional competence necessary to review specific research activities, the IRB shall be able to ascertain the acceptability of proposed research in terms of institutional commitments and regulations, applicable law, and standards of professional conduct and practice. The IRB shall therefore include persons knowledgeable in these areas. If an IRB regularly reviews research that involves a vulnerable category of subjects, such as children, prisoners, pregnant women, or handicapped or mentally disabled persons, consideration shall be given to the inclusion of one or more individuals who are knowledgeable about and experienced in working with these subjects.
| (b) |
Every nondiscriminatory effort will be made to ensure that no IRB consists entirely of men or entirely of women, including the institution’s consideration of qualified persons of both sexes, so long as no selection is made to the IRB on the basis of gender. No IRB may consist entirely of members of one profession. |
| (c) |
Each IRB shall include at least one member whose primary concerns are in scientific areas and at least one member whose primary concerns are in nonsci-entific areas. |
| (d) |
Each IRB shall include at least one member who is not otherwise affiliated with the institution and who is not part of the immediate family of a person who is affiliated with the institution. |
| (e) |
No IRB may have a member participate in the IRB’s initial or continuing review of any project in which the member has a conflicting interest, except to provide information requested by the IRB. |
| (f) |
An IRB may, in its discretion, invite individuals with competence in special areas to assist in the review of issues which require expertise beyond or in addition to that available on the IRB. These individuals may not vote with the IRB. |
173
Appendix E: Code of Federal Regulations, Title 45 Part 46
§ 46.108 IRB functions and operations.
In order to fulfill the requirements of this policy each IRB shall:
| (a) |
Follow written procedures in the same detail as described in §46.103(b)(4) and to the extent required by §46.103(b)(5). |
| (b) |
Except when an expedited review procedure is used (see §46.110), review proposed research at convened meetings at which a majority of the members of the IRB are present, including at least one member whose primary concerns are in nonscientific areas. In order for the research to be approved, it shall receive the approval of a majority of those members present at the meeting |
§ 46.109 IRB review of research.
| (a) |
An IRB shall review and have authority to approve, require modifications in (to secure approval), or disapprove all research activities covered by this policy. |
| (b) |
An IRB shall require that information given to subjects as part of informed consent is in accordance with §46.116. The IRB may require that information, in addition to that specifically mentioned in §46.116, be given to the subjects when in the IRB’s judgment the information would meaningfully add to the protection of the rights and welfare of subjects. |
| (c) |
An IRB shall require documentation of informed consent or may waive documentation in accordance with §46.117. |
| (d) |
An IRB shall notify investigators and the institution in writing of its decision to approve or disapprove the proposed research activity, or of modifications required to secure IRB approval of the research activity. If the IRB decides to disapprove a research activity, it shall include in its written notification a statement of the reasons for its decision and give the investigator an opportunity to respond in person or in writing. |
| (e) |
An IRB shall conduct continuing review of research covered by this policy at intervals appropriate to the degree of risk, but not less than once per year, and shall have authority to observe or have a third party observe the consent process and the research. (Approved by the Office of Management and Budget under Control Number 9999-0020.) |
§46.110 Expedited review procedures for certain kinds of research involving no more than minimal risk, and for minor changes in approved research.
| (a) |
The Secretary, HHS, has established, and published as a Notice in the Federal Register, a list of categories of research that may be reviewed by the IRB through an expedited review procedure. The list will be amended, as appropriate, after consultation with other departments and agencies, through periodic republication by the Secretary, HHS, in the Federal Register. A copy of the list is available from the Office for Protection from Research Risks, National Institutes of Health, DHHS, Bethesda, Maryland 20892. |
|
| (b) |
An IRB may use the expedited review procedure to review either or both of the following: |
|
| (1) |
some or all of the research appearing on the list and found by the reviewer(s) to involve no more than minimal risk, |
|
| (2) |
minor changes in previously approved research during the period (of one year or less) for which approval is authorized. |
|
Under an expedited review procedure, the review may be carried out by the IRB chairperson or by one or more experienced reviewers designated by the chairperson from among members of the IRB. In reviewing the research, the reviewers may exercise all of the authorities of the IRB except that the reviewers may not disapprove the research. A research activity may be disapproved only after review in accordance with the non-expedited procedure set forth in §46.108(b).
| (c) |
Each IRB which uses an expedited review procedure shall adopt a method for keeping all members advised of research proposals which have been approved under the procedure. |
| (d) |
The Department or Agency head may restrict, suspend, terminate, or choose not to authorize an institution’s or IRB’s use of the expedited review procedure. |
§ 46.111 Criteria for IRB approval of research.
| (a) |
In order to approve research covered by this policy the IRB shall determine that all of the following requirements are satisfied: |
174
| (1) |
Risks to subjects are minimized: (i) by using procedures which are consistent with sound research design and which do not unnecessarily expose subjects to risk, and (ii) whenever appropriate, by using procedures already being performed on the subjects for diagnostic or treatment purposes. |
|
| (2) |
Risks to subjects are reasonable in relation to anticipated benefits, if any, to subjects, and the importance of the knowledge that may reasonably be expected to result. In evaluating risks and benefits, the IRB should consider only those risks and benefits that may result from the research (as distinguished from risks and benefits of therapies subjects would receive even if not participating in the research). The IRB should not consider possible long-range effects of applying knowledge gained in the research (for example, the possible effects of the research on public policy) as among those research risks that fall within the purview of its responsibility. |
|
| (3) |
Selection of subjects is equitable. In making this assessment the IRB should take into account the purposes of the research and the setting in which the research will be conducted and should be particularly cognizant of the special problems of research involving vulnerable populations, such as children, prisoners, pregnant women, mentally disabled persons, or economically or educationally disadvantaged persons. |
|
| (4) |
Informed consent will be sought from each prospective subject or the subject’s legally authorized representative, in accordance with, and to the extent required by §46.116. |
|
| (5) |
Informed consent will be appropriately documented, in accordance with, and to the extent required by §46.117. |
|
| (6) |
When appropriate, the research plan makes adequate provision for monitoring the data collected to ensure the safety of subjects. |
|
| (7) |
When appropriate, there are adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data. |
|
| (b) |
When some or all of the subjects are likely to be vulnerable to coercion or undue influence, such as children, prisoners, pregnant women, mentally disabled persons, or economically or educationally |
|
National Bioethics Advisory Commission
disadvantaged persons, additional safeguards have been included in the study to protect the rights and welfare of these subjects.
§ 46.112 Review by institution.
Research covered by this policy that has been approved by an IRB may be subject to further appropriate review and approval or disapproval by officials of the institution. However, those officials may not approve the research if it has not been approved by an IRB.
§46.113 Suspension or termination of IRB approval of research.
An IRB shall have authority to suspend or terminate approval of research that is not being conducted in accordance with the IRB’s requirements or that has been associated with unexpected serious harm to subjects. Any suspension or termination of approval shall include a statement of the reasons for the IRB’s action and shall be reported promptly to the investigator, appropriate institutional officials, and the Department or Agency head.
(Approved by the Office of Management and Budget under Control Number 9999-0020.)
§ 46.114 Cooperative research.
Cooperative research projects are those projects covered by this policy which involve more than one institution. In the conduct of cooperative research projects, each institution is responsible for safeguarding the rights and welfare of human subjects and for complying with this policy. With the approval of the Department or Agency head, an institution participating in a cooperative project may enter into a joint review arrangement, rely upon the review of another qualified IRB, or make similar arrangements for avoiding duplication of effort.
§ 46.115 IRB records.
| (a) |
An institution, or when appropriate an IRB, shall prepare and maintain adequate documentation of IRB activities, including the following: |
|
| (1) |
Copies of all research proposals reviewed, scientific evaluations, if any, that accompany the proposals, approved sample consent documents, progress reports submitted by investigators, and reports of injuries to subjects. |
|
175
Appendix E: Code of Federal Regulations, Title 45 Part 46
| (2) |
Minutes of IRB meetings which shall be in sufficient detail to show attendance at the meetings; actions taken by the IRB; the vote on these actions including the number of members voting for, against, and abstaining; the basis for requiring changes in or disapproving research; and a written summary of the discussion of controverted issues and their resolution. |
|
| (3) |
Records of continuing review activities. |
|
| (4) |
Copies of all correspondence between the IRB and the investigators. |
|
| (5) |
A list of IRB members in the same detail as described in §46.103(b)(3). |
|
| (6) |
Written procedures for the IRB in the same detail as described in §46.103(b)(4) and § 46.103(b)(5). |
|
| (7) |
Statements of significant new findings provided to subjects, as required by §46.116(b)(5). |
|
| (b) |
The records required by this policy shall be retained for at least 3 years, and records relating to research which is conducted shall be retained for at least 3 years after completion of the research. All records shall be accessible for inspection and copying by authorized representatives of the Department or Agency at reasonable times and in a reasonable manner. (Approved by the Office of Management and Budget under Control Number 9999-0020.) |
|
§46.116 General requirements for informed consent.
Except as provided elsewhere in this policy, no investigator may involve a human being as a subject in research covered by this policy unless the investigator has obtained the legally effective informed consent of the subject or the subject’s legally authorized representative. An investigator shall seek such consent only under circumstances that provide the prospective subject or the representative sufficient opportunity to consider whether or not to participate and that minimize the possibility of coercion or undue influence. The information that is given to the subject or the representative shall be in language understandable to the subject or the representative. No informed consent, whether oral or written, may include any exculpatory language through which the subject or the representative is made
to waive or appear to waive any of the subject’s legal rights, or releases or appears to release the investigator, the sponsor, the institution or its agents from liability for negligence.
| (a) |
Basic elements of informed consent. Except as provided in paragraph (c) or (d) of this section, in seeking informed consent the following information shall be provided to each subject: |
|
| (1) |
a statement that the study involves research, an explanation of the purposes of the research and the expected duration of the subject’s participation, a description of the procedures to be followed, and identification of any procedures which are experimental; |
|
| (2) |
a description of any reasonably foreseeable risks or discomforts to the subject; |
|
| (3) |
a description of any benefits to the subject or to others which may reasonably be expected from the research; |
|
| (4) |
a disclosure of appropriate alternative procedures or courses of treatment, if any, that might be advantageous to the subject; |
|
| (5) |
a statement describing the extent, if any, to which confidentiality of records identifying the subject will be maintained; |
|
| (6) |
for research involving more than minimal risk, an explanation as to whether any compensation and an explanation as to whether any medical treatments are available if injury occurs and, if so, what they consist of, or where further information may be obtained; |
|
| (7) |
an explanation of whom to contact for answers to pertinent questions about the research and research subjects’ rights, and whom to contact in the event of a research-related injury to the subject; and |
|
| (8) |
a statement that participation is voluntary, refusal to participate will involve no penalty or loss of benefits to which the subject is otherwise entitled, and the subject may discontinue participation at any time without penalty or loss of benefits to which the subject is otherwise entitled. |
|
| (b) |
additional elements of informed consent. When appropriate, one or more of the following elements |
|
176
of information shall also be provided to each subject:
| (1) |
a statement that the particular treatment or procedure may involve risks to the subject (or to the embryo or fetus, if the subject is or may become pregnant) which are currently unforeseeable; |
|
| (2) |
anticipated circumstances under which the subject’s participation may be terminated by the investigator without regard to the subject’s consent; |
|
| (3) |
any additional costs to the subject that may result from participation in the research; |
|
| (4) |
the consequences of a subject’s decision to withdraw from the research and procedures for orderly termination of participation by the subject; |
|
| (5) |
A statement that significant new findings developed during the course of the research which may relate to the subject’s willingness to continue participation will be provided to the subject; and |
|
| (6) |
the approximate number of subjects involved in the study. |
|
| (c) |
An IRB may approve a consent procedure which does not include, or which alters, some or all of the elements of informed consent set forth above, or waive the requirement to obtain informed consent provided the IRB finds and documents that: |
|
| (1) |
the research or demonstration project is to be conducted by or subject to the approval of state or local government officials and is designed to study, evaluate, or otherwise examine: (i) public benefit or service programs; (ii) procedures for obtaining benefits or services under those programs; (iii) possible changes in or alternatives to those programs or procedures; or (iv) possible changes in methods or levels of payment for benefits or services under those programs; and |
|
| (2) |
the research could not practicably be carried out without the waiver or alteration. |
|
| (d) |
An IRB may approve a consent procedure which does not include, or which alters, some or all of the elements of informed consent set forth in this section, or waive the requirements to obtain informed consent provided the IRB finds and documents that: |
|
National Bioethics Advisory Commission
| (1) |
the research involves no more than minimal risk to the subjects; |
|
| (2) |
the waiver or alteration will not adversely affect the rights and welfare of the subjects; |
|
| (3) |
the research could not practicably be carried out without the waiver or alteration; and |
|
| (4) |
whenever appropriate, the subjects will be provided with additional pertinent information after participation. |
|
| (e) |
The informed consent requirements in this policy are not intended to preempt any applicable Federal, State, or local laws which require additional information to be disclosed in order for informed consent to be legally effective. |
|
| (f) |
Nothing in this policy is intended to limit the authority of a physician to provide emergency medical care, to the extent the physician is permitted to do so under applicable Federal, State, or local law. (Approved by the Office of Management and Budget under Control Number 9999-0020.) |
|
§ 46.117 Documentation of informed consent.
| (a) |
Except as provided in paragraph (c) of this section, informed consent shall be documented by the use of a written consent form approved by the IRB and signed by the subject or the subject’s legally authorized representative. A copy shall be given to the person signing the form. |
|
| (b) |
Except as provided in paragraph (c) of this section, the consent form may be either of the following: |
|
| (1) |
A written consent document that embodies the elements of informed consent required by §46.116. This form may be read to the subject or the subject’s legally authorized representative, but in any event, the investigator shall give either the subject or the representative adequate opportunity to read it before it is signed; or |
|
| (2) |
A short form written consent document stating that the elements of informed consent required by §46.116 have been presented orally to the subject or the subject’s legally authorized representative. When this method is used, there shall be a witness to the oral presentation. Also, the IRB shall approve a written summary of what is to be said to the subject or the representative. |
|
177
Appendix E: Code of Federal Regulations, Title 45 Part 46
Only the short form itself is to be signed by the subject or the representative. However, the witness shall sign both the short form and a copy of the summary, and the person actually obtaining consent shall sign a copy of the summary. A copy of the summary shall be given to the subject or the representative, in addition to a copy of the short form.
| (c) |
An IRB may waive the requirement for the investigator to obtain a signed consent form for some or all subjects if it finds either: |
|
| (1) |
That the only record linking the subject and the research would be the consent document and the principal risk would be potential harm resulting from a breach of confidentiality. Each subject will be asked whether the subject wants documentation linking the subject with the research, and the subject’s wishes will govern; or |
|
| (2) |
That the research presents no more than minimal risk of harm to subjects and involves no procedures for which written consent is normally required outside of the research context. |
|
In cases in which the documentation requirement is waived, the IRB may require the investigator to provide subjects with a written statement regarding the research. (Approved by the Office of Management and Budget under Control Number 9999-0020.)
§46.118 Applications and proposals lacking definite plans for involvement of human subjects.
Certain types of applications for grants, cooperative agreements, or contracts are submitted to departments or agencies with the knowledge that subjects may be involved within the period of support, but definite plans would not normally be set forth in the application or proposal. These include activities such as institutional type grants when selection of specific projects is the institution’s responsibility; research training grants in which the activities involving subjects remain to be selected; and projects in which human subjects’ involvement will depend upon completion of instruments, prior animal studies, or purification of compounds. These applications need not be reviewed by an IRB before an award may be made. However, except for research exempted or waived under §46.101 (b) or (i), no human subjects may be involved in any project supported by these awards until the project has been
reviewed and approved by the IRB, as provided in this policy, and certification submitted, by the institution, to the Department or Agency.
§46.119 Research undertaken without the intention of involving human subjects.
In the event research is undertaken without the intention of involving human subjects, but it is later proposed to involve human subjects in the research, the research shall first be reviewed and approved by an IRB, as provided in this policy, a certification submitted, by the institution, to the Department or Agency, and final approval given to the proposed change by the Department or Agency.
§46.120 Evaluation and disposition of applications and proposals for research to be conducted or supported by a Federal Department or Agency.
| (a) |
The Department or Agency head will evaluate all applications and proposals involving human subjects submitted to the Department or Agency through such officers and employees of the Department or Agency and such experts and consultants as the Department or Agency head determines to be appropriate. This evaluation will take into consideration the risks to the subjects, the adequacy of protection against these risks, the potential benefits of the research to the subjects and others, and the importance of the knowledge gained or to be gained. |
| (b) |
On the basis of this evaluation, the Department or Agency head may approve or disapprove the application or proposal, or enter into negotiations to develop an approvable one. |
§ 46.121 [Reserved]
§ 46.122 Use of Federal funds.
Federal funds administered by a Department or Agency may not be expended for research involving human subjects unless the requirements of this policy have been satisfied.
§46.123 Early termination of research support: Evaluation of applications and proposals.
| (a) |
The Department or Agency head may require that Department or Agency support for any project be terminated or suspended in the manner prescribed |
178
in applicable program requirements, when the Department or Agency head finds an institution has materially failed to comply with the terms of this policy.
| (b) |
In making decisions about supporting or approving applications or proposals covered by this policy the Department or Agency head may take into account, in addition to all other eligibility requirements and program criteria, factors such as whether the applicant has been subject to a termination or suspension under paragraph (a) of this section and whether the applicant or the person or persons who would direct or has/have directed the scientific and technical aspects of an activity has/have, in the judgment of the Department or Agency head, materially failed to discharge responsibility for the protection of the rights and welfare of human subjects (whether or not the research was subject to Federal regulation). |
§ 46.124 Conditions.
With respect to any research project or any class of research projects the Department or Agency head may impose additional conditions prior to or at the time of approval when in the judgment of the Department or Agency head additional conditions are necessary for the protection of human subjects.
Subpart B: Additional DHHS Protections Pertaining to Research, Development, and Related Activities Involving Fetuses, Pregnant Women, and Human In Vitro Fertilization
Source: 40 FR 33528, Aug. 8, 1975; 43 FR 1758, January 11, 1978; 43 FR 51559, November 3, 1978.
§ 46.201 Applicability.
| (a) |
The regulations in this subpart are applicable to all Department of Health and Human Services grants and contracts supporting research, development, and related activities involving: (1) the fetus, (2) pregnant women, and (3) human in vitro fertilization. |
| (b) |
Nothing in this subpart shall be construed as indicating that compliance with the procedures set forth herein will in any way render inapplicable pertinent |
National Bioethics Advisory Commission
State or local laws bearing upon activities covered by this subpart.
| (c) |
The requirements of this subpart are in addition to those imposed under the other subparts of this part. |
§ 46.202 Purpose.
It is the purpose of this subpart to provide additional safeguards in reviewing activities to which this subpart is applicable to assure that they conform to appropriate ethical standards and relate to important societal needs.
§ 46.203 Definitions.
As used in this subpart:
| (a) |
“Secretary” means the Secretary of Health and Human Services and any other officer or employee of the Department of Health and Human Services (DHHS) to whom authority has been delegated. |
| (b) |
“Pregnancy” encompasses the period of time from confirmation of implantation (through any of the presumptive signs of pregnancy, such as missed menses, or by a medically acceptable pregnancy test), until expulsion or extraction of the fetus. |
| (c) |
“Fetus” means the product of conception from the time of implantation (as evidenced by any of the presumptive signs of pregnancy, such as missed menses, or a medically acceptable pregnancy test), until a determination is made, following expulsion or extraction of the fetus, that it is viable. |
| (d) |
“Viable” as it pertains to the fetus means being able, after either spontaneous or induced delivery, to survive (given the benefit of available medical therapy) to the point of independently maintaining heart beat and respiration. The Secretary may from time to time, taking into account medical advances, publish in the Federal Register guidelines to assist in determining whether a fetus is viable for purposes of this subpart. If a fetus is viable after delivery, it is a premature infant. |
| (e) |
“Nonviable fetus” means a fetus ex utero which, although living, is not viable. |
| (f) |
“Dead fetus” means a fetus ex utero which exhibits neither heart beat, spontaneous respiratory activity, spontaneous movement of voluntary muscles, nor pulsation of the umbilical cord (if still attached). |
| (g) |
“In vitro fertilization” means any fertilization of human ova which occurs outside the body of a |
179
Appendix E: Code of Federal Regulations, Title 45 Part 46
female, either through admixture of donor human sperm and ova or by any other means.
§ 46.204 Ethical Advisory Boards.
| (a) |
One or more Ethical Advisory Boards shall be established by the Secretary. Members of these Board(s) shall be so selected that the Board(s) will be competent to deal with medical, legal, social, ethical, and related issues and may include, for example, research scientists, physicians, psychologists, sociologists, educators, lawyers, and ethicists, as well as representatives of the general public. No Board member may be a regular, full-time employee of the Department of Health and Human Services. |
|
| (b) |
At the request of the Secretary, the Ethical Advisory Board shall render advice consistent with the policies and requirements of this part as to ethical issues, involving activities covered by this subpart, raised by individual applications or proposals. In addition, upon request by the Secretary, the Board shall render advice as to classes of applications or proposals and general policies, guidelines, and procedures. |
|
| (c) |
A Board may establish, with the approval of the Secretary, classes of applications or proposals which: |
|
| (1) |
must be submitted to the Board, or (2) need not be submitted to the Board. Where the Board so establishes a class of applications or proposals which must be submitted, no application or proposal within the class may be funded by the Department or any component thereof until the application or proposal has been reviewed by the Board and the Board has rendered advice as to its acceptability from an ethical standpoint. |
|
| (d) |
[Nullified under Public Law 103-43, June 10, 1993] |
|
§46.205 Additional duties of the Institutional Review Boards in connection with activities involving fetuses, pregnant women, or human in vitro fertilization.
| (a) |
In addition to the responsibilities prescribed for Institutional Review Boards under Subpart A of this part, the applicant’s or offeror’s Board shall, with respect to activities covered by this subpart, carry out the following additional duties: |
| (1) |
determine that all aspects of the activity meet the requirements of this subpart; |
|
| (2) |
determine that adequate consideration has been given to the manner in which potential subjects will be selected, and adequate provision has been made by the applicant or offeror for monitoring the actual informed consent process (e.g., through such mechanisms, when appropriate, as participation by the Institutional Review Board or subject advocates in: (i) overseeing the actual process by which individual consents required by this subpart are secured either by approving induction of each individual into the activity or verifying, perhaps through sampling, that approved procedures for induction of individuals into the activity are being followed, and (ii) monitoring the progress of the activity and intervening as necessary through such steps as visits to the activity site and continuing evaluation to determine if any unanticipated risks have arisen); |
|
| (3) |
carry out such other responsibilities as may be assigned by the Secretary. |
|
| (b) |
No award may be issued until the applicant or offeror has certified to the Secretary that the Institutional Review Board has made the determinations required under paragraph (a) of this section and the Secretary has approved these determinations, as provided in §46.120 of Subpart A of this part. |
|
| (c) |
Applicants or offerors seeking support for activities covered by this subpart must provide for the designation of an Institutional Review Board, subject to approval by the Secretary, where no such Board has been established under Subpart A of this part. |
|
§ 46.206 General limitations.
| (a) |
No activity to which this subpart is applicable may be undertaken unless: |
|
| (1) |
appropriate studies on animals and nonpregnant individuals have been completed; |
|
| (2) |
except where the purpose of the activity is to meet the health needs of the mother or the particular fetus, the risk to the fetus is minimal and, in all cases, is the least possible risk for achieving the objectives of the activity; |
|
180
| (3) |
individuals engaged in the activity will have no part in: (i) any decisions as to the timing, method, and procedures used to terminate the pregnancy, and (ii) determining the viability of the fetus at the termination of the pregnancy; and |
|
| (4) |
no procedural changes which may cause greater than minimal risk to the fetus or the pregnant woman will be introduced into the procedure for terminating the pregnancy solely in the interest of the activity. |
|
| (b) |
No inducements, monetary or otherwise, may be offered to terminate pregnancy for purposes of the activity. Source: 40 FR 33528, Aug. 8, 1975, as amended at 40 FR 51638, Nov. 6, 1975. |
|
§46.207 Activities directed toward pregnant women as subjects.
| (a) |
No pregnant woman may be involved as a subject in an activity covered by this subpart unless: (1) the purpose of the activity is to meet the health needs of the mother and the fetus will be placed at risk only to the minimum extent necessary to meet such needs, or (2) the risk to the fetus is minimal. |
|
| (b) |
An activity permitted under paragraph (a) of this section may be conducted only if the mother and father are legally competent and have given their informed consent after having been fully informed regarding possible impact on the fetus, except that the father’s informed consent need not be secured if: |
|
| (1) |
the purpose of the activity is to meet the health needs of the mother; (2) his identity or whereabouts cannot reasonably be ascertained; (3) he is not reasonably available; or (4) the pregnancy resulted from rape. |
|
§46.208 Activities directed toward fetuses in utero as subjects.
| (a) |
No fetus in utero may be involved as a subject in any activity covered by this subpart unless: (1) the purpose of the activity is to meet the health needs of the particular fetus and the fetus will be placed at risk only to the minimum extent necessary to meet such needs, or (2) the risk to the fetus imposed by the research is minimal and the purpose of the |
National Bioethics Advisory Commission
activity is the development of important biomedical knowledge which cannot be obtained by other means.
| (b) |
An activity permitted under paragraph (a) of this section may be conducted only if the mother and father are legally competent and have given their informed consent, except that the father’s consent need not be secured if: (1) his identity or whereabouts cannot reasonably be ascertained, (2) he is not reasonably available, or (3) the pregnancy resulted from rape. |
§46.209 Activities directed toward fetuses ex utero, including nonviable fetuses, as subjects.
| (a) |
Until it has been ascertained whether or not a fetus ex utero is viable, a fetus ex utero may not be involved as a subject in an activity covered by this subpart unless: |
|
| (1) |
there will be no added risk to the fetus resulting from the activity, and the purpose of the activity is the development of important biomedical knowledge which cannot be obtained by other means, or |
|
| (2) |
the purpose of the activity is to enhance the possibility of survival of the particular fetus to the point of viability. |
|
| (b) |
No nonviable fetus may be involved as a subject in an activity covered by this subpart unless: |
|
| (1) |
vital functions of the fetus will not be artificially maintained, |
|
| (2) |
experimental activities which of themselves would terminate the heartbeat or respiration of the fetus will not be employed, and |
|
| (3) |
the purpose of the activity is the development of important biomedical knowledge which cannot be obtained by other means. |
|
| (c) |
In the event the fetus ex utero is found to be viable, it may be included as a subject in the activity only to the extent permitted by and in accordance with the requirements of other subparts of this part. |
|
| (d) |
An activity permitted under paragraph (a) or (b) of this section may be conducted only if the mother and father are legally competent and have given their informed consent, except that the father’s informed consent need not be secured if: (1) his |
|
181
Appendix E: Code of Federal Regulations, Title 45 Part 46
identity or whereabouts cannot reasonably be ascertained, (2) he is not reasonably available, or (3) the pregnancy resulted from rape.
§46.210 Activities involving the dead fetus, fetal material, or the placenta.
Activities involving the dead fetus, mascerated fetal material, or cells, tissue, or organs excised from a dead fetus shall be conducted only in accordance with any applicable State or local laws regarding such activities.
§46.211 Modification or waiver of specific requirements.
Upon the request of an applicant or offeror (with the approval of its Institutional Review Board), the Secretary may modify or waive specific requirements of this subpart, with the approval of the Ethical Advisory Board after such opportunity for public comment as the Ethical Advisory Board considers appropriate in the particular instance. In making such decisions, the Secretary will consider whether the risks to the subject are so outweighed by the sum of the benefit to the subject and the importance of the knowledge to be gained as to warrant such modification or waiver and that such benefits cannot be gained except through a modification or waiver. Any such modifications or waivers will be published as notices in the Federal Register.
Subpart C: Additional DHHS Protections Pertaining to Biomedical and Behavioral Research Involving Prisoners as Subjects
Source: 43 FR 53655, Nov. 16, 1978.
§ 46.301 Applicability.
| (a) |
The regulations in this subpart are applicable to all biomedical and behavioral research conducted or supported by the Department of Health and Human Services involving prisoners as subjects. |
| (b) |
Nothing in this subpart shall be construed as indicating that compliance with the procedures set forth herein will authorize research involving prisoners as subjects, to the extent such research is limited or barred by applicable State or local law. |
| (c) |
The requirements of this subpart are in addition to those imposed under the other subparts of this part. |
§ 46.302 Purpose.
Inasmuch as prisoners may be under constraints because of their incarceration which could affect their ability to make a truly voluntary and uncoerced decision whether or not to participate as subjects in research, it is the purpose of this subpart to provide additional safeguards for the protection of prisoners involved in activities to which this subpart is applicable.
§ 46.303 Definitions.
As used in this subpart:
| (a) |
“Secretary” means the Secretary of Health and Human Services and any other officer or employee of the Department of Health and Human Services to whom authority has been delegated. |
| (b) |
“DHHS” means the Department of Health and Human Services. |
| (c) |
“Prisoner” means any individual involuntarily confined or detained in a penal institution. The term is intended to encompass individuals sentenced to such an institution under a criminal or civil statute, individuals detained in other facilities by virtue of statutes or commitment procedures which provide alternatives to criminal prosecution or incarceration in a penal institution, and individuals detained pending arraignment, trial, or sentencing. |
| (d) |
“Minimal risk” is the probability and magnitude of physical or psychological harm that is normally encountered in the daily lives, or in the routine medical, dental, or psychological examination of healthy persons. |
§46.304 Composition of Institutional Review Boards where prisoners are involved.
In addition to satisfying the requirements in §46.107 of this part, an Institutional Review Board, carrying out responsibilities under this part with respect to research covered by this subpart, shall also meet the following specific requirements:
| (a) |
A majority of the Board (exclusive of prisoner members) shall have no association with the prison(s) involved, apart from their membership on the Board. |
| (b) |
At least one member of the Board shall be a prisoner, or a prisoner representative with appropriate background and experience to serve in that capacity, |
182
except that where a particular research project is reviewed by more than one Board only one Board need satisfy this requirement.
§46.305 Additional duties of the Institutional Review Boards where prisoners are involved.
| (a) |
In addition to all other responsibilities prescribed for Institutional Review Boards under this part, the Board shall review research covered by this subpart and approve such research only if it finds that: |
|
| (1) |
the research under review represents one of the categories of research permissible under § 46.306(a)(2); |
|
| (2) |
any possible advantages accruing to the prisoner through his or her participation in the research, when compared to the general living conditions, medical care, quality of food, amenities and opportunity for earnings in the prison, are not of such a magnitude that his or her ability to weigh the risks of the research against the value of such advantages in the limited choice environment of the prison is impaired; |
|
| (3) |
the risks involved in the research are commensurate with risks that would be accepted by non-prisoner volunteers; |
|
| (4) |
procedures for the selection of subjects within the prison are fair to all prisoners and immune from arbitrary intervention by prison authorities or prisoners. Unless the principal investigator provides to the Board justification in writing for following some other procedures, control subjects must be selected randomly from the group of available prisoners who meet the characteristics needed for that particular research project; |
|
| (5) |
the information is presented in language which is understandable to the subject population; |
|
| (6) |
adequate assurance exists that parole boards will not take into account a prisoner’s participation in the research in making decisions regarding parole, and each prisoner is clearly informed in advance that participation in the research will have no effect on his or her parole; and |
|
| (7) |
where the Board finds there may be a need for follow-up examination or care of participants after the end of their participation, adequate provision has been made for such examination |
|
National Bioethics Advisory Commission
or care, taking into account the varying lengths of individual prisoners’ sentences, and for informing participants of this fact.
| (b) |
The Board shall carry out such other duties as may be assigned by the Secretary. |
| (c) |
The institution shall certify to the Secretary, in such form and manner as the Secretary may require, that the duties of the Board under this section have been fulfilled. |
§ 46.306 Permitted research involving prisoners.
| (a) |
Biomedical or behavioral research conducted or supported by DHHS may involve prisoners as subjects only if: |
||
| (1) |
the institution responsible for the conduct of the research has certified to the Secretary that the Institutional Review Board has approved the research under §46.305 of this subpart; and |
||
| (2) |
in the judgment of the Secretary the proposed research involves solely the following: |
||
| (A) |
study of the possible causes, effects, and processes of incarceration, and of criminal behavior, provided that the study presents no more than minimal risk and no more than inconvenience to the subjects; |
||
| (B) |
study of prisons as institutional structures or of prisoners as incarcerated persons, provided that the study presents no more than minimal risk and no more than inconvenience to the subjects; |
||
| (C) |
research on conditions particularly affecting prisoners as a class (for example, vaccine trials and other research on hepatitis which is much more prevalent in prisons than elsewhere; and research on social and psychological problems such as alcoholism, drug addiction, and sexual assaults) provided that the study may proceed only after the Secretary has consulted with appropriate experts including experts in penology, medicine, and ethics, and published notice, in the Federal Register, of his intent to approve such research; or |
||
| (D) |
research on practices, both innovative and accepted, which have the intent and |
||
183
Appendix E: Code of Federal Regulations, Title 45 Part 46
reasonable probability of improving the health or well-being of the subject. In cases in which those studies require the assignment of prisoners in a manner consistent with protocols approved by the IRB to control groups which may not benefit from the research, the study may proceed only after the Secretary has consulted with appropriate experts, including experts in penology, medicine, and ethics, and published notice, in the Federal Register, of the intent to approve such research.
| (b) |
Except as provided in paragraph (a) of this section, biomedical or behavioral research conducted or supported by DHHS shall not involve prisoners as subjects. |
Subpart D: Additional DHHS Protections for Children Involved as Subjects in Research
Source: 48 FR 9818, March 8, 1983; 56 FR 28032, June 18, 1991.
§ 46.401 To what do these regulations apply?
| (a) |
This subpart applies to all research involving children as subjects, conducted or supported by the Department of Health and Human Services. |
|
| (1) |
This includes research conducted by Department employees, except that each head of an Operating Division of the Department may adopt such nonsubstantive, procedural modifications as may be appropriate from an administrative standpoint. |
|
| (2) |
It also includes research conducted or supported by the Department of Health and Human Services outside the United States, but in appropriate circumstances, the Secretary may, under paragraph (i) of §46.101 of Subpart A, waive the applicability of some or all of the requirements of these regulations for research of this type. |
|
| (b) |
Exemptions at §46.101(b)(1) and (b)(3) through (b)(6) are applicable to this subpart. The exemption at §46.101(b)(2) regarding educational tests is also applicable to this subpart. However, the exemption at §46.101(b)(2) for research involving survey or |
|
interview procedures or observations of public behavior does not apply to research covered by this subpart, except for research involving observation of public behavior when the investigator(s) do not participate in the activities being observed.
| (c) |
The exceptions, additions, and provisions for waiver as they appear in paragraphs (c) through (i) of § 46.101 of Subpart A are applicable to this subpart. |
§ 46.402 Definitions.
The definitions in §46.102 of Subpart A shall be applicable to this subpart as well. In addition, as used in this subpart:
| (a) |
“Children” are persons who have not attained the legal age for consent to treatments or procedures involved in the research, under the applicable law of the jurisdiction in which the research will be conducted. |
| (b) |
“Assent” means a child’s affirmative agreement to participate in research. Mere failure to object should not, absent affirmative agreement, be construed as assent. |
| (c) |
“Permission” means the agreement of parent(s) or guardian to the participation of their child or ward in research. |
| (d) |
“Parent” means a child’s biological or adoptive parent. |
| (e) |
“Guardian” means an individual who is authorized under applicable State or local law to consent on behalf of a child to general medical care. |
§ 46.403 IRB duties.
In addition to other responsibilities assigned to IRBs under this part, each IRB shall review research covered by this subpart and approve only research which satisfies the conditions of all applicable sections of this subpart.
§46.404 Research not involving greater than minimal risk.
DHHS will conduct or fund research in which the IRB finds that no greater than minimal risk to children is presented, only if the IRB finds that adequate provisions are made for soliciting the assent of the children and the permission of their parents or guardians, as set forth in
§ 46.408.
184
§46.405 Research involving greater than minimal risk but presenting the prospect of direct benefit to the individual subjects.
DHHS will conduct or fund research in which the IRB finds that more than minimal risk to children is presented by an intervention or procedure that holds out the prospect of direct benefit for the individual subject, or by a monitoring procedure that is likely to contribute to the subject’s well-being, only if the IRB finds that:
| (a) |
the risk is justified by the anticipated benefit to the subjects; |
| (b) |
the relation of the anticipated benefit to the risk is at least as favorable to the subjects as that presented by available alternative approaches; and |
| (c) |
adequate provisions are made for soliciting the assent of the children and permission of their parents or guardians, as set forth in §46.408. |
§46.406 Research involving greater than minimal risk and no prospect of direct benefit to individual subjects, but likely to yield generaliz-able knowledge about the subject’s disorder or condition.
DHHS will conduct or fund research in which the IRB finds that more than minimal risk to children is presented by an intervention or procedure that does not hold out the prospect of direct benefit for the individual subject, or by a monitoring procedure which is not likely to contribute to the well-being of the subject, only if the IRB finds that:
| (a) |
the risk represents a minor increase over minimal risk; |
| (b) |
the intervention or procedure presents experiences to subjects that are reasonably commensurate with those inherent in their actual or expected medical, dental, psychological, social, or educational situations; |
| (c) |
the intervention or procedure is likely to yield generalizable knowledge about the subjects’ disorder or condition which is of vital importance for the understanding or amelioration of the subjects’ disorder or condition; and |
| (d) |
adequate provisions are made for soliciting assent of the children and permission of their parents or guardians, as set forth in §46.408. |
National Bioethics Advisory Commission
§46.407 Research not otherwise approvable which presents an opportunity to understand, prevent, or alleviate a serious problem affecting the health or welfare of children.
DHHS will conduct or fund research that the IRB does not believe meets the requirements of §46.404,
§ 46.405, or §46.406 only if:
| (a) |
the IRB finds that the research presents a reasonable opportunity to further the understanding, prevention, or alleviation of a serious problem affecting the health or welfare of children; and |
|
| (b) |
the Secretary, after consultation with a panel of experts in pertinent disciplines (for example: science, medicine, education, ethics, law) and following opportunity for public review and comment, has determined either: |
|
| (1) |
that the research in fact satisfies the conditions of §46.404, §46.405, or §46.406, as applicable, or (2) the following: |
|
| (i) |
the research presents a reasonable opportunity to further the understanding, prevention, or alleviation of a serious problem affecting the health or welfare of children; (ii) the research will be conducted in accordance with sound ethical principles; (iii)adequate provisions are made for soliciting the assent of children and the permission of their parents or guardians, as set forth in §46.408. |
|
§46.408 Requirements for permission by parents or guardians and for assent by children.
| (a) |
In addition to the determinations required under other applicable sections of this subpart, the IRB shall determine that adequate provisions are made for soliciting the assent of the children, when in the judgment of the IRB the children are capable of providing assent. In determining whether children are capable of assenting, the IRB shall take into account the ages, maturity, and psychological state of the children involved. This judgment may be made for all children to be involved in research under a particular protocol, or for each child, as the IRB deems appropriate. If the IRB determines that the capability of some or all of the children is so limited that they cannot reasonably be consulted or that the intervention or procedure involved in the research |
185
Appendix E: Code of Federal Regulations, Title 45 Part 46
holds out a prospect of direct benefit that is important to the health or well-being of the children and is available only in the context of the research, the assent of the children is not a necessary condition for proceeding with the research. Even where the IRB determines that the subjects are capable of assenting, the IRB may still waive the assent requirement under circumstances in which consent may be waived in accord with §46.116 of Subpart A.
| (b) |
In addition to the determinations required under other applicable sections of this subpart, the IRB shall determine, in accordance with and to the extent that consent is required by §46.116 of Subpart A, that adequate provisions are made for soliciting the permission of each child’s parents or guardian. Where parental permission is to be obtained, the IRB may find that the permission of one parent is sufficient for research to be conducted under §46.404 or §46.405. Where research is covered by §46.406 and §46.407 and permission is to be obtained from parents, both parents must give their permission unless one parent is deceased, unknown, incompetent, or not reasonably available, or when only one parent has legal responsibility for the care and custody of the child. |
| (c) |
In addition to the provisions for waiver contained in §46.116 of Subpart A, if the IRB determines that a research protocol is designed for conditions or for a subject population for which parental or guardian permission is not a reasonable requirement to protect the subjects (for example, neglected or abused children), it may waive the consent requirements in Subpart A of this part and paragraph (b) of this section, provided an appropriate mechanism for protecting the children who will participate as subjects in the research is substituted, and provided further that the waiver is not inconsistent with Federal, |
186
State, or local law. The choice of anappropriate mechanism would depend upon the nature and purpose of the activities described in the protocol, the risk and anticipated benefit to the research subjects, and their age, maturity, status, and condition.
| (d) |
Permission by parents or guardians shall be documented in accordance with and to the extent required by §46.117 of Subpart A. |
| (e) |
When the IRB determines that assent is required, it shall also determine whether and how assent must be documented. |
§ 46.409 Wards.
| (a) |
Children who are wards of the State or any other agency, institution, or entity can be included in research approved under §46.406 or §46.407 only if such research is: |
|
| (1) |
related to their status as wards; or |
|
| (2) |
conducted in schools, camps, hospitals, institutions, or similar settings in which the majority of children involved as subjects are not wards. |
|
| (b) |
If the research is approved under paragraph (a) of this section, the IRB shall require appointment of an advocate for each child who is a ward, in addition to any other individual acting on behalf of the child as guardian or in loco parentis. One individual may serve as advocate for more than one child. The advocate shall be an individual who has the background and experience to act in, and agrees to act in, the best interests of the child for the duration of the child’s participation in the research and who is not associated in any way (except in the role as advocate or member of the IRB) with the research, the investigator(s), or the guardian organization. |
|
Appendix F
Code of Federal Regulations, Title 21 Parts 50 and 56
Part 50: Protection of Human Subjects Subpart A: General Provisions
Source: 45 FR 36390, May 30, 1980, unless otherwise noted.
§ 50.1 Scope.
| (a) |
This part applies to all clinical investigations regulated by the Food and Drug Administration under sections 505(i) and 520(g) of the Federal Food, Drug, and Cosmetic Act, as well as clinical investigations that support applications for research or marketing permits for products regulated by the Food and Drug Administration, including food and color additives, drugs for human use, medical devices for human use, biological products for human use, and electronic products. Additional specific obligations and commitments of, and standards of conduct for, persons who sponsor or monitor clinical investigations involving particular test articles may also be found in other parts (e.g., parts 312 and 812). Compliance with these parts is intended to protect the rights and safety of subjects involved in investigations filed with the Food and Drug Administration pursuant to sections 406, 409, 502, 503, 505, 510, 513–516, 518–520, 721, and 801 of the Federal Food, Drug, and Cosmetic Act and sections 351 and 354–360F of the Public Health Service Act. |
| (b) |
References in this part to regulatory sections of the Code of Federal Regulations are to chapter I of title 21, unless otherwise noted. |
§ 50.3 Definitions.
As used in this part:
| (a) |
Act means the Federal Food, Drug, and Cosmetic Act, as amended (secs. 201–902, 52 Stat. 1040 et seq. as amended (21 U.S.C. 321–392)). |
| (b) |
Application for research or marketing permit includes: |
|
| (1) |
A color additive petition, described in part 71. |
|
| (2) |
A food additive petition, described in parts 171 and 571. |
|
| (3) |
Data and information about a substance submitted as part of the procedures for establishing that the substance is generally recognized as safe for use that results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food, described in §170.30 and § 570.30. |
|
| (4) |
Data and information about a food additive submitted as part of the procedures for food additives permitted to be used on an interim basis pending additional study, described in §180.1. |
|
| (5) |
Data and information about a substance submitted as part of the procedures for establishing a tolerance for unavoidable contaminants in food and food-packaging materials, described in section 406 of the act. |
|
| (6) |
An investigational new drug application, described in part 312 of this chapter. |
|
| (7) |
A new drug application, described in part 314. |
|
| (8) |
Data and information about the bioavailability or bioequivalence of drugs for human use submitted as part of the procedures for issuing, amending, or repealing a bioequivalence requirement, described in part 320. |
|
| (9) |
Data and information about an over-the-counter drug for human use submitted as part of the procedures for classifying these drugs as generally recognized as safe and effective and not mis-branded, described in part 330. |
|
187
Appendix F: Code of Federal Regulations, Title 21 Parts 50 and 56
| (10) |
Data and information about a prescription drug for human use submitted as part of the procedures for classifying these drugs as generally recognized as safe and effective and not misbranded, described in this chapter. |
| (11) |
Data and information about an antibiotic drug submitted as part of the procedures for issuing, amending, or repealing regulations for these drugs, described in §314.300 of this chapter. |
| (12) |
An application for a biologics license, described in part 601 of this chapter. |
| (13) |
Data and information about a biological product submitted as part of the procedures for determining that licensed biological products are safe and effective and not misbranded, described in part 601. |
| (14) |
Data and information about an in vitro diagnostic product submitted as part of the procedures for establishing, amending, or repealing a standard for these products, described in part 809. |
| (15) |
An Application for an Investigational Device Exemption, described in part 812. |
| (16) |
Data and information about a medical device submitted as part of the procedures for classifying these devices, described in section 513. |
| (17) |
Data and information about a medical device submitted as part of the procedures for establishing, amending, or repealing a standard for these devices, described in section 514. |
| (18) |
An application for premarket approval of a medical device, described in section 515. |
| (19) |
A product development protocol for a medical device, described in section 515. |
| (20) |
Data and information about an electronic product submitted as part of the procedures for establishing, amending, or repealing a standard for these products, described in section 358 of the Public Health Service Act. |
| (21) |
Data and information about an electronic product submitted as part of the procedures for obtaining a variance from any electronic product performance standard, as described in § 1010.4. |
| (22) |
Data and information about an electronic product submitted as part of the procedures for granting, amending, or extending an exemption from a radiation safety performance standard, as described in §1010.5. |
|
| (c) |
Clinical investigation means any experiment that involves a test article and one or more human subjects and that either is subject to requirements for prior submission to the Food and Drug Administration under section 505(i) or 520(g) of the act, or is not subject to requirements for prior submission to the Food and Drug Administration under these sections of the act, but the results of which are intended to be submitted later to, or held for inspection by, the Food and Drug Administration as part of an application for a research or marketing permit. The term does not include experiments that are subject to the provisions of part 58 of this chapter, regarding nonclinical laboratory studies. |
|
| (d) |
Investigator means an individual who actually conducts a clinical investigation, i.e., under whose immediate direction the test article is administered or dispensed to, or used involving, a subject, or, in the event of an investigation conducted by a team of individuals, is the responsible leader of that team. |
|
| (e) |
Sponsor means a person who initiates a clinical investigation, but who does not actually conduct the investigation, i.e., the test article is administered or dispensed to or used involving, a subject under the immediate direction of another individual. A person other than an individual (e.g., corporation or agency) that uses one or more of its own employees to conduct a clinical investigation it has initiated is considered to be a sponsor (not a sponsor-investigator), and the employees are considered to be investigators. |
|
| (f) |
Sponsor-investigator means an individual who both initiates and actually conducts, alone or with others, a clinical investigation, i.e., under whose immediate direction the test article is administered or dispensed to, or used involving, a subject. The term does not include any person other than an individual, e.g., corporation or agency. |
|
| (g) |
Human subject means an individual who is or becomes a participant in research, either as a recipient of the test article or as a control. A subject may be either a healthy human or a patient. |
|
188
| (h) |
Institution means any public or private entity or agency (including Federal, State, and other agencies). The word facility as used in section 520(g) of the act is deemed to be synonymous with the term institution for purposes of this part. |
| (i) |
Institutional review board (IRB) means any board, committee, or other group formally designated by an institution to review biomedical research involving humans as subjects, to approve the initiation of and conduct periodic review of such research. The term has the same meaning as the phrase institutional review committee as used in section 520(g) of the act. |
| (j) |
Test article means any drug (including a biological product for human use), medical device for human use, human food additive, color additive, electronic product, or any other article subject to regulation under the act or under sections 351 and 354-360F of the Public Health Service Act (42 U.S.C. 262 and 263b–263n). |
| (k) |
Minimal risk means that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests. |
| (l) |
Legally authorized representative means an individual or judicial or other body authorized under applicable law to consent on behalf of a prospective subject to the subject’s particpation in the procedure(s) involved in the research. |
(m)Family member means any one of the following legally competent persons: Spouse; parents; children (including adopted children); brothers, sisters, and spouses of brothers and sisters; and any individual related by blood or affinity whose close association with the subject is the equivalent of a family relationship.
National Bioethics Advisory Commission
Subpart B: Informed Consent of Human Subjects
Source: 46 FR 8951, Jan. 27, 1981, unless otherwise noted.
§50.20 General requirements for informed consent.
Except as provided in §50.23 and §50.24, no investigator may involve a human being as a subject in research covered by these regulations unless the investigator has obtained the legally effective informed consent of the subject or the subject’s legally authorized representative. An investigator shall seek such consent only under circumstances that provide the prospective subject or the representative sufficient opportunity to consider whether or not to participate and that minimize the possibility of coercion or undue influence. The information that is given to the subject or the representative shall be in language understandable to the subject or the representative. No informed consent, whether oral or written, may include any exculpatory language through which the subject or the representative is made to waive or appear to waive any of the subject’s legal rights, or releases or appears to release the investigator, the sponsor, the institution, or its agents from liability for negligence.
§ 50.23 Exception from general requirements.
| (a) |
The obtaining of informed consent shall be deemed feasible unless, before use of the test article (except as provided in paragraph (b) of this section), both the investigator and a physician who is not otherwise participating in the clinical investigation certify in writing all of the following: |
|
| (1) |
The human subject is confronted by a life-threatening situation necessitating the use of the test article. |
|
| (2) |
Informed consent cannot be obtained from the subject because of an inability to communicate with, or obtain legally effective consent from, the subject. |
|
| (3) |
Time is not sufficient to obtain consent from the subject’s legal representative. |
|
189
Appendix F: Code of Federal Regulations, Title 21 Parts 50 and 56
| (4) |
There is available no alternative method of approved or generally recognized therapy that provides an equal or greater likelihood of saving the life of the subject. |
|
| (b) |
If immediate use of the test article is, in the investigator’s opinion, required to preserve the life of the subject, and time is not sufficient to obtain the independent determination required in paragraph (a) of this section in advance of using the test article, the determinations of the clinical investigator shall be made and, within 5 working days after the use of the article, be reviewed and evaluated in writing by a physician who is not participating in the clinical investigation. |
|
| (c) |
The documentation required in paragraph (a) or (b) of this section shall be submitted to the IRB within 5 working days after the use of the test article. |
|
(d)(1) Under 10 U.S.C. 1107(f) the President may waive the prior consent requirement for the administration of an investigational new drug to a member of the armed forces in connection with the member’s participation in a particular military operation. The statute specifies that only the President may waive informed consent in this connection and the President may grant such a waiver only if the President determines in writing that obtaining consent: Is not feasible; is contrary to the best interests of the military member; or is not in the interests of national security. The statute further provides that in making a determination to waive prior informed consent on the ground that it is not feasible or the ground that it is contrary to the best interests of the military members involved, the President shall apply the standards and criteria that are set forth in the relevant FDA regulations for a waiver of the prior informed consent requirements of section 505(i)(4) of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. 355(i)(4)). Before such a determination may be made that obtaining informed consent from military personnel prior to the use of an investigational drug (including an antibiotic or biological product) in a specific protocol under an investigational new drug application (IND) sponsored by the Department of Defense (DOD) and limited to specific military personnel involved in a particular military operation is not feasible or is contrary to the best interests of the military members involved the Secretary of
Defense must first request such a determination from the President, and certify and document to the President that the following standards and criteria contained in paragraphs (d)(1) through (d)(4) of this section have been met.
| (i) |
The extent and strength of evidence of the safety and effectiveness of the investigational new drug in relation to the medical risk that could be encountered during the military operation supports the drug’s administration under an IND. |
(ii) The military operation presents a substantial risk that military personnel may be subject to a chemical, biological, nuclear, or other exposure likely to produce death or serious or life-threatening injury or illness.
(iii)There is no available satisfactory alternative therapeutic or preventive treatment in relation to the intended use of the investigational new drug.
(iv) Conditioning use of the investigational new drug on the voluntary participation of each member could significantly risk the safety and health of any individual member who would decline its use, the safety of other military personnel, and the accomplishment of the military mission.
| (v) |
A duly constituted institutional review board (IRB) established and operated in accordance with the requirements of paragraphs (d)(2) and (d)(3) of this section, responsible for review of the study, has reviewed and approved the investigational new drug protocol and the administration of the investigational new drug without informed consent. DOD’s request is to include the documentation required by §56.115(a)(2) of this chapter. |
(vi) DOD has explained:
| (A) |
The context in which the investigational drug will be administered, e.g., the setting or whether it will be self-administered or it will be administered by a health professional; |
| (B) |
The nature of the disease or condition for which the preventive or therapeutic treatment is intended; and |
190
(C)To the extent there are existing data or information available, information on conditions that could alter the effects of the investigational drug.
(vii) DOD’s recordkeeping system is capable of tracking and will be used to track the proposed treatment from supplier to the individual recipient.
(viii) Each member involved in the military operation will be given, prior to the administration of the investigational new drug, a specific written information sheet (including information required by 10 U.S.C. 1107(d)) concerning the investigational new drug, the risks and benefits of its use, potential side effects, and other pertinent information about the appropriate use of the product.
(ix) Medical records of members involved in the military operation will accurately document the receipt by members of the notification required by paragraph (d)(1)(viii) of this section.
| (x) |
Medical records of members involved in the military operation will accurately document the receipt by members of any investigational new drugs in accordance with FDA regulations including part 312 of this chapter. |
(xi) DOD will provide adequate followup to assess whether there are beneficial or adverse health consequences that result from the use of the investigational product. (xii) DOD is pursuing drug development, including a time line, and marketing approval with due diligence.
(xiii)FDA has concluded that the investigational new drug protocol may proceed subject to a decision by the President on the informed consent waiver request.
(xiv) DOD will provide training to the appropriate medical personnel and potential recipients on the specific investigational new drug to be administered prior to its use. (xv) DOD has stated and justified the time period for which the waiver is needed, not to exceed one year, unless separately renewed under these standards and criteria.
National Bioethics Advisory Commission
(xvi) DOD shall have a continuing obligation to report to the FDA and to the President any changed circumstances relating to these standards and criteria (including the time period referred to in paragraph (d)(1)(xv) of this section) or that otherwise might affect the determination to use an investigational new drug without informed consent. (xvii)DOD is to provide public notice as soon as practicable and consistent with classification requirements through notice in the Federal Register describing each waiver of informed consent determination, a summary of the most updated scientific information on the products used, and other pertinent information.
(xviii)Use of the investigational drug without informed consent otherwise conforms with applicable law.
| (2) |
The duly constituted institutional review board, described in paragraph (d)(1)(v) of this section, must include at least 3 nonaffiliated members who shall not be employees or officers of the Federal Government (other than for purposes of membership on the IRB) and shall be required to obtain any necessary security clearances. This IRB shall review the proposed IND protocol at a convened meeting at which a majority of the members are present including at least one member whose primary concerns are in nonsci-entific areas and, if feasible, including a majority of the nonaffiliated members. The information required by §56.115(a)(2) of this chapter is to be provided to the Secretary of Defense for further review. |
|
| (3) |
The duly constituted institutional review board, described in paragraph (d)(1)(v) of this section, must review and approve: |
|
| (i) |
The required information sheet; (ii) The adequacy of the plan to disseminate information, including distribution of the information sheet to potential recipients, on the investigational product (e.g., in forms other than written); (iii)The adequacy of the information and plans for its dissemination to health care providers, including potential side effects, |
|
191
Appendix F: Code of Federal Regulations, Title 21 Parts 50 and 56
contraindications, potential interactions, and other pertinent considerations; and (iv) An informed consent form as required by part 50 of this chapter, in those circumstances in which DOD determines that informed consent may be obtained from some or all personnel involved.
| (4) |
DOD is to submit to FDA summaries of institutional review board meetings at which the proposed protocol has been reviewed. |
| (5) |
Nothing in these criteria or standards is intended to preempt or limit FDA’s and DOD’s authority or obligations under applicable statutes and regulations. |
§50.24 Exception from informed consent requirements for emergency research.
| (a) |
The IRB responsible for the review, approval, and continuing review of the clinical investigation described in this section may approve that investigation without requiring that informed consent of all research subjects be obtained if the IRB (with the concurrence of a licensed physician who is a member of or consultant to the IRB and who is not otherwise participating in the clinical investigation) finds and documents each of the following: |
||
| (1) |
The human subjects are in a life-threatening situation, available treatments are unproven or unsatisfactory, and the collection of valid scientific evidence, which may include evidence obtained through randomized placebo-controlled investigations, is necessary to determine the safety and effectiveness of particular interventions. |
||
| (2) |
Obtaining informed consent is not feasible because: |
||
| (i) |
The subjects will not be able to give their informed consent as a result of their medical condition; (ii) The intervention under investigation must be administered before consent from the subjects’ legally authorized representatives is feasible; and (iii)There is no reasonable way to identify prospectively the individuals likely to become eligible for participation in the clinical investigation. |
||
| (3) |
Participation in the research holds out the prospect of direct benefit to the subjects because: |
|
| (i) |
Subjects are facing a life-threatening situation that necessitates intervention; (ii) Appropriate animal and other preclinical studies have been conducted, and the information derived from those studies and related evidence support the potential for the intervention to provide a direct benefit to the individual subjects; and (iii)Risks associated with the investigation are reasonable in relation to what is known about the medical condition of the potential class of subjects, the risks and benefits of standard therapy, if any, and what is known about the risks and benefits of the proposed intervention or activity. |
|
| (4) |
The clinical investigation could not practicably be carried out without the waiver. |
|
| (5) |
The proposed investigational plan defines the length of the potential therapeutic window based on scientific evidence, and the investigator has committed to attempting to contact a legally authorized representative for each subject within that window of time and, if feasible, to asking the legally authorized representative contacted for consent within that window rather than proceeding without consent. The investigator will summarize efforts made to contact legally authorized representatives and make this information available to the IRB at the time of continuing review. |
|
| (6) |
The IRB has reviewed and approved informed consent procedures and an informed consent document consistent with §50.25. These procedures and the informed consent document are to be used with subjects or their legally authorized representatives in situations where use of such procedures and documents is feasible. The IRB has reviewed and approved procedures and information to be used when providing an opportunity for a family member to object to a subject’s participation in the clinical investigation consistent with paragraph (a)(7)(v) of this section. |
|
192
| (7) |
Additional protections of the rights and welfare of the subjects will be provided, including, at least: |
||
| (i) |
Consultation (including, where appropriate, consultation carried out by the IRB) with representatives of the communities in which the clinical investigation will be conducted and from which the subjects will be drawn; (ii) Public disclosure to the communities in which the clinical investigation will be conducted and from which the subjects will be drawn, prior to initiation of the clinical investigation, of plans for the investigation and its risks and expected benefits; (iii)Public disclosure of sufficient information following completion of the clinical investigation to apprise the community and researchers of the study, including the demographic characteristics of the research population, and its results; (iv) Establishment of an independent data monitoring committee to exercise oversight of the clinical investigation; and |
||
| (v) |
If obtaining informed consent is not feasible and a legally authorized representative is not reasonably available, the investigator has committed, if feasible, to attempting to contact within the therapeutic window the subject’s family member who is not a legally authorized representative, and asking whether he or she objects to the subject’s participation in the clinical investigation. The investigator will summarize efforts made to contact family members and make this information available to the IRB at the time of continuing review. |
||
| (b) |
The IRB is responsible for ensuring that procedures are in place to inform, at the earliest feasible opportunity, each subject, or if the subject remains incapacitated, a legally authorized representative of the subject, or if such a representative is not reasonably available, a family member, of the subject’s inclusion in the clinical investigation, the details of the investigation and other information contained in the informed consent document. The IRB shall also ensure that there is a procedure to inform the |
||
National Bioethics Advisory Commission
subject, or if the subject remains incapacitated, a legally authorized representative of the subject, or if such a representative is not reasonably available, a family member, that he or she may discontinue the subject’s participation at any time without penalty or loss of benefits to which the subject is otherwise entitled. If a legally authorized representative or family member is told about the clinical investigation and the subject’s condition improves, the subject is also to be informed as soon as feasible. If a subject is entered into a clinical investigation with waived consent and the subject dies before a legally authorized representative or family member can be contacted, information about the clinical investigation is to be provided to the subject’s legally authorized representative or family member, if feasible.
| (c) |
The IRB determinations required by paragraph (a) of this section and the documentation required by paragraph (e) of this section are to be retained by the IRB for at least 3 years after completion of the clinical investigation, and the records shall be accessible for inspection and copying by FDA in accordance with §56.115(b) of this chapter. |
| (d) |
Protocols involving an exception to the informed consent requirement under this section must be performed under a separate investigational new drug application (IND) or investigational device exemption (IDE) that clearly identifies such protocols as protocols that may include subjects who are unable to consent. The submission of those protocols in a separate IND/IDE is required even if an IND for the same drug product or an IDE for the same device already exists. Applications for investigations under this section may not be submitted as amendments under §312.30 or §812.35 of this chapter. |
| (e) |
If an IRB determines that it cannot approve a clinical investigation because the investigation does not meet the criteria in the exception provided under paragraph (a) of this section or because of other relevant ethical concerns, the IRB must document its findings and provide these findings promptly in writing to the clinical investigator and to the sponsor of the clinical investigation. The sponsor of the clinical investigation must promptly disclose this information to FDA and to the sponsor’s clinical investigators who are participating or are asked to participate in this or a substantially |
193
Appendix F: Code of Federal Regulations, Title 21 Parts 50 and 56
equivalent clinical investigation of the sponsor, and to other IRB’s that have been, or are, asked to review this or a substantially equivalent investigation by that sponsor.
§ 50.25 Elements of informed consent.
| (a) |
Basic elements of informed consent. In seeking informed consent, the following information shall be provided to each subject: |
|
| (1) |
A statement that the study involves research, an explanation of the purposes of the research and the expected duration of the subject’s participation, a description of the procedures to be followed, and identification of any procedures which are experimental. |
|
| (2) |
A description of any reasonably foreseeable risks or discomforts to the subject. |
|
| (3) |
A description of any benefits to the subject or to others which may reasonably be expected from the research. |
|
| (4) |
A disclosure of appropriate alternative procedures or courses of treatment, if any, that might be advantageous to the subject. |
|
| (5) |
A statement describing the extent, if any, to which confidentiality of records identifying the subject will be maintained and that notes the possibility that the Food and Drug Administration may inspect the records. |
|
| (6) |
For research involving more than minimal risk, an explanation as to whether any compensation and an explanation as to whether any medical treatments are available if injury occurs and, if so, what they consist of, or where further information may be obtained. |
|
| (7) |
An explanation of whom to contact for answers to pertinent questions about the research and research subjects’ rights, and whom to contact in the event of a research-related injury to the subject. |
|
| (8) |
A statement that participation is voluntary, that refusal to participate will involve no penalty or loss of benefits to which the subject is otherwise entitled, and that the subject may discontinue participation at any time without penalty or loss of benefits to which the subject is otherwise entitled. |
|
| (b) |
Additional elements of informed consent. When appropriate, one or more of the following elements of information shall also be provided to each subject: |
|
| (1) |
A statement that the particular treatment or procedure may involve risks to the subject (or to the embryo or fetus, if the subject is or may become pregnant) which are currently unforeseeable. |
|
| (2) |
Anticipated circumstances under which the subject’s participation may be terminated by the investigator without regard to the subject’s consent. |
|
| (3) |
Any additional costs to the subject that may result from participation in the research. |
|
| (4) |
The consequences of a subject’s decision to withdraw from the research and procedures for orderly termination of participation by the subject. |
|
| (5) |
A statement that significant new findings developed during the course of the research which may relate to the subject’s willingness to continue participation will be provided to the subject. |
|
| (6) |
The approximate number of subjects involved in the study. |
|
| (c) |
The informed consent requirements in these regulations are not intended to preempt any applicable Federal, State, or local laws which require additional information to be disclosed for informed consent to be legally effective. |
|
| (d) |
Nothing in these regulations is intended to limit the authority of a physician to provide emergency medical care to the extent the physician is permitted to do so under applicable Federal, State, or local law. |
|
§ 50.27 Documentation of informed consent.
| (a) |
Except as provided in §56.109(c), informed consent shall be documented by the use of a written consent form approved by the IRB and signed and dated by the subject or the subject’s legally authorized representative at the time of consent. A copy shall be given to the person signing the form. |
| (b) |
Except as provided in §56.109(c), the consent form may be either of the following: |
194
| (1) |
A written consent document that embodies the elements of informed consent required by §50.25. This form may be read to the subject or the subject’s legally authorized representative, but, in any event, the investigator shall give either the subject or the representative adequate opportunity to read it before it is signed. |
| (2) |
A short form written consent document stating that the elements of informed consent required by §50.25 have been presented orally to the subject or the subject’s legally authorized representative. When this method is used, there shall be a witness to the oral presentation. Also, the IRB shall approve a written summary of what is to be said to the subject or the representative. Only the short form itself is to be signed by the subject or the representative. However, the witness shall sign both the short form and a copy of the summary, and the person actually obtaining the consent shall sign a copy of the summary. A copy of the summary shall be given to the subject or the representative in addition to a copy of the short form. |
Part 56: Institutional Review Boards Subpart A: General Provisions
§ 56.101 Scope.
| (a) |
This part contains the general standards for the composition, operation, and responsibility of an Institutional Review Board (IRB) that reviews clinical investigations regulated by the Food and Drug Administration under section 505(i) and 520(g) of the act, as well as clinical investigations that support applications for research or marketing permits for products regulated by the Food and Drug Administration, including food and color additives, drugs for human use, medical devices for human use, biological products for human use, and electronic products. Compliance with this part is intended to protect the rights and welfare of human subjects involved in such investigations. |
| (b) |
References in this part to regulatory sections of the Code of Federal Regulations are to chapter I of title 21, unless otherwise noted. |
National Bioethics Advisory Commission
§ 56.102 Definitions.
As used in this part:
| (a) |
Act means the Federal Food, Drug, and Cosmetic Act, as amended (secs. 201-902, 52 Stat. 1040 et seq., as amended (21 U.S.C. 321-392)). |
|
| (b) |
Application for research or marketing permit includes: |
|
| (1) |
A color additive petition, described in part 71. |
|
| (2) |
Data and information regarding a substance submitted as part of the procedures for establishing that a substance is generally recognized as safe for a use which results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food, described in § 170.35. |
|
| (3) |
A food additive petition, described in part 171. |
|
| (4) |
Data and information regarding a food additive submitted as part of the procedures regarding food additives permitted to be used on an interim basis pending additional study, described in §180.1. |
|
| (5) |
Data and information regarding a substance submitted as part of the procedures for establishing a tolerance for unavoidable contaminants in food and food-packaging materials, described in section 406 of the act. |
|
| (6) |
An investigational new drug application, described in part 312 of this chapter. |
|
| (7) |
A new drug application, described in part 314. |
|
| (8) |
Data and information regarding the bioavailability or bioequivalence of drugs for human use submitted as part of the procedures for issuing, amending, or repealing a bioequivalence requirement, described in part 320. |
|
| (9) |
Data and information regarding an over-the-counter drug for human use submitted as part of the procedures for classifying such drugs as generally recognized as safe and effective and not misbranded, described in part 330. (10)An application for a biological product license, described in part 601. (11)An application for a biologics license, described in part 601 of this chapter. |
|
195
Appendix F: Code of Federal Regulations, Title 21 Parts 50 and 56
| (12) |
An Application for an Investigational Device Exemption, described in parts 812 and 813. |
|
| (13) |
Data and information regarding a medical device for human use submitted as part of the procedures for classifying such devices, described in part 860. |
|
| (14) |
Data and information regarding a medical device for human use submitted as part of the procedures for establishing, amending, or repealing a standard for such device, described in part 861. |
|
| (15) |
An application for premarket approval of a medical device for human use, described in section 515 of the act. |
|
| (16) |
A product development protocol for a medical device for human use, described in section 515 of the act. |
|
| (17) |
Data and information regarding an electronic product submitted as part of the procedures for establishing, amending, or repealing a standard for such products, described in section 358 of the Public Health Service Act. |
|
| (18) |
Data and information regarding an electronic product submitted as part of the procedures for obtaining a variance from any electronic product performance standard, as described in §1010.4. |
|
| (19) |
Data and information regarding an electronic product submitted as part of the procedures for granting, amending, or extending an exemption from a radiation safety performance standard, as described in §1010.5. |
|
| (20) |
Data and information regarding an electronic product submitted as part of the procedures for obtaining an exemption from notification of a radiation safety defect or failure of compliance with a radiation safety performance standard, described in subpart D of part 1003. |
|
| (c) |
Clinical investigation means any experiment that involves a test article and one or more human subjects, and that either must meet the requirements for prior submission to the Food and Drug Administration under section 505(i) or 520(g) of the act, or need not meet the requirements for prior submission to the Food and Drug Administration under these sections of the act, but the results of |
|
which are intended to be later submitted to, or held for inspection by, the Food and Drug Administration as part of an application for a research or marketing permit. The term does not include experiments that must meet the provisions of part 58, regarding nonclinical laboratory studies. The terms research, clinical research, clinical study, study, and clinical investigation are deemed to be synonymous for purposes of this part.
| (d) |
Emergency use means the use of a test article on a human subject in a life-threatening situation in which no standard acceptable treatment is available, and in which there is not sufficient time to obtain IRB approval. |
| (e) |
Human subject means an individual who is or becomes a participant in research, either as a recipient of the test article or as a control. A subject may be either a healthy individual or a patient. |
| (f) |
Institution means any public or private entity or agency (including Federal, State, and other agencies). The term facility as used in section 520(g) of the act is deemed to be synonymous with the term institution for purposes of this part. |
| (g) |
Institutional Review Board (IRB) means any board, committee, or other group formally designated by an institution to review, to approve the initiation of, and to conduct periodic review of, biomedical research involving human subjects. The primary purpose of such review is to assure the protection of the rights and welfare of the human subjects. The term has the same meaning as the phrase institutional review committee as used in section 520(g) of the act. |
| (h) |
Investigator means an individual who actually conducts a clinical investigation (i.e., under whose immediate direction the test article is administered or dispensed to, or used involving, a subject) or, in the event of an investigation conducted by a team of individuals, is the responsible leader of that team. |
| (i) |
Minimal risk means that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests. |
196
| (j) |
Sponsor means a person or other entity that initiates a clinical investigation, but that does not actually conduct the investigation, i.e., the test article is administered or dispensed to, or used involving, a subject under the immediate direction of another individual. A person other than an individual (e.g., a corporation or agency) that uses one or more of its own employees to conduct an investigation that it has initiated is considered to be a sponsor (not a sponsor-investigator), and the employees are considered to be investigators. |
| (k) |
Sponsor-investigator means an individual who both initiates and actually conducts, alone or with others, a clinical investigation, i.e., under whose immediate direction the test article is administered or dispensed to, or used involving, a subject. The term does not include any person other than an individual, e.g., it does not include a corporation or agency. The obligations of a sponsor-investigator under this part include both those of a sponsor and those of an investigator. |
| (l) |
Test article means any drug for human use, biological product for human use, medical device for human use, human food additive, color additive, electronic product, or any other article subject to regulation under the act or under sections 351 or 354–360F of the Public Health Service Act. |
(m)IRB approval means the determination of the IRB that the clinical investigation has been reviewed and may be conducted at an institution within the constraints set forth by the IRB and by other institutional and Federal requirements.
§56.103 Circumstances in which IRB review is required.
| (a) |
Except as provided in §56.104 and §56.105, any clinical investigation which must meet the requirements for prior submission (as required in parts 312, 812, and 813) to the Food and Drug Administration shall not be initiated unless that investigation has been reviewed and approved by, and remains subject to continuing review by, an IRB meeting the requirements of this part. |
National Bioethics Advisory Commission
| (b) |
Except as provided in §56.104 and §56.105, the Food and Drug Administration may decide not to consider in support of an application for a research or marketing permit any data or information that has been derived from a clinical investigation that has not been approved by, and that was not subject to initial and continuing review by, an IRB meeting the requirements of this part. The determination that a clinical investigation may not be considered in support of an application for a research or marketing permit does not, however, relieve the applicant for such a permit of any obligation under any other applicable regulations to submit the results of the investigation to the Food and Drug Administration. |
| (c) |
Compliance with these regulations will in no way render inapplicable pertinent Federal, State, or local laws or regulations. |
§ 56.104 Exemptions from IRB requirement.
The following categories of clinical investigations are exempt from the requirements of this part for IRB review:
| (a) |
Any investigation which commenced before July 27, 1981 and was subject to requirements for IRB review under FDA regulations before that date, provided that the investigation remains subject to review of an IRB which meets the FDA requirements in effect before July 27, 1981. |
| (b) |
Any investigation commenced before July 27, 1981 and was not otherwise subject to requirements for IRB review under Food and Drug Administration regulations before that date. |
| (c) |
Emergency use of a test article, provided that such emergency use is reported to the IRB within 5 working days. Any subsequent use of the test article at the institution is subject to IRB review. |
| (d) |
Taste and food quality evaluations and consumer acceptance studies, if wholesome foods without additives are consumed or if a food is consumed that contains a food ingredient at or below the level and for a use found to be safe, or agricultural, chemical, or environmental contaminant at or below the level found to be safe, by the Food and Drug Administration or approved by the Environmental Protection Agency or the Food Safety and Inspection Service of the U.S. Department of Agriculture. |
197
Appendix F: Code of Federal Regulations, Title 21 Parts 50 and 56
§ 56.105 Waiver of IRB requirement.
On the application of a sponsor or sponsor-investigator, the Food and Drug Administration may waive any of the requirements contained in these regulations, including the requirements for IRB review, for specific research activities or for classes of research activities, otherwise covered by these regulations.
Subpart B: Organization and Personnel
§ 56.107 IRB membership.
| (a) |
Each IRB shall have at least five members, with varying backgrounds to promote complete and adequate review of research activities commonly conducted by the institution. The IRB shall be sufficiently qualified through the experience and expertise of its members, and the diversity of the members, including consideration of race, gender, cultural backgrounds, and sensitivity to such issues as community attitudes, to promote respect for its advice and counsel in safeguarding the rights and welfare of human subjects. In addition to possessing the professional competence necessary to review the specific research activities, the IRB shall be able to ascertain the acceptability of proposed research in terms of institutional commitments and regulations, applicable law, and standards or professional conduct and practice. The IRB shall therefore include persons knowledgeable in these areas. If an IRB regularly reviews research that involves a vulnerable category of subjects, such as children, prisoners, pregnant women, or handicapped or mentally disabled persons, consideration shall be given to the inclusion of one or more individuals who are knowledgeable about and experienced in working with those subjects. |
| (b) |
Every nondiscriminatory effort will be made to ensure that no IRB consists entirely of men or entirely of women, including the institution’s consideration of qualified persons of both sexes, so long as no selection is made to the IRB on the basis of gender. No IRB may consist entirely of members of one profession. |
| (c) |
Each IRB shall include at least one member whose primary concerns are in the scientific area and at least one member whose primary concerns are in nonscientific areas. |
| (d) |
Each IRB shall include at least one member who is not otherwise affiliated with the institution and who is not part of the immediate family of a person who is affiliated with the institution. |
| (e) |
No IRB may have a member participate in the IRB’s initial or continuing review of any project in which the member has a conflicting interest, except to provide information requested by the IRB. |
| (f) |
An IRB may, in its discretion, invite individuals with competence in special areas to assist in the review of complex issues which require expertise beyond or in addition to that available on the IRB. These individuals may not vote with the IRB. |
Subpart C: IRB Functions and Operations
§ 56.108 IRB functions and operations.
In order to fulfill the requirements of these regulations, each IRB shall:
| (a) |
Follow written procedures: (1) For conducting its initial and continuing review of research and for reporting its findings and actions to the investigator and the institution; (2) for determining which projects require review more often than annually and which projects need verification from sources other than the investigator that no material changes have occurred since previous IRB review; (3) for ensuring prompt reporting to the IRB of changes in research activity; and (4) for ensuring that changes in approved research, during the period for which IRB approval has already been given, may not be initiated without IRB review and approval except where necessary to eliminate apparent immediate hazards to the human subjects. |
|
| (b) |
Follow written procedures for ensuring prompt reporting to the IRB, appropriate institutional officials, and the Food and Drug Administration of: (1) Any unanticipated problems involving risks to human subjects or others; (2) any instance of serious or continuing noncompliance with these regulations or the requirements or determinations of the IRB; or |
|
| (3) |
any suspension or termination of IRB approval. |
|
198
| (c) |
Except when an expedited review procedure is used (see §56.110), review proposed research at convened meetings at which a majority of the members of the IRB are present, including at least one member whose primary concerns are in nonscientific areas. In order for the research to be approved, it shall receive the approval of a majority of those members present at the meeting. |
(Information collection requirements in this section were approved by the Office of Management and Budget (OMB) and assigned OMB control number 0910-0130)
§ 56.109 IRB review of research.
| (a) |
An IRB shall review and have authority to approve, require modifications in (to secure approval), or disapprove all research activities covered by these regulations. |
|
| (b) |
An IRB shall require that information given to subjects as part of informed consent is in accordance with §50.25. The IRB may require that information, in addition to that specifically mentioned in §50.25, be given to the subjects when in the IRB’s judgment the information would meaningfully add to the protection of the rights and welfare of subjects. |
|
| (c) |
An IRB shall require documentation of informed consent in accordance with §50.27 of this chapter, except as follows: |
|
| (1) |
The IRB may, for some or all subjects, waive the requirement that the subject, or the subject’s legally authorized representative, sign a written consent form if it finds that the research presents no more than minimal risk of harm to subjects and involves no procedures for which written consent is normally required outside the research context; or |
|
| (2) |
The IRB may, for some or all subjects, find that the requirements in §50.24 of this chapter for an exception from informed consent for emergency research are met. |
|
| (d) |
In cases where the documentation requirement is waived under paragraph (c)(1) of this section, the IRB may require the investigator to provide subjects with a written statement regarding the research. |
|
National Bioethics Advisory Commission
| (e) |
An IRB shall notify investigators and the institution in writing of its decision to approve or disapprove the proposed research activity, or of modifications required to secure IRB approval of the research activity. If the IRB decides to disapprove a research activity, it shall include in its written notification a statement of the reasons for its decision and give the investigator an opportunity to respond in person or in writing. For investigations involving an exception to informed consent under §50.24 of this chapter, an IRB shall promptly notify in writing the investigator and the sponsor of the research when an IRB determines that it cannot approve the research because it does not meet the criteria in the exception provided under §50.24(a) of this chapter or because of other relevant ethical concerns. The written notification shall include a statement of the reasons for the IRB’s determination. |
| (f) |
An IRB shall conduct continuing review of research covered by these regulations at intervals appropriate to the degree of risk, but not less than once per year, and shall have authority to observe or have a third party observe the consent process and the research. |
| (g) |
An IRB shall provide in writing to the sponsor of research involving an exception to informed consent under §50.24 of this chapter a copy of information that has been publicly disclosed under §50.24(a)(7)(ii) and (a)(7)(iii) of this chapter. The IRB shall provide this information to the sponsor promptly so that the sponsor is aware that such disclosure has occurred. Upon receipt, the sponsor shall provide copies of the information disclosed to FDA. |
§56.110 Expedited review procedures for certain kinds of research involving no more than minimal risk, and for minor changes in approved research.
| (a) |
The Food and Drug Administration has established, and published in the Federal Register, a list of categories of research that may be reviewed by the IRB through an expedited review procedure. The list will be amended, as appropriate, through periodic republication in the Federal Register. |
199
Appendix F: Code of Federal Regulations, Title 21 Parts 50 and 56
| (b) |
An IRB may use the expedited review procedure to review either or both of the following: (1) Some or all of the research appearing on the list and found by the reviewer(s) to involve no more than minimal risk, (2) minor changes in previously approved research during the period (of 1 year or less) for which approval is authorized. Under an expedited review procedure, the review may be carried out by the IRB chairperson or by one or more experienced reviewers designated by the IRB chairperson from among the members of the IRB. In reviewing the research, the reviewers may exercise all of the authorities of the IRB except that the reviewers may not disapprove the research. A research activity may be disapproved only after review in accordance with the nonexpedited review procedure set forth in § 56.108(c). |
| (c) |
Each IRB which uses an expedited review procedure shall adopt a method for keeping all members advised of research proposals which have been approved under the procedure. |
| (d) |
The Food and Drug Administration may restrict, suspend, or terminate an institution’s or IRB’s use of the expedited review procedure when necessary to protect the rights or welfare of subjects. |
§ 56.111 Criteria for IRB approval of research.
| (a) |
In order to approve research covered by these regulations the IRB shall determine that all of the following requirements are satisfied: |
|
| (1) |
Risks to subjects are minimized: (i) By using procedures which are consistent with sound research design and which do not unnecessarily expose subjects to risk, and (ii) whenever appropriate, by using procedures already being performed on the subjects for diagnostic or treatment purposes. |
|
| (2) |
Risks to subjects are reasonable in relation to anticipated benefits, if any, to subjects, and the importance of the knowledge that may be expected to result. In evaluating risks and benefits, the IRB should consider only those risks and benefits that may result from the research (as distinguished from risks and benefits of therapies that subjects would receive even if not participating in the research). The IRB |
|
should not consider possible long-range effects of applying knowledge gained in the research (for example, the possible effects of the research on public policy) as among those research risks that fall within the purview of its responsibility.
| (3) |
Selection of subjects is equitable. In making this assessment the IRB should take into account the purposes of the research and the setting in which the research will be conducted and should be particularly cognizant of the special problems of research involving vulnerable populations, such as children, prisoners, pregnant women, handicapped, or mentally disabled persons, or economically or educationally disadvantaged persons. |
|
| (4) |
Informed consent will be sought from each prospective subject or the subject’s legally authorized representative, in accordance with and to the extent required by part 50. |
|
| (5) |
Informed consent will be appropriately documented, in accordance with and to the extent required by §50.27. |
|
| (6) |
Where appropriate, the research plan makes adequate provision for monitoring the data collected to ensure the safety of subjects. |
|
| (7) |
Where appropriate, there are adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data. |
|
| (b) |
When some or all of the subjects, such as children, prisoners, pregnant women, handicapped, or mentally disabled persons, or economically or educationally disadvantaged persons, are likely to be vulnerable to coercion or undue influence additional safeguards have been included in the study to protect the rights and welfare of these subjects. |
|
§ 56.112 Review by institution.
Research covered by these regulations that has been approved by an IRB may be subject to further appropriate review and approval or disapproval by officials of the institution. However, those officials may not approve the research if it has not been approved by an IRB.
200
§56.113 Suspension or termination of IRB approval of research.
An IRB shall have authority to suspend or terminate approval of research that is not being conducted in accordance with the IRB’s requirements or that has been associated with unexpected serious harm to subjects. Any suspension or termination of approval shall include a statement of the reasons for the IRB’s action and shall be reported promptly to the investigator, appropriate institutional officials, and the Food and Drug Administration.
§ 56.114 Cooperative research.
In complying with these regulations, institutions involved in multi-institutional studies may use joint review, reliance upon the review of another qualified IRB, or similar arrangements aimed at avoidance of duplication of effort.
Subpart D: Records and Reports
§ 56.115 IRB records.
| (a) |
An institution, or where appropriate an IRB, shall prepare and maintain adequate documentation of IRB activities, including the following: |
|
| (1) |
Copies of all research proposals reviewed, scientific evaluations, if any, that accompany the proposals, approved sample consent documents, progress reports submitted by investigators, and reports of injuries to subjects. |
|
| (2) |
Minutes of IRB meetings which shall be in sufficient detail to show attendance at the meetings; actions taken by the IRB; the vote on these actions including the number of members voting for, against, and abstaining; the basis for requiring changes in or disapproving research; and a written summary of the discussion of controverted issues and their resolution. |
|
| (3) |
Records of continuing review activities. |
|
| (4) |
Copies of all correspondence between the IRB and the investigators. |
|
National Bioethics Advisory Commission
| (5) |
A list of IRB members identified by name; earned degrees; representative capacity; indications of experience such as board certifications, licenses, etc., sufficient to describe each member’s chief anticipated contributions to IRB deliberations; and any employment or other relationship between each member and the institution; for example: full-time employee, part-time employee, a member of governing panel or board, stockholder, paid or unpaid consultant. |
|
| (6) |
Written procedures for the IRB as required by § 56.108 (a) and (b). |
|
| (7) |
Statements of significant new findings provided to subjects, as required by §50.25. |
|
| (b) |
The records required by this regulation shall be retained for at least 3 years after completion of the research, and the records shall be accessible for inspection and copying by authorized representatives of the Food and Drug Administration at reasonable times and in a reasonable manner. |
|
| (c) |
The Food and Drug Administration may refuse to consider a clinical investigation in support of an application for a research or marketing permit if the institution or the IRB that reviewed the investigation refuses to allow an inspection under this section. (Information collection requirements in this section were approved by the Office of Management and Budget (OMB) and assigned OMB control number 0910-0130) |
|
Subpart E: Administrative Actions for Noncompliance
§ 56.120 Lesser administrative actions.
| (a) |
If apparent noncompliance with these regulations in the operation of an IRB is observed by an FDA investigator during an inspection, the inspector will present an oral or written summary of observations to an appropriate representative of the IRB. The Food and Drug Administration may subsequently send a letter describing the noncompliance to the IRB and to the parent institution. The agency will require that the IRB or the parent institution respond to this letter within a time period specified by FDA and describe the corrective actions that will be taken by the IRB, the institution, or both to achieve compliance with these regulations. |
201
Appendix F: Code of Federal Regulations, Title 21 Parts 50 and 56
| (b) |
On the basis of the IRB’s or the institution’s response, FDA may schedule a reinspection to confirm the adequacy of corrective actions. In addition, until the IRB or the parent institution takes appropriate corrective action, the agency may: |
|
| (1) |
Withhold approval of new studies subject to the requirements of this part that are conducted at the institution or reviewed by the IRB; |
|
| (2) |
Direct that no new subjects be added to ongoing studies subject to this part; |
|
| (3) |
Terminate ongoing studies subject to this part when doing so would not endanger the subjects; or |
|
| (4) |
When the apparent noncompliance creates a significant threat to the rights and welfare of human subjects, notify relevant State and Federal regulatory agencies and other parties with a direct interest in the agency’s action of the deficiencies in the operation of the IRB. |
|
| (c) |
The parent institution is presumed to be responsible for the operation of an IRB, and the Food and Drug Administration will ordinarily direct any administrative action under this subpart against the institution. However, depending on the evidence of responsibility for deficiencies, determined during the investigation, the Food and Drug Administration may restrict its administrative actions to the IRB or to a component of the parent institution determined to be responsible for formal designation of the IRB. |
|
§56.121 Disqualification of an IRB or an institution.
| (a) |
Whenever the IRB or the institution has failed to take adequate steps to correct the noncompliance stated in the letter sent by the agency under §56.120(a), and the Commissioner of Food and Drugs determines that this noncompliance may justify the disqualification of the IRB or of the parent institution, the Commissioner will institute proceedings in accordance with the requirements for a regulatory hearing set forth in part 16. |
|
| (b) |
The Commissioner may disqualify an IRB or the parent institution if the Commissioner determines that: |
|
| (1) |
The IRB has refused or repeatedly failed to comply with any of the regulations set forth in this part, and |
|
| (2) |
The noncompliance adversely affects the rights or welfare of the human subjects in a clinical investigation. |
|
| (c) |
If the Commissioner determines that disqualification is appropriate, the Commissioner will issue an order that explains the basis for the determination and that prescribes any actions to be taken with regard to ongoing clinical research conducted under the review of the IRB. The Food and Drug Administration will send notice of the disqualification to the IRB and the parent institution. Other parties with a direct interest, such as sponsors and clinical investigators, may also be sent a notice of the disqualification. In addition, the agency may elect to publish a notice of its action in the Federal Register. |
|
| (d) |
The Food and Drug Administration will not approve an application for a research permit for a clinical investigation that is to be under the review of a disqualified IRB or that is to be conducted at a disqualified institution, and it may refuse to consider in support of a marketing permit the data from a clinical investigation that was reviewed by a disqualified IRB as conducted at a disqualified institution, unless the IRB or the parent institution is reinstated as provided in §56.123. |
|
§56.122 Public disclosure of information regarding revocation.
A determination that the Food and Drug Administration has disqualified an institution and the administrative record regarding that determination are disclosable to the public under part 20.
§56.123 Reinstatement of an IRB or an institution.
An IRB or an institution may be reinstated if the Commissioner determines, upon an evaluation of a written submission from the IRB or institution that explains the corrective action that the institution or IRB plans to take, that the IRB or institution has provided adequate assurance that it will operate in compliance with the standards set forth in this part. Notification of reinstatement shall be provided to all persons notified under §56.121(c).
202
§56.124 Actions alternative or additional to disqualification.
Disqualification of an IRB or of an institution is independent of, and neither in lieu of nor a precondition to, other proceedings or actions authorized by the act. The Food and Drug Administration may, at any time, through the Department of Justice institute any appropriate judicial proceedings (civil or criminal) and any other appropriate regulatory action, in addition to or in lieu of, and before, at the time of, or after, disqualification. The agency may also refer pertinent matters to another Federal, State, or local government agency for any action that that agency determines to be appropriate.
National Bioethics Advisory Commission
203
Appendix G
Statutory Authority for Agency Human Subjects Protection Regulations (Common Rule)
| Department | Regulation | |
| Department of Agriculture: | 7 CFR 1c | Protection of human subjects |
| Department of Energy: | 10 CFR 745 | Protection of human subjects |
| National Aeronautics and Space Administration: | 14 CFR 1230 | Protection of human subjects |
| Department of Commerce: | 15 CFR 27 | Protection of human subjects |
| Consumer Product Safety Commission: | 16 CFR 1028 | Protection of human subjects |
| Agency for International Development: | 22 CFR 225 | Protection of human subjects |
| Department of Housing and Urban Development: | 24 CFR 60 | Protection of human subjects |
| Department of Justice: | 28 CFR 46 | Protection of human subjects |
| Department of Defense: | 32 CFR 219 | Protection of human subjects |
| Department of Education: | 34 CFR 97 | Protection of human subjects |
| Department of Veterans Affairs: | 38 CFR 16 | Protection of human subjects |
| Environmental Protection Agency: | 40 CFR 26 | Protection of human subjects |
| Department of Health and Human Services: | 45 CFR 46 | Protection of human subjects |
| National Science Foundation: | 45 CFR 690 | Protection of human subjects |
| Department of Transportation: | 49 CFR 11 | Protection of human subjects |
| Statutory Authority Cited | ||
5 USC 301 Departmental regulations.
The head of an Executive department or military department may prescribe regulations for the government of his department, the conduct of its employees, the distribution and performance of its business, and the custody, use, and preservation of its records, papers, and property. This section does not authorize withholding information from the public or limiting the availability of records to the public.
42 USC 300v-1(b) Duties of Commission.
| (b) |
Recommendations to agencies; subsequent administrative requirements |
| (1) |
Within 60 days of the date a Federal agency receives a recommendation from the Commission that the agency |
take any action with respect to its rules, policies, guidelines, or regulations, the agency shall publish such recommendation in the Federal Register and shall provide opportunity for interested persons to submit written data, views, and arguments with respect to adoption of the recommendation.
205
Appendix G: Statutory Authority for Agency Human Subjects Protection Regulations (Common Rule)
(2) Within the 180-day period beginning on the date of such publication, the agency shall determine whether the action proposed by such recommendation is appropriate, and, to the extent that it determines that - (A) such action is not appropriate, the agency shall, within such time period, provide the Commission with, and publish in the Federal Register, a notice of such determination (including an adequate statement of the reasons for the determination), or (B) such action is appropriate, the agency shall undertake such action as expeditiously as feasible and shall notify the Commission of the determination and the action undertaken.
Additional Statutory Authority Cited for Agency Human Subject Protection Regulations (Common Rule)
Department of Energy
42 USC 7254 Rules and regulations.
The Secretary is authorized to prescribe such procedural and administrative rules and regulations as he may deem necessary or appropriate to administer and manage the functions now or hereafter vested in him.
Department of Housing and Urban Development
42 USC 3535 Administrative provisions.
(d) Delegation of authority; rules and regulations
The Secretary may delegate any of his functions, powers, and duties to such officers and employees of the Department as he may designate, may authorize such successive redelegations of such functions, powers, and duties as he may deem desirable, and may make such rules and regulations as may be necessary to carry out his functions, powers, and duties.
Department of Justice
28 USC 509 Functions of the Attorney General.
All functions of other officers of the Department of Justice and all functions of agencies and employees of the Department of Justice are vested in the Attorney General except the functions -(1) vested by subchapter II of chapter 5 of title 5 in administrative law judges employed by the Department of Justice;
| (2) |
of the Federal Prison Industries, Inc.; and |
| (3) |
of the Board of Directors and officers of the Federal Prison Industries, Inc.. |
28 USC 510 Delegation of authority.
The Attorney General may from time to time make such provisions as he considers appropriate authorizing the performance by any other officer, employee, or agency of the Department of Justice of any function of the Attorney General.
Department of Education
20 USC 1221e-3 General authority of Secretary.
The Secretary, in order to carry out functions otherwise vested in the Secretary by law or by delegation of authority pursuant to law, and subject to limitations as may be otherwise imposed by law, is authorized to make, promulgate, issue, rescind, and amend rules and regulations governing the manner of operation of, and governing the applicable programs administered by, the Department.
20 USC 3474 Rules and regulations.
The Secretary is authorized to prescribe such rules and regulations as the Secretary determines necessary or appropriate to administer and manage the functions of the Secretary or the Department.
206
National Bioethics Advisory Commission
Department of Veterans Affairs
38 USC 501 Rules and regulations.
(a) The Secretary has authority to prescribe all rules and regulations which are necessary or appropriate to carry out the laws administered by the Department and are consistent with those laws, including - (1) regulations with respect to the nature and extent of proof and evidence and the method of taking and furnishing them in order to establish the right to benefits under such laws;… (c) In applying section 552(a)(1) of title 5 to the Department, the Secretary shall ensure that subparagraphs (C), (D), and (E) of that section are complied with, particularly with respect to opinions and interpretations of the General Counsel.
38 USC 7331 Informed consent.
The Secretary, upon the recommendation of the Under Secretary for Health and pursuant to the provisions of section 7334 of this title, shall prescribe regulations establishing procedures to ensure that all medical and prosthetic research carried out and, to the maximum extent practicable, all patient care furnished under this title shall be carried out only with the full and informed consent of the patient or subject or, in appropriate cases, a representative thereof.
38 USC 7334 Regulations.
(a) Regulations prescribed by the Secretary under section 7331 of this title, section 7332 of this title with respect to the confidentiality of alcohol and drug abuse medical records, and section 7333 of this title with respect to alcohol or drug abusers shall, to the maximum extent feasible consistent with other provisions of this title, make applicable the regulations described in subsection (b) to the conduct of research and to the provision of hospital care, nursing home care, domiciliary care, and medical services under this title.
| (b) |
The regulations referred to in subsection (a) are - |
| (1) |
regulations governing human experimentation and informed consent prescribed by the Secretary of Health |
and Human Services, based on the recommendations of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research, established by section 201 of the National Research Act (Public Law 93-348; 88 Stat. 348); and (2) regulations governing (A) the confidentiality of drug and alcohol abuse medical records, and (B) the admission of drug and alcohol abusers to private and public hospitals, prescribed pursuant to the Comprehensive Alcohol Abuse and Alcoholism Prevention, Treatment, and Rehabilitation Act of 1970 (42 USC 4551 et seq.) and the Drug Abuse Office and Treatment Act of 1972 (21 USC 1101 et seq.).
(c) Regulations prescribed by the Secretary under sections 7331, 7332, and 7333 of this title may contain such definitions, and may provide for such safeguards and procedures (including procedures and criteria for the issuance and scope of court orders undersection 7332(b)(2)(C) [1] of this title), as are necessary to prevent circumvention or evasion of such regulations or to facilitate compliance with such regulations.
(d) In prescribing and implementing such regulations, the Secretary shall, from time to time, consult with the Secretary of Health and Human Services and, as appropriate, with the President (or the delegate of the President) in order to achieve the maximum possible coordination of the regulations, and the implementation of the regulations, which they and the Secretary prescribe.
Department of Health and Human Services
42 USC 289 Institutional review boards; ethics guidance program.
(a) The Secretary shall by regulation require that each entity which applies for a grant, contract, or cooperative agreement under this chapter for any project or program which involves the conduct of biomedical or behavioral research involving human subjects submit in or with its application for such grant, contract, or cooperative agreement assurances satisfactory to the Secretary that it has established (in accordance with regulations which the Secretary shall prescribe) a board (to be known as an “Institutional Review Board”) to review biomedical and
207
Appendix G: Statutory Authority for Agency Human Subjects Protection Regulations (Common Rule)
behavioral research involving human subjects conducted at or supported by such entity in order to protect the rights of the human subjects of such research.
(b)(1) The Secretary shall establish a program within the Department of Health and Human Services under which requests for clarification and guidance with respect to ethical issues raised in connection with biomedical or behavioral research involving human subjects are responded to promptly and appropriately.
(2) The Secretary shall establish a process for the prompt and appropriate response to information provided to the Director of NIH respecting incidences of violations of the rights of human subjects of research for which funds have been made available under this chapter. The process shall include procedures for the receiving of reports of such information from recipients of funds under this chapter and taking appropriate action with respect to such violations.
Related Human Subjects Protection Regulations (Not Common Rule) and Statutory Authority Cited
| Department of Justice | |
| Bureau of Prisons | |
| 28 CFR 512 Research | |
| 5 USC 301 | Departmental regulations. |
| 18 USC 3621 | Imprisonment of a convicted person. |
| 18 USC 3622 | Temporary release of a prisoner. |
| 18 USC 3624 | Release of a prisoner. |
| 18 USC 4001 | Limitation on detention; control of prisons. |
| 18 USC 4042 | Duties of Bureau of Prisons. |
| 18 USC 4081 | Classification and treatment of prisoners. |
| 18 USC 4082 | Commitment to Attorney General; residential treatment centers; extension of limits of |
| confinement; work furlough (repealed in part as to offenses committed on or after | |
| November 1, 1987). | |
| 18 USC 5006 - 5024 | Chapter 402 of Part IV Correction of youthful offenders (Repealed October 12, 1984 |
| as to offenses committed after that date). | |
| 18 USC 5039 | Commitment (of youthful offenders). |
| 28 USC 509 | Functions of the Attorney General. |
| 28 USC 510 | Delegation of authority. |
| Department of Education | |
| 34 CFR 97, Subpart D | Additional ED protections for children who are subjects in research |
| 5 USC 301 | Departmental regulations. |
| 20 USC 1221e-3 | General authority of Secretary. |
| 20 USC 3474 | Rules and regulations. |
| 42 USC 300v-1(b) | Duties of Commission. |
208
| National Bioethics Advisory Commission | ||
| 34 CFR 98 Student rights in research, experimental programs, and testing. | ||
| 20 USC 1221e-3 | General authority of Secretary. | |
| 20 USC 1232h | Protection of pupil rights (a) Inspection of instructional materials by parents or guardians. | |
| 20 USC 1232h | Protection of pupil rights (b) Limits on survey, analysis, or evaluations. | |
| 20 USC 3474 | Rules and regulations. | |
| 34 CFR 99 Family educational rights and privacy. | ||
| 20 USC 1232g | Family educational and privacy rights. | |
| Office of Special Education and Rehabilitative Services | ||
| 34 CFR 350.4(c) What regulations apply? | ||
| 29 USC 761a | Authorization of appropriations. | |
| 29 USC 762 | National Institute on Disability and Rehabilitation Research. | |
| 42 USC 300v-1(b) | Duties of Commission. | |
| 34 CFR 356.3(c) Disability and rehabilitation research: Research fellowships. | ||
| 29 USC 761a | Authorization of appropriations. | |
| 42 USC 300v-1(b) | Duties of Commission. | |
| Department of Veterans Affairs | ||
| 38 CFR 17.85 Treatment of research-related injuries to human subjects. | ||
| 38 USC 501 | Rules and regulations. | |
| 38 USC 7303 | Functions of Veterans Health Administration: research programs. | |
Department of Health and Human Services
45 CFR 46 Subpart B Additional Protections Pertaining to Research, Development, and Related Activities Involving Fetuses, Pregnant Women, and Human In Vitro Fertilization.
| 5 USC 301 | Departmental regulations. |
| 42 USC 289 | Institutional review boards; ethics guidance program. |
| 42 USC 300v-1(b) | Duties of Commission. |
45 CFR 46 Subpart C Additional Protections Pertaining to Biomedical and Behavioral Research Involving Prisoners as Subjects
| 5 USC 301 | Departmental regulations. |
| 42 USC 289 | Institutional review boards; ethics guidance program. |
| 42 USC 300v-1(b) | Duties of Commission. |
| 45 CFR 46 Subpart D Additional protections for children involved as subjects in research | |
| 5 USC 301 | Departmental regulations. |
| 42 USC 289 | Institutional review boards; ethics guidance program. |
| 42 USC 300v-1(b) | Duties of Commission. |
209
Appendix G: Statutory Authority for Agency Human Subjects Protection Regulations (Common Rule)
Food and Drug Administration
21 CFR 50 Protection of human subjects
21 USC § 321 21 USC § 346
Definitions; generally.
Tolerances for poisonous or deleterious substances in food; regulations.
21 USC § 346a Tolerances and exemptions for pesticide chemical residues.
21 USC § 348 21 USC § 352 21 USC § 353 21 USC § 355 21 USC § 360
Food additives.
Misbranded drugs and devices.
Exemptions and consideration for certain drugs, devices, and biological products. New drugs.
Registration of producers of drugs or devices.
21 USC § 360c Classification of devices intended for human use. 21 USC § 360d Performance standards.
21 USC § 360e Premarket approval.
21 USC § 360f
Banned devices.
21 USC § 360h Notification and other remedies.
21 USC § 360i 21 USC § 360j 21 USC § 371
Records and reports on devices.
General provisions respecting control of devices intended for human use. Regulations and hearings.
21 USC § 379e Listing and certification of color additives for foods, drugs, devices, and cosmetics.
21 USC § 381 42 USC § 216 42 USC § 241 42 USC § 262
Imports and exports. Regulations.
Research and investigations generally. Regulation of biological products.
42 USC § 263b Certification of mammography facilities.
21 CFR 56 Institutional review boards
21 USC § 321. 21 USC § 346
Definitions; generally.
Tolerances for poisonous or deleterious substances in food; regulations.
21 USC § 346a Tolerances and exemptions for pesticide chemical residues.
21 USC § 348 21 USC § 351 21 USC § 352 21 USC § 353 21 USC § 355 21 USC § 360
Food additives.
Adulterated drugs and devices. Misbranded drugs and devices.
Exemptions and consideration for certain drugs, devices, and biological products. New drugs.
Registration of producers of drugs or devices.
21 USC § 360c Classification of devices intended for human use. 21 USC § 360d Performance standards.
210
| National Bioethics Advisory Commission | ||
| 21 USC § 360e | Premarket approval. | |
| 21 USC § 360f | Banned devices. | |
| 21 USC § 360h | Notification and other remedies. | |
| 21 USC § 360i | Records and reports on devices. | |
| 21 USC § 360j | General provisions respecting control of devices intended for human use. | |
| 21 USC § 371 | Regulations and hearings. | |
| 21 USC § 379e | Listing and certification of color additives for foods, drugs, devices, and cosmetics. | |
| 21 USC § 381 | Imports and exports. | |
| 42 USC § 216 | Regulations. | |
| 42 USC § 241 | Research and investigations generally. | |
| 42 USC § 262 | Regulation of biological products. | |
| 42 USC § 263b | Certification of mammography facilities. | |
211
Appendix H
Food and Drug Administration Regulations Requiring Reporting of Adverse Events
| The regulations containing provisions related to adverse | |
| event reporting include: | |
| 21 CFR Part 56 | Institutional Review Boards |
| 21 CFR Part 310 | New Drugs |
| 21 CFR Part 312 | Investigational New Drug |
| Application | |
| 21 CFR Part 314 | Applications for FDA Approval |
| to Market a New Drug | |
| 21 CFR Part 600 | Biological Products: General |
| 21 CFR Part 803 | Medical Device Reporting |
| 21 CFR Part 812 | Investigational Device Exemptions |
| Regulatory Text1 | |
| Investigator Regulatory Reporting | |
| Requirements | |
| Investigator Reporting to Sponsor | |
| 21 CFR 312.64 | Investigator Reports. |
(b) Safety reports. An investigator shall promptly report to the sponsor any adverse effect that may reasonably be regarded as caused by, or probably caused by, the drug. If the adverse effect is alarming, the investigator shall report the effect immediately.
[312.64(b) requires the investigator to report certain adverse effect to the sponsor. Life-threatening adverse drug experience, serious adverse drug experience and unexpected adverse drug experience are defined in 312.32(a), but adverse effect is not defined in Part 312. Therefore, the reporting requirement is open-ended.]
1 Annotations provided by P. Goebel, Office for Human Research Protections.
[The investigator is required to notify the sponsor of only probably drug-caused adverse effects. However, most sponsors require the investigator to report all adverse effects to the sponsor, no matter what the cause. This is probably because 21 CFR 312.32(b) and 21 CFR 812.3(s) appear to require the sponsor to know about all events that happen anywhere in the world both to people on study or just taking the drug. Recent FDA enforcement actions have reinforced this view.]
21 CFR 812.150 Reports.
(a) Investigator reports. An investigator shall prepare and submit he following complete, accurate, and timely reports:
(1) Unanticipated adverse device effects. An investigator shall submit to the sponsor and to the reviewing IRB a report of any unanticipated adverse device effect occurring during an investigation as soon as possible, but in no event later than 10 working days after the investigator first learns of the effect.
[Investigator informs the sponsor and his/her IRB, not all of the IRBs of a multi-site study. The Part 312 regulations do not include a time limit.]
21 CFR 812.3 Definitions.
(s) Unanticipated adverse device effect means any serious adverse effect on health or safety or any life-threatening problem or death caused by, or associated with, a device, if that effect, problem, or death was not previously identified in nature, severity, or degree of incidence in the investigational plan or application (including a supplementary plan or application), or any other unanticipated serious problem associated with a device that relates to the rights, safety, or welfare of subjects.
213
Appendix H: Food and Drug Administration Regulations Requiring Reporting of Adverse Events
[The unanticipated adverse device effect definition (21 CFR 812.3(s)) requires reporting of certain unexpected effects.]
Investigator Reporting to IRB
21 CFR 312.66 Assurance of IRB review.
An investigator shall assure that an IRB that complies with the requirements set forth in part 56 will be responsible for the initial and continuing review and approval of the proposed clinical study. The investigator shall also assure that he or she will promptly report to the IRB all changes in the research activity [this wording is identical to 21 CFR 56.108(a)(3)] and all unanticipated problems involving risk to human subjects or others, [This wording is identical to 21 CFR 56.108(b)(1), which follows] and that he or she will not make any changes in the research without IRB approval, except where necessary to eliminate apparent immediate hazards to human subjects.
21 CFR 56.108 IRB functions and operations.
In order to fulfill the requirements of these regulations, each IRB shall: (b) Follow written procedures for ensuring prompt reporting to the IRB, appropriate institutional officials, and the Food and Drug Administration of: (1) Any unanticipated problems involving risks to human subjects or others; (2) any instance of serious or continuing non-compliance with these regulations or the requirements or determinations of the IRB; or (3) any suspension or termination of IRB approval.
[Any unanticipated problem is all-inclusive. It includes adverse events that occur at the investigator’s study site, and adverse events that occur at all other institutions that are reported to the investigator by the sponsor. This section seems to require the investigator to notify the IRB of all unanticipated problems that occurred anyplace in the world.]
21 CFR 812.150 Reports.
(a) Investigator reports. An investigator shall prepare and submit the following complete, accurate, and timely reports: (1) Unanticipated adverse device effects. An investigator shall submit to the sponsor and to the reviewing IRB a report of any unanticipated adverse device effect occurring
during an investigation as soon as possible, but in no event later than 10 working days after the investigator first learns of the effect.
Sponsor Evaluation of Adverse Event Reports Requirements [Annotated]
21 CFR 312.32 IND safety reports.
(b) Review of safety information. The sponsor shall promptly review all information relevant to the safety of the drug obtained or otherwise received by the sponsor from any source, foreign or domestic, including information derived from any clinical or epidemiological investigations, animal investigations, commercial marketing experience, reports in the scientific literature, and unpublished scientific papers, as well as reports from foreign regulatory authorities that have not already been previously reported to the agency by the sponsor.
[This review requirement is not limited to “serious and unexpected” or to “adverse events that may be caused by the drug,” but includes “all information relevant to the safety of the drug....” Also, the requirement to review information “from any source” is interpreted by sponsors to mean they are required to aggressively search worldwide for any event that may remotely be related to use of the drug.]
21 CFR 312.32 IND safety reports. d) Followup. (1) The sponsor shall promptly investigate all safety information received by it.
(2) Followup information to a safety report shall be submitted as soon as the relevant information is available. (3) If the results of a sponsor’s investigation show that an adverse drug experience not initially determined to be reportable under paragraph (c) of this section is so reportable, the sponsor shall report such experience in a written safety report as soon as possible, but in no event later than 15 calendar days after the determination is made. (4) Results of a sponsor’s investigation of other safety information shall be submitted, as appropriate, in an information amendment or annual report.
21 CFR 812.46 Monitoring investigations.
(b) Unanticipated adverse device effects (1) A sponsor shall immediately evaluate any unanticipated adverse device effect.
214
Sponsor Reporting Requirements [Annotated]
Sponsor Reporting to Investigators
21 CFR 312.32 IND safety reports.
(c) IND safety reports. (1) Written reports - (i) The sponsor shall notify FDA and all participating investigators in a written IND safety report of: (A) Any adverse experience associated with the use of the drug that is both serious and unexpected; or (B) Any finding from tests in laboratory animals that suggests a significant risk for human subjects including reports of mutagenicity, teratogenicity, or carcinogenicity. Each notification shall be made as soon as possible and in no event later than 15 calendar days after the sponsor’s initial receipt of the information. Each written notification may be submitted on FDA Form 3500A or in a narrative format (foreign events may be submitted either on an FDA Form 3500A or, if preferred, on a CIOMS I form; reports from animal or epidemiological studies shall be submitted in a narrative format) and shall bear prominent identification of its contents, i.e., “IND Safety Report.” Each written notification to FDA shall be transmitted to the FDA new drug review division in the Center for Drug Evaluation and Research or the product review division in the Center for Biologics Evaluation and Research that has responsibility for review of the IND. If FDA determines that additional data are needed, the agency may require further data to be submitted.
[The regulations require the sponsor to notify all investigators within 15 days. This may not allow time for the sponsor to analyze the significance of the adverse event. The adverse event report would be of greater value to the clinical investigator and the IRB if it included a judgement as to whether a change in the protocol or the informed consent should be made.]
(ii) In each written IND safety report, the sponsor shall identify all safety reports previously filed with the IND concerning a similar adverse experience, and shall analyze the significance of the adverse experience in light of the previous, similar reports.
[The adverse event reports that sponsors presently send to investigators and IRBs do not include an evaluation or if they do, the evaluation is so general as to be of little use to the IRBs in attempting to determine whether changes in the study or consent should be made.]
National Bioethics Advisory Commission
21 CFR 312.55 Informing Investigators.
(b) The sponsor shall, as the overall investigation proceeds, keep each participating investigator informed of new observations discovered by or reported to the sponsor on the drug, particularly with respect to adverse effects and safe use. Such information may be distributed to investigators by means of periodically revised investigator brochures, reprints or published studies, reports or letters to clinical investigators, or other appropriate means. Important safety information is required to be relayed to investigators in accordance with Sec. 312.32.
[This section requires sponsors to notify all investigators in a multi-site study of all adverse events or “new observations.” New observations is a very general all-inclusive term.]
21 CFR 812.150 Reports (b) Sponsor reports.
(1) Unanticipated adverse device effects. A sponsor who conducts an evaluation of a unanticipated adverse device effect under 21 CFR 812.46(b) shall report the results to FDA and to all reviewing IRBs and participating investigators within 10 working days after the sponsor first receives notice of the effect. Thereafter the sponsor shall submit such additional reports concerning the effect as FDA requests.
Sponsor Reporting to IRBs
21 CFR 812.150 Reports (b) Sponsor reports.
(1) Unanticipated adverse device effects. A sponsor who conducts an evaluation of a unanticipated adverse device effect under 21 CFR 812.46(b) shall report the results to FDA and to all reviewing IRBs and participating investigators within 10 working days after the sponsor first receives notice of the effect. Thereafter the sponsor shall submit such additional reports concerning the effect as FDA requests.
Sponsor Reporting to FDA
21 CFR 312.32 IND safety reports (c) IND safety reports. (1) Written reports - (i) The sponsor shall notify FDA and all participating investigators in a written IND safety report of: (A) Any adverse experience associated with the use of the drug that is both serious and unexpected; or
215
Appendix H: Food and Drug Administration Regulations Requiring Reporting of Adverse Events
(B) Any finding from tests in laboratory animals that suggests a significant risk for human subjects including reports of mutagenicity, teratogenicity, or carcinogenicity. Each notification shall be made as soon as possible and in no event later than 15 calendar days after the sponsor’s initial receipt of the information. Each written notification may be submitted on FDA Form 3500A or in a narrative format (foreign events may be submitted either on an FDA Form 3500A or, if preferred, on a CIOMS I form; reports from animal or epidemiological studies shall be submitted in a narrative format) and shall bear prominent identification of its contents, i.e., “IND Safety Report.” Each written notification to FDA shall be transmitted to the FDA new drug review division in the Center for Drug Evaluation and Research or the product review division in the Center for Biologics Evaluation and Research that has responsibility for review of the IND. If FDA determines that additional data are needed, the agency may require further data to be submitted.
[The regulations require the sponsor to notify all investigators within 15 days. This may not allow time for the sponsor to analyze the significance of the adverse event. The adverse event report would be of greater value to the clinical investigator and the IRB if it included a judgement as to whether a change in the protocol or the informed consent should be made.]
(ii) In each written IND safety report, the sponsor shall identify all safety reports previously filed with the IND concerning a similar adverse experience, and shall analyze the significance of the adverse experience in light of the previous, similar reports.
[The adverse event reports that sponsors presently send to investigators and IRBs do not include an evaluation or if they do, the evaluation is so general as to be of little use to the IRBs in attempting to determine whether changes in the study or consent should be made.]
21 CFR 312.32
IND safety reports (c) IND safety reports.
(2) Telephone and facsimile transmission safety reports. The sponsor shall also notify FDA by telephone or by facsimile transmission of any unexpected fatal or
life-threatening experience associated with the use of the drug as soon as possible but in no event later than 7 calendar days after the sponsor’s initial receipt of the information. Each telephone call or facsimile transmission to FDA shall be transmitted to the FDA new drug review division in the Center for Drug Evaluation and Research or the product review division in the Center for Biologics Evaluation and Research that has responsibility for review of the IND.
[Inconsistency: No requirement to notify each investigator of the 7 day reports, but each investigator must get a copy of the 15 day report. The 7 day report is for reporting more alarming situations.]
21 CFR 312.33 Annual reports.
(b) Summary information. Information obtained during the previous year’s clinical and nonclinical investigations, including: (1) A narrative or tabular summary showing the most frequent and most serious adverse experiences by body system.
(2) A summary of all IND safety reports submitted during the past year.
(3) A list of subjects who died during participation in the investigation, with the cause of death for each subject. (4) A list of subjects who dropped out during the course of the investigation in association with any adverse experience, whether or not thought to be drug related.
21 CFR 812.150 Reports (b) Sponsor reports.
(1) Unanticipated adverse device effects. A sponsor who conducts an evaluation of a unanticipated adverse device effect under 21 CFR 812.46(b) shall report the results to FDA and to all reviewing IRBs and participating investigators within 10 working days after the sponsor first receives notice of the effect. Thereafter the sponsor shall submit such additional reports concerning the effect as FDA requests.
[Sponsors are required to send unanticipated adverse device effects directly to the IRBs, and to the investigators. The 10 working day time frame is inconsistent with the IND requirement of 15 calendar days.]
216
Appendix I
Public Comments on NBAC’s December 19, 2000, Draft
Alaska Division of Public Health (Juneau, AK)
Albany Medical College (Albany, NY)
Alzheimer’s Association (Washington, D.C.)
American Anthropological Association (Arlington, VA)
American Association of Physical Anthropologists (Chapel Hill, NC)
American College of Obstetricians and Gynecologists (Washington, D.C.)
American Geriatrics Society (New York, NY)
American Medical Association (Chicago, IL)
American Medical Directors Association (Columbia, MD)
American Political Science Organization (Washington, D.C.)
American Psychiatric Association (Washington, D.C.)
American Psychological Association (Washington, D.C.)
American Red Cross, Jerome H. Holland Laboratory (Rockville, MD)
American Sociological Association (Washington, D.C.)
American Thoracic Society (New York, NY)
Appalachian State University (Boone, NC)
| Applied Research Ethics National | Michael Birnbaum, California State |
| Association (Boston, MA) | University at Fullerton (Fullerton, CA) |
| Mark H. Ashcraft, Cleveland State | Paula A. Bistak, Newark Beth Israel |
| University Institutional Review Board | Medical Center (Newark, NJ) |
| (Cleveland, OH) | |
| Melissa Bottrell, New York University | |
| Association of American Medical | Division of Nursing (New York, NY) |
| Colleges (Washington, D.C.) | |
| S. Gaylen Bradley, Penn State College | |
| Association of Independent Research | of Medicine (Hershey, PA) |
| Institutes (Westminster, MD) | |
| Mark L. Brenner, Indiana University | |
| John A. Balint, Albany Medical College | Purdue University-Indianapolis |
| (Albany, NY) | (Indianapolis, IN) |
| Deborah R. Barnbaum, Kent State | Bristol-Myers Squibb Pharmaceutical |
| University (Kent, OH) | Research Institute (Princeton, NJ) |
| Jonathan Baron, University of | Kerwin Brook, San Francisco State |
| Pennsylvania (Philadelphia, PA) | University (San Francisco, CA) |
| Paul Batalden, Center for the | Bill Burman, Denver Health Authority |
| Evaluative Clinical Sciences, | (Denver, CO) |
| Dartmouth College (Hanover, NH) | |
| Corrinne Caldwell, Temple University | |
| Lisa Bellantoni, Quincy University | (Philadelphia, PA) |
| (Quincy, IL) | |
| Case Western Reserve University | |
| Daniel O. Benson, “Eye Care” | (Cleveland, OH) |
| (Klamath Falls, OR) | |
| David Cassaratt, University of | |
| Malcolm P. Berger, Armstrong County | Pennsylvania (Philadelphia, PA) |
| Memorial Hospital (Kittanning, PA) | |
| Oswaldo Castro, Victor R. Gordeuk, | |
| Don Berwick, University of | and Fitzroy W. Dawkins, Howard |
| Pennsylvania (Philadelphia, PA) | University (Washington, D.C.) |
| Bethany College (Bethany, WV) | Centers for Disease Control and |
| Prevention (Atlanta, GA) | |
| Biotechnology Industry Organization | |
| (Washington, D.C.) | Center for Regulatory Effectiveness |
| (Washington, D.C.) | |
| Gretchen Birbeck, Michigan State | |
| University (East Lansing, MI) | Chicago State University (Chicago, IL) |
217
Appendix I: Public Comments on NBAC’s December 19, 2000, Draft
Citizens for Responsible Care & Research (New York, NY)
Thomas Coggins, University of South Carolina (Columbia, SC)
Consortium of Social Science Associations (Washington, D.C.)
Consumer Product Safety Commission (Bethesda, MD)
Council of State and Territorial Epidemiologists (CSTE) (Atlanta, GA)
Shan Cretin, RAND Health (Santa Monica, CA)
Richard D. Culbertson (Fort Worth, TX)
Virginia Dato, University of Pittsburgh, Graduate School of Pubic Health (Pittsburgh, PA)
Robyn M. Dawes, Carnegie Mellon University (Pittsburgh, PA)
Linda Degh, Indiana University, Folklore Institute (Bloomington, IN)
Department of Agriculture, Agricultural Research Service (Washington, D.C.)
Department of Commerce, National Institutes of Standards and Technology (Gaithersburg, MD)
Department of Education (Washington, D.C.)
Department of Energy (Germantown, MD)
Department of Veterans Affairs, Office of Research and Development (Washington, D.C.)
Department of Veterans Affairs, Veterans Health Administration National Center for Ethics (Washington, D.C.)
Department of Veterans Affairs, Veterans Health Administration Office of Research Compliance and Assurance (Washington, D.C.)
Evan G. DeRenzo, Washington Hospital Center (Washington, D.C.)
Rebecca Dresser, Washington University (St. Louis, MO)
Nancy Dubler, Montefiore Medical Center (Bronx, NY)
East Carolina University, University and Medical Center Institutional Review Board (Greenville, NC)
The Endocrine Society (Bethesda, MD)
Environmental Protection Agency (Washington, D.C.)
Katherine Ewing and Linda K. George, Duke University (Durham, NC)
Federation of American Societies for Experimental Biology (Bethesda, MD)
Sandra Finestone,
Hoag Memorial Hospital Presbyterian (Newport Beach, CA)
John C. Fletcher, University of Virginia School of Medicine (Charlottesville, VA)
Food and Drug Administration (Rockville, MD)
Elmar H. Frangenberg, Finger Lakes DDSO, MDC Medical Clinic (Rochester, NY)
Edward F. Gabriele (Germantown, MD)
Leonard H. Glantz, George Annas, and Michael Grodin, Boston University School of Public Health (Boston, MA)
Jacquelyn Gnuse, Deaconess College of Nursing (St. Louis, MO)
Goodwyn Institutional Review Board (Cincinnati, OH)
Robert M. Hauser, Wisconsin University (Madison, WI)
Harvard University, Committee on the Use of Human Subjects in Research (Boston, MA)
Health Privacy Project, Institute for Health Care Research, Georgetown University (Washington, D.C.)
Sherri Heitner-Margalit (New York, NY)
218
Hellenic National Bioethics Commission (Athens, Greece)
Shirley Y. Hill, University of Pittsburgh School of Medicine (Pittsburgh, PA)
Søren Holm, University of Manchester (Manchester, United Kingdom)
Frank C. Hood, Battelle Columbus Operations (Columbus, OH)
Human Factors and Ergonomics Society (Santa Monica, CA)
Illinois Department of Public Health (Springfield, IL)
Indiana University at Bloomington (Bloomington, IN)
David Introcaso, Center for Clinical Bioethics at Georgetown University (Washington, D.C.)
Anne Janin, Hopital Saint Louis (Paris, France)
Bruce Jennings, The Hastings Center (Garrison, NY)
Timothy Jones, Tennessee Department of Health (Nashville, TN)
Eric T. Juengst, Case Western Reserve University (Cleveland, OH)
Neil Jumonville, Florida State University (Tallahassee, FL)
Juvenile Diabetes Research Foundation International (New York, NY)
Jonathan Kaplan, U.S. Army Research Institute for Behavioral and Social Sciences (Alexandria, VA)
Jason H. T. Karlawish, University of Pennsylvania (Philadelphia, PA)
Kenneth Kipnis, University of Hawaii at Manoa (Honolulu, HI)
Donald S. Kornfeld, Columbia Presbyterian Medical Center (New York, NY)
Michael Kubovy, University of Virginia (Charlottesville, VA)
David Leder, Crystalion Publishing (Las Vegas, NV)
Lewis and Clark College (Portland, OR)
Fernando Lolas, Pan American
Health Organization, Regional Program on Bioethics (Santiago, Chile)
Aurolyn Luykx, Universidad Mayor de San Simón (Cochabamba, Bolivia)
Joanne Lynn, RAND Center to Improve Care of the Dying (Washington, D.C.)
Howard Mann, Intermountain Health Care (Salt Lake City, UT)
Peter J. Marshall, Cleveland VAMC (Cleveland, OH)
Dennis J. Mazur, Department of Veterans Affairs Medical Center Institutional Review Board (Portland, OR)
A. Thomas McLellan, Treatment Research Institute (Philadelphia, PA)
Elizabeth S. Menkin (San Jose, CA)
Merck & Co., Inc. (Whitehouse Station, NJ)
Minnesota Center for Health Care Ethics (Minneapolis, MN)
Ethel Mitty, New York University Division of Nursing (New York, NY)
Naomi Modeste, Loma Linda University (Loma Linda, CA)
Torin Monahan, Rensselaer Polytechnic Institute (Troy, NY)
Jane Mostoller, Florida State University (Tallahassee, FL)
Roxanne D. Mountford, University of Arizona (Tucson, AZ)
Cynthia F. Mulliken, Pikeville College (Pikeville, KY)
Frances M. Murphy, Office for Under Secretary for Health Veterans Affairs (Washington, D.C.)
National Aeronautics and Space Administration (Washington, D.C.)
National Association of School Psychologists (Bethesda, MD)
National Breast Cancer Coalition (Washington, D.C.)
National Institutes of Health (Bethesda, MD)
National Organization for Rare Disorders, Inc. (New Fairfield, CT)
National Science Foundation (Arlington, VA)
National Society of Genetic Counselors (Wallingford, PA)
Karl M. Nelson, Aultman Health Foundation (Canton, OH)
Robert M. Nelson, The Children’s Hospital of Philadelphia (Philadelphia, PA)
Newark Beth Israel Medical Center Institutional Review Board (Newark, NJ)
North Dakota State University (Fargo, ND)
John Nurnberger, Indiana University School of Medicine (Indianapolis, IN)
John O’Shea
OSF Healthcare (Peoria, IL)
Malcolm I. Page, University Hospital Institutional Review Board (Augusta, GA)
Pediatric Pharmacology Research Network (PPRU) (Rockville, MD)
Pennsylvania State University, College of Medicine, Milton S. Hershey Medical Center (Hershey, PA)
Pharmaceutical Research and Manufacturers of America (Washington, D.C.)
Kenneth W. Phifer, First Unitarian Universalist Church of Ann Arbor, University of Michigan IRBMED (Ann Arbor, MI)
219
National Bioethics Advisory Commission
Rexann G. Pickering, Methodist Healthcare Institutional Review Board (Memphis, TN)
Donald Pinkel
Public Citizen (Washington, D.C.)
David Quadagno, Florida State University (Tallahassee, FL)
Quintiles Transnational Corp. (Research Triangle Park, NC)
Gary E. Raskob, University of Oklahoma Health Sciences Center (Oklahoma City, OH)
Research Foundation for Mental Hygiene, Inc. (Albany, NY)
Robert Richardson, Kaiser-Permanente North West (Portland, OR)
Jennifer L. Rosato, Brooklyn Law School (Brooklyn, NY)
Adam A. Rosenberg, University of Colorado School of Medicine (Denver, CO)
Nancy Sander, Allergy and Asthma Network, Mothers of Asthmatics, Inc. (Fairfax, VA)
Naomi Scheman, University of Minnesota (Minneapolis, MN)
Karen L. Schmidt, University of Pittsburgh (Pittsburgh, PA)
George S. Schuster, Medical College of Georgia Human Assurance Committee (Augusta, GA)
Larry D. Scott, University of Texas at Houston, Health Science Center, Texas Medical Center (Houston, TX)
Jeanne M. Sears, University of California at San Francisco (San Francisco, CA)
Lauren S. Seifert, Malone College (Canton, OH)
Shannon Health System, Human Subjects Committee (San Angelo, TX)
Appendix I: Public Comments on NBAC’s December 19, 2000, Draft
James Shelton, U.S. Agency for International Development (Washington, D.C.)
Richard C. Shelton, Vanderbilt University Medical Center (Nashville, TN)
Linda Shopes, Pennsylvania Historical & Museum Commission (Harrisburg, PA)
Wayne Silverman (Holmdel, NJ)
John Simmons
Social Security Administration (Washington, D.C.)
Southern Polytechnic State University (Marietta, GA)
Southern University at New Orleans (New Orleans, LA)
Stuart Spicker, Massachusetts College of Pharmacy and Health Sciences (Boston, MA)
State University of New York at Fredonia (Fredonia, NY)
Kenneth A. Suarez, Midwestern University (Downers Grove, IL)
Dennis M. Sullivan, Cedarville University (Cedarville, OH)
Syracuse University (Syracuse, NY)
Leon Thal, University of California at San Diego (San Diego, CA)
Cecilia Tiller, Abilene Intercollegiate School of Nursing (Abilene, TX)
James A. Tulsky, VA Medical Center and Duke University (Durham, NC)
University of California (Oakland, CA)
University of California at Santa Barbara (Santa Barbara, CA)
University of California at
San Diego, Human Subjects Program (San Diego, CA)
University of Colorado at Boulder (Boulder, CO)
University of Iowa (Iowa City, IA)
University of Maryland, Baltimore County (Baltimore, MD)
University of Southern California (Los Angeles, CA)
University of Texas at El Paso, Office of Research and Sponsored Projects (El Paso, TX)
University of Texas at Houston, Health Science Center Institutional Review Board (Houston, TX)
Valdosta State University Institutional Review Board (Valdosta, GA)
Stephen Vanderslice, Christus St. Frances Cabrini Hospital Institutional Review Board (Alexandria, LA)
Washington State Department of Health (Olympia, WA)
Stanley Way, Myasthenia Gravis Foundation of America (Rockville, MD)
David Webb, Kutztown University (Kutztown, PA)
Jan Weber, Huron University (Huron, SD)
Charles Weijer, Dalhousie University (Halifax, Canada)
Dorothy C. Wertz, University of Massachusetts Medical School, Eunice Kennedy Shriver Center (Worcester, MA)
Shelia L. White, Office of Clinical Trials Compliance, Medical College of Georgia (Augusta, GA)
Anne M. Wilkinson, RAND (Washington, D.C.)
Tina Wininger, University of Nevada at Las Vegas (Las Vegas, NV)
Wisconsin Preventive Medicine Society (Madison, WI)
Beverly Woodward, Brandeis University (Waltham, MA)
World Health Organization, Communicable Diseases (Geneva, Switzerland)
Shirley J. Wright, University of Dayton (Dayton, OH)
Peter Zilgalvis, Council of Europe (Strasbourg, France)
220
Listed below are those who provided comments after the public comment deadline. These comments were not included in the analysis.
American Academy of Pediatrics (Elk Grove Village, IL)
Marie-Christine Bercot United Nations Educational, Scientific and Cultural Organization Devision of Human Sciences, Philosophy and the Ethics of Science and Technology (Paris, France)
Marisela Vargas Cortes,
Ministry of Health, Epidemiologia (Mexico City, Mexico)
Department of Justice (Washington, D.C.)
Antonio M. Gotto, Cornell University (New York, NY)
Henry Harpending
University of Utah, Department of Anthropology (Salt Lake City, UT)
Melvin M. Mathias, U.S. Department of Agriculture (Washington, D.C.)
John S. Meyer, St. Luke’s Hospital (Chesterfield, MO)
National Association of State Mental Health Program Directors (Alexandria, VA)
Donald Pasquale
Stratton Veterans Affairs Medical Center Institutional Review Board (Albany, NY)
Julie Simpson, University of New Hampshire (Durham, NH)
State University of New York College at Old Westbury (Old Westbury, NY)
Rubye Prigmore Torrey,
Tennessee Technological University (Cookeville, TN)
Virginia State University (Petersburg, VA)
Joe E. Winstead, Southern Arkansas University (Magnolia, AK)
Appendix J
Public and Expert Testimony
October 4, 1996 (Bethesda, Maryland)
Public:
Gwendon Plair, Task Force on Radiation and Human Rights Jeffrey Cossman, College of American Pathologists and Georgetown University Medical Center Charles MacKay Suzanne Thomlinson, Biotechnology Industry Association Acie Byrd, Atomic Veterans Robert McMurrough, Centers for Disease Control and Prevention, HIV/AIDS Prevention and Community Planning
Expert:
John H. Gibbons, Assistant to the President for Science and Technology Aaron Meinkoff, Legislative Assistant, Senator Mark O. Hatfield, U.S. Senate Leonard Weiss, Minority Staff Director, U.S. Senate Committee on Governmental Affairs Francis S. Collins, National Human Genome Research Institute Gary B. Ellis, Office for Protection from Research Risks
November 21, 1996 (San Francisco, California)
Public:
John Cavanaugh O’Keefe, American Life League
December 16, 1996 (Bethesda, Maryland)
Public:
Joan Rachlin, Public Responsibility in Medicine and Research Susan Rose, Department of Energy
Expert:
Gary B. Ellis, Office for Protection from Research Risks J. Thomas Puglisi, Office for Protection from Research Risks Sandy Titus, Food and Drug Administration Allison Wichman, National Institutes of Health Janice Stoklosa, National Aeronautics and Space Administration Amanda Norton, Food and Drug Administration Ronald Wilson, Food and Drug Administration Joan Rachlin, Public Responsibility in Medicine and Research
January 9–10, 1997 (Washington, D.C.)
Public:
Chris Kline, Minority Staff Member, U.S. Senate Committee on Governmental Affairs
February 24, 1997 (Bethesda, Maryland)
Public:
James Shelton, Joint United Nations Programme on HIV/AIDS
Expert:
Charles McKay, National Institutes of Health
April 12–13, 1997 (Arlington, Virginia)
Expert:
Ruth Faden, Advisory Committee on Human Radiation Experiments Helen McGough, Applied Research Ethics National Association
November 23, 1997 (Bethesda, Maryland)
Expert:
Charles R. McCarthy, Georgetown University John C. Fletcher, University of Virginia
Joan Porter, Presidential Advisory Committee on Gulf War Veterans Illnesses
December 9, 1997 (Arlington, Virginia)
Public:
Mark Sobel, National Cancer Institute
Expert:
Debra Saslow, National Action Plan for Breast Cancer
January 6–8, 1998 (Arlington, Virginia)
Expert:
John C. Fletcher, University of Virginia C.K. Gunsalus, University of Illinois
Joan Porter, Office of Science and Technology Policy Gary B. Ellis, Office for Protection from Research Risks
February 5–6, 1998 (Los Angeles, California)
Public:
Art Ablin, University of California, San Francisco Robert Aller, Patients Rights Network Con Hopper, University of California System
Expert:
Rachel Levinson, Office of Science and Technology Policy
221
Appendix J: Public and Expert Testimony
May 19–20, 1998 (Cleveland, Ohio)
Public:
Tillman Bauknight
July 14–15, 1998 (Portland, Oregon)
Public:
Karen Hansen, Public Responsibility in Medicine and Research
Expert:
Albert R. Jonsen, University of Washington
January 19–20, 1999 (Miami, Florida)
Public:
John Price
May 11–12, 1999 (Northbrook, Illinois)
Public:
Peggy Connelly
June 28–29, 1999 (Washington, D.C.)
Expert:
Roger Cortesi, Environmental Protection Agency Timothy Gerrity, Department of Veterans Affairs
Barbara C. Levin, National Institute on Standards and Technology Beth McCormick, National Aeronautics and Space Administration Stuart Plattner, National Science Foundation Blanca Rosa Rodriguez, Department of Education James D. Shelton, U.S. Agency for International Development Lana Skirboll, National Institutes of Health Marjorie Speers, Centers for Disease Control and Prevention Nancy Dubler, Albert Einstein College of Medicine Renee M. Landers, Ropes & Gray Jonathan D. Moreno, University of Virginia
October 21–22, 1999 (Washington, D.C.)
Expert:
Neal F. Lane, Assistant to the President for Science and Technology Policy
December 2–3, 1999 (Baltimore, Maryland)
Expert:
Jonathan D. Moreno, University of Virginia
January 13–14, 2000 (Washington, D.C.)
Expert:
Robert J. Levine, Yale University School of Medicine
Dixie E. Snider, Centers for Disease Control and Prevention Duane Alexander, National Institute of Child Health and Human Development Paul W. Goebel, Jr., Food and Drug Administration Douglas Forcino, Department of Defense Ed Lane, Department of Defense Helene Deramond, Department of Education Peter Wathen-Dunn, Department of Education James Burris, Department of Veterans Affairs Joan Porter, Department of Veterans Affairs Ellen Fox, Department of Veterans Affairs
February 29–March 1, 2000 (Herndon, Virginia)
Expert:
Claudia Mickelson, Recombination DNA Advisory Committee Lana Skirboll, National Institutes of Health Kathryn C. Zoon, Food and Drug Administration Michele Russell-Einhorn, Office for Protection from Research Risks J. Thomas Puglisi, Office for Protection from Research Risks Gary B. Ellis, Office for Protection from Research Risks Diane Flack, Nuclear Regulatory Commission Jane Ley, Office of Government Ethics Andrew Nelson, Health Partners and HMO Research Network Mary Durham, Kaiser Foundation Hospitals Paul T. Kim, Counsel, Congressman Henry A. Waxman, U.S. House of Representatives Souheila Al-Jadda, Legislative Aide, Congressman Dennis J.Kucinich, U.S. House of Representatives
April 6–7, 2000 (Washington, D.C.)
Expert:
Shirley Fry, Oak Ridge Associate Universities, Oak Ridge National Laboratory IRB
John Eisenberg, Agency for Health Care and Research Quality Norman M. Bradburn, National Science Foundation Murray Wax, Washington University John M. Abowd, Cornell University Joan E. Sieber, California State University, Hayward Linda Shopes, Organization of American Historians and American Historical Society Bert A. Spilker, Pharmaceutical Research and Manufacturers of America R. Sebastian Wanless, Bristol-Myers Squibb Company Rose G. Snipes, Glaxo Wellcome, Inc.
Bernice R. Welles, Genentech, Inc. Thomas W. McKenna, Westat James G. Ross, Macro International, Inc. Inderjit Kaul, Abt Associates, Inc. Anne Coletti, Abt Associates, Inc.
May 4–5, 2000 (Madison, Wisconsin)
Expert:
Bernard M. Dickens, University of Toronto
June 5–6, 2000 (San Francisco, California)
Expert:
Jeffrey Kahn, University of Minnesota
Richard Schreck, General Motors Corporation Henry B. Chin, National Food Processors Association Donald Chalmers, University of Tasmania Erica Heath, Independent Review Consulting, Inc. Harold Y. Vanderpool, University of Texas Medical Branch at Galveston Jonathan D. Moreno, University of Virginia Donald Magnus, University of Pennsylvania
222
July 10–11, 2000 (Bethesda, Maryland)
Public:
Jim Tozzi, Center for Regulatory Effectiveness
Expert:
Vincent T. Francisco, University of Kansas Edison J. Trickett, University of Maryland Timmeca Wilson Susan May Linda Smith Kenneth Kipnis, University of Hawaii at Manoa
Ernest D. Prentice, University of Nebraska Medical Center Daniel K. Nelson, University of North Carolina at Chapel Hill Moira A. Keene, University of Minnesota Health Center E. Ray Stinson, Wayne State University Robert M. Nelson, The Children’s Hospital of Philadelphia Sharon B. Murphy, Northwestern University School of Medicine William Burman, Denver Department of Public Health Monica M. Farley, Emory University School of Medicine, Atlanta VA Medical Center Samuel A. Wells, Jr., American College of Surgeons
September 12–13, 2000 (Washington, D.C.)
Public:
Lee Zwanziger, Institute of Medicine, National Academy of Sciences
Expert:
Joan E. Sieber, California State University, Hayward Janlori Goldman, Georgetown University Greg Koski, Office for Human Research Protections David Lepay, Food and Drug Administration Daniel P. Schuster, Washington University in St. Louis Steven Peckman, University of California, Los Angeles Søren Holm, University of Manchester, England Michael S. Hamm, Michael Hamm & Associates
National Bioethics Advisory Commission
October 24–25, 2000 (Salt Lake City, Utah)
Public:
Howard Mann, Intermountain Health Care
Colin Thomson, Australian Health Ethics Committee Kateri Harnetiaux
December 7–8, 2000 (Washington, D.C.)
Public:
Vera Hassner Sharav, Citizens for Responsible Care in Research
January 18–19, 2001 (Washington, D.C.)
Public:
Kate Louise Gottfried, National Human Research Protections Advisory Committee Mary Faith Marshall, National Human Research Protections Advisory Committee
March 15–16, 2001 (Atlanta, Georgia)
Public:
Erica Frank, Emory University School of Medicine
223
Appendix K
Commissioned Papers and Staff Analysis
Research Ethics in Australia
Donald Chalmers
University of Tasmania
Location of the Office for Protection from Research Risks Within the National Institutes of Health: Problems of Status and Independent Authority
John C. Fletcher
University of Virginia
Privacy and Confidentiality in Health Research
Janlori Goldman and Angela Choy
Georgetown University
An Examination of Issues Presented by Proposals to Unify and Expand Federal Oversight of Human Subject Research
C.K. Gunsalus
University of Illinois at Urbana-Champaign
The History, Function, and Future of Independent Institutional Review Boards
Erica Heath
Independent Review Consulting, Inc.
The Danish Research Ethics Committee System—Overview and Critical Assessment
Søren Holm
University of Manchester
Vulnerability in Research Subjects: A Bioethical Taxonomy
Kenneth Kipnis
University of Hawaii at Manoa
Reflections on the Organizational Locus of the Office for Protection from Research Risks
Charles R. McCarthy
225
Appendix K: Commissioned Papers and Staff Analysis
Protectionism in Research Involving Human Subjects
Jonathan D. Moreno
University of Virginia
Federal Agency Survey on Policies and Procedures for the Protection of Human Subjects in Research
National Bioethics Advisory Commission
Local Institutional Review Boards
Steven Peckman
University of California-Los Angeles
Institutional Review Board Assessment of Risks and Benefits Associated with Research
Ernest D. Prentice and Bruce G. Gordon
University of Nebraska Medical Center
Oversight of Human Subject Research: The Role of the States
Jack Schwartz
Office of the Maryland Attorney General
Privacy and Confidentiality: As Related to Human Research in Social and Behavioral Science
Joan E. Sieber
California State University, Hayward
Unfulfilled Promise: How the Belmont Report Can Amend the Code of Federal Regulations Title 45 Part 46—Protection of Human Subjects
Harold Y. Vanderpool
University of Texas Medical Branch, Galveston
The Ethical Analysis of Risks and Potential Benefits in Human Subjects Research: History, Theory, and Implications for U.S. Regulation
Charles Weijer
Dalhousie University
Protecting Communities in Research: Philosophical and Pragmatic Challenges
Charles Weijer
Dalhousie University
226
Index
A
AAALAC. See Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)
AAAS. See American Association for the Advancement of Science (AAAS)
AAHRPP. See Association for the Accreditation of Human Research Protection Programs (AAHRPP)
AAMC. See Association of American Medical Colleges (AAMC)
Accountability public, of human participants research, oversight system proposal for, xii, 26
Accreditation organizations, for human participants research, oversight responsibilities of, 27 research ethics competence of institutions/IRBs, measures for demonstrating, vii, xiv, 49–50
ACHRE. See Advisory Committee on Human Radiation Experiments (ACHRE)
ADAMHA. See Alcohol, Drug Abuse and Mental Health Administration (ADAMHA)
Adaptability human participants research and, oversight issues, 11–12
Administration for Children and Families, 11, 132
See also Department of Health and Human Services (DHHS)
Administration on Aging, 11
See also Department of Health and Human Services (DHHS)
Adverse reactions research, FDA reporting requirements for, 113–114, 115, 116 research, FDA reporting requirements for, regulatory text, 213–216 research, reporting requirements for local institutions/IRBs, xix–xx, 113–114, 115–117 research, review requirements for local institutions/IRBs, 114, 115–116 See also Drugs and drug testing
Advisory Committee on Human Radiation Experiments (ACHRE), 45, 46, 99, 124, 156
Final Report, 124 human participants research, recommendations of, 158
Advocacy organizations, for human participants research, oversight responsibilities of, 27
Africa vaccine death rates in, 85
African Americans
Tuskegee Syphilis Study, 46, 153, 164
Agency for Healthcare Research and Quality, 11, 132
See also Department of Health and Human Services (DHHS)
Agency for International Development (USAID), 11, 132, 205 HIV/AIDS and Pre-Adolescent Awareness Programs in Africa, 5 human participants research, regulations of, 157
Alabama. See Macon County, Alabama; Tuskegee, Alabama; University of Alabama at Birmingham
Alcohol, Drug Abuse and Mental Health Administration (ADAMHA), 155
AMA. See American Medical Association (AMA)
American Association for the Advancement of Science (AAAS), 47 American Medical Association (AMA), 151 American Psychological Association, 33 Applied Research Ethics National Association, 15, 33 Armed Forces Epidemiological Board, 153
Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC), 49
Association for the Accreditation of Human Research Protection Programs (AAHRPP), 49
Association of American Medical Colleges (AAMC), 15, 46, 60 Association of American Universities, 15 Association of American University Professors, 13
Assurances of compliance federal agencies practices regarding, listing by agency, 51 research standards compliance by institutions/IRBs, mechanisms for, xiv, 50–51, 52 See also Compliance; Cooperative Project Assurance (CPA); Federalwide Assurance (FWA); Multiple Project Assurances (MPAs); Single Project Assurances (SPAs)
Audits institutional, for research standards compliance of institutions/IRBs, xiv, 53
Auschwitz, Germany interview, face-to-face, risks/potential benefits analysis example, 81
Australia human participants research, terminology for, 33 research-related injuries, compensation for, 123
Autonomy human participants research and, ethical principles, 162
227
Index
B
Backlar, Patricia, 33
Balanced Budget Act of 1997, 4
Beecher, Henry, 153
Behavior psychological experiment on, risk/potential benefits analysis, 82 state law effects on, study of, risks/potential benefits analysis, 81
Behavioral research prisoners as subjects, protections for, 45 CFR 46, Subpart C, in entirety, 182–184
See also Research
Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research, ii, v, 35, 69, 97, 98, 154 applications of principles, 164–167 beneficence, relevance for, 162–163, 165–167 discussion of principles, 162–164 informed consent for, considerations, 164–165 justice, relevance for, 163–164, 167 research subject selection for, considerations, 167 research v. practice, distinction between, 161–162 respect for persons, relevance for, 162, 164–165 risks/benefits assessment for, considerations, 165–167 text, in entirety, 161–167 See also National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission)
Beneficence, 71
See also Ethics
Benefits research, potential, from Belmont Report, 162–163, 165–167 research, potential, to others, 74 research, potential, to participants, 73–74 research, potential, to society, 73 research, potential, types, 73–74 research, potential, v. risks, analysis in IRB review process, ii–iv, 79 research, potential, v. risks, analysis of, new technologies effects on, 7 research, potential, v. risks, analysis of, xvi–xvii, 69–85, 93 research, potential, v. risks, assessment of, ethical guidelines for, 165–167 research, potential, v. risks, balance between, assessment procedure, 78–80 research, potential, v. risks, component-based framework for analysis, 76–77, 78, 81–82 research, potential, v. risks, historical perspective, 75–76 research, potential, v. risks, relationship, assessment of, 74–75
Bernard, Claude, 163
Bioethics Commission. See National Bioethics Advisory Commission (NBAC)
Biomedical research prisoners as subjects, protections for, 45 CFR 46, Subpart C, in entirety, 182–184
See also Research
Brazil compensation for research-related injuries, 123
Brooklyn Jewish Chronic Disease Hospital, 152
Bureau of Census, 5, 10, 39 Disclosure Review Board, 10 Policy Office, 10
C
California, 3
See also Los Angeles, California; University of California, Irvine; University of California, Los Angeles; University of California, San Diego; University of California, San Francisco
Capron, Alexander M., 33
Cassell, Eric J., 33
CCIP. See Council for Certification of IRB Professionals (CCIP) CDC. See Centers for Disease Control and Prevention (CDC) Census Bureau. See Bureau of Census
Center for Molecular Medicine and Immunology research compliance investigations resulting in restrictions/actions, 55
Centers for Disease Control and Prevention (CDC), 5, 11, 36, 39, 105, 107, 132 research compliance investigations resulting in restrictions/actions, 55 See also Department of Health and Human Services (DHHS)
Central Intelligence Agency (CIA), 9, 11, 85, 132, 156
Certificate of Confidentiality, 106, 107
Certification organizations, for human participants research, oversight responsibilities of, 27 research ethics competence of individuals, measures for demonstrating, vii, xiv, 48–49
21 CFR 50. See Code of Federal Regulations, Title 21, Part 50 (21 CFR 50)
21 CFR 56. See Code of Federal Regulations, Title 21, Part 56 (21 CFR 56)
45 CFR 46. See Code of Federal Regulations, Title 45, Part 46 (45 CFR 46)
Chicago, Illinois. See University of Illinois at Chicago
Children research involving human participants, possible exemptions from oversight, 39 research participants with vulnerabilities targeted, listing by sponsoring agency, 86 research use of, protections for, 45 CFR 46, Subpart D, in entirety, 184–186
228
Children’s Research Act of 2000, 156
China human research participants protection, regulations of, 157 research-related injuries, compensation for, 123
CIA. See Central Intelligence Agency (CIA)
CIOMS. See Council for International Organizations of Medical Sciences (CIOMS)
City University of New York research compliance investigations resulting in restrictions/actions, 55
Clinical trials drug efficacy, risks/potential benefits analysis example, 82 vaccine safety/immunogenicity, risk/potential benefits analysis example, 82
See also Research
Cloning Human Beings, vii
Code of Federal Regulations, Title 21, Part 50 (21 CFR 50), 28, 98, 100 human research participants protection, agency regulations, statutory authority for, 205–208 human research participants protection, regulations of, 157 Subpart A: General Provisions, for human subjects protection, in entirety, 187–189 Subpart B: Informed Consent of Human Subjects, in entirety, 189–195 See also Federal regulation
Code of Federal Regulations, Title 21, Part 56 (21 CFR 56), 28, 53, 87, 98 human research participants protection, regulations of, 157 Subpart A: General Provisions, for IRBs, in entirety, 195–198 Subpart B: Organization and Personnel, for IRBs, in entirety, 198 Subpart C: IRB Functions and Operations, in entirety, 198–201 Subpart D: Records and Reports, for IRBs, in entirety, 201 Subpart E: Administrative Actions for Noncompliance, for IRBs, in entirety, 201–203 See also Federal regulation
Code of Federal Regulations, Title 45, Part 46 (45 CFR 46), 2, 11, 28, 35, 87, 98, 100 human research participants protection, Common Rule and agency-specific regulations, 157 Subpart A: Federal Policy for Protection of Human Subjects, in entirety, 169–179 Subpart B: Additional DHHS Protections Pertaining to Research, Development, and Related Activities Involving Fetuses, Pregnant Women, and Human In Vitro Fertilization, in entirety, 179–182 Subpart C: Additional DHHS Protections Pertaining to Biomedical and Behavioral Research Involving Prisoners as Subjects, in entirety, 182–184 Subpart D: Additional DHHS Protections for Children Involved as Subjects in Research, in entirety, 184–186 See also Federal regulation
National Bioethics Advisory Commission
Coercion human participants research, requirements for, 45 CFR 46, Subpart A, 176–178 voluntariness of, ethical guidelines from Belmont Report, 165 vulnerability in human participants research, definition of, NBAC recommendation, xvii, 92 vulnerable individuals protection in human participants research and, iv–v, xvii, 9, 11, 85–93
Cognitive ability psychological experiment on, risk/potential benefits analysis example, 82 vulnerability, capacity- and situation-related, in research participants, ethical considerations, 88
Colleges. See Universities and colleges
Comments. See Public comments
Commission. See National Bioethics Advisory Commission (NBAC)
Commissioned papers. See Papers commissioned
Common Rule. See Code of Federal Regulations, Title 45, Part 46 (45 CFR 46); Federal Policy for the Protection of Human Subjects (Federal Policy)
Communication vulnerability, in research participants, ethical considerations, 88
Communities research involving human participants, consideration of, 7–8
Compensating for Research Injuries, 124
See also President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission)
Compensation research-related harm and, current regulatory requirements, 123–124 research-related harm and, current situation, 125 research-related harm and, NBAC recommendations, xx, 126 research-related harm and, need for, v, 125–126 research-related harm and, previous recommendations for program, 124–125 research-related harm and, role of local institutions, xx, 123–126
Competence research ethics, accreditation for institutions/IRBs, vii, xiv,
49–50 research ethics, certification of individuals, vii, xiv, 48–49 research ethics, measures for demonstrating, xiv, 48–50
Compliance research oversight enforcement weaknesses, 12–13 research oversight investigations resulting in restrictions/actions, listing by institution, 54–56 research standards and, by institutions/IRBs/investigators, mechanisms for ensuring, xiv, 50–58 research standards and, institutional audits for, xiv, 53 research standards and, site inspections by FDA for, xiv, 52–53
229
Index
research standards and, site inspections by other than FDA for, xiv, 53 See also Assurances of compliance; Cooperative Project Assurance (CPA); Federalwide Assurance (FWA); Multiple Project Assurances (MPAs); Single Project Assurances (SPAs)
Comprehensive Alcohol Abuse and Alcoholism Prevention, Treatment, Rehabilitation Act of 1970, 207
Confidentiality definition for human participants research, proposed by NBAC, 107 research involving human participants, current regulations on, 104–105 research involving human participants, issues for, xviii, 106–107 research risk/potential benefit analysis, new technologies effects on, 7
See also Privacy
Conflict of interest
IRB as institutional body and, protection of research participants, xv, 61–63, 64
IRB composition issues for, protection of research participants,
63 –64
IRB membership and, protection of research participants, xv–xvi, 60–61, 63–64 research institutions/investigators/IRBs and, managing, NBAC recommendations, xv–xvi, 64 research investigators/IRBs and protection of human participants, measures for managing, xiv–xv, 58–60
See also Ethics
Congress, U.S. See U.S. Congress
Consent. See Informed consent
Consent forms. See Documentation; Informed consent
Consent process. See Informed consent
Consumer Product Safety Commission, 11, 132, 205 human participants research, regulations of, 157
Contract research organizations (CROs), 6, 23, 25
Cook County Hospital Hektoen Institute for Medical Research research compliance investigations resulting in restrictions/actions, 55
Cooperative Project Assurance (CPA), 52
See also Assurances of compliance; Compliance; Office for Human Research Protections (OHRP)
Cooperative research human participants research, and IRB multi-site research studies, 14 range of, summary, 117 review of, by IRBs, foreign models of, 121–122 review of, by local IRBs, considerations, 118–122 review of, current requirements and problems, xx, 118
See also Research
Cornell University Medical Center research compliance investigations resulting in restrictions/actions, 55
Council for Certification of IRB Professionals (CCIP), 48
Council for International Organizations of Medical Sciences (CIOMS), 123, 215, 216
Council of Europe, 123
CPA. See Cooperative Project Assurance (CPA) CROs. See Contract research organizations (CROs) Current Population Survey, 1988, 81
D
Data existing, research risks/potential benefits analysis of, example, 81
Data and Safety Monitoring Board (DSMB), 25, 54, 134 research adverse events reporting requirements for, xix–xx, 113, 115–116, 117
Death research involving deceased individuals, possible exemptions from oversight, 39–40
Deception research informed consent requirements for human participants in, 101
Decisionmaking capacity mentally impaired research participants and, informed consent and limitations on, 101
Denmark cooperative research, IRB review models in, 121
Department of Commerce, 11, 132, 205 human participants research, regulations of, 157
Department of Defense (DOD), 5, 11, 132, 190, 191, 192, 205 human participants research, regulations of, 157 Secretary, 151 Top Secret policy, 151–152
Department of Education, 9, 11, 39, 85, 104, 107, 132 human participants research, regulations of, 157 human participants research, regulations of, statutory authority for, 205, 206, 208–209 Secretary, 206 site inspections to ensure compliance with research standards, 53
Department of Energy (DOE), 5, 11, 12, 132 human participants research, regulations of, 157 human participants research, regulations of, statutory authority for, 205, 206 Secretary, 206 site inspections to ensure compliance with research standards, 53 See also Nuclear Regulatory Commission (NRC)
Department of Health and Human Services (DHHS), vi, xiv, 2, 4, 8, 9, 10, 11, 12, 15, 28, 29, 30, 31–32, 37, 39, 50, 51, 55, 56, 71, 75, 76, 83, 85, 87, 88, 91, 105, 106, 116, 124, 131, 132, 152, 154, 155, 156 children as research subjects, protections for, 45 CFR 46, Subpart D, in entirety, 184–186 Ethics Advisory Board, 155
230
human participants research, 45 CFR 46, Subpart A, in entirety, 169–179 human participants research, regulations of, 157 human participants research, regulations of, statutory authority for, 205, 207–208, 209 Office of Inspector General (OIG), 8, 57, 112, 116, 131 Office of the Secretary, 15, 30 pregnant women, fetuses, and in vitro fertilization in research, protections for, 45 CFR 46, Subpart B, in entirety, 179–182 prisoners in biomedical/behavioral research, protections for, 45 CFR 46, Subpart C, in entirety, 182–184 Secretary, 152, 154, 155, 179, 180, 182, 183, 185, 207, 208 “Standards for Privacy of Individually Identifiable Health Information,” 105 See also Administration for Children and Families; Administration on Aging; Agency for Healthcare Research and Quality; Centers for Disease Control and Prevention (CDC); Department of Health, Education, and Welfare (DHEW), Food and Drug Administration (FDA); Health Care Financing Administration; Health Resources and Services Administration (HRSA); Indian Health Service; National Institutes of Health (NIH); Substance Abuse and Mental Health Services Administration Department of Health, Education, and Welfare (DHEW), 124, 153, 154 Ethics Advisory Board, 124 Medical Malpractice Commission, 124 Secretary, 124 Secretary’s Task Force on the Compensation of Injured Research Subjects, 124 “Yellow Book,” 153 See also Department of Health and Human Services (DHHS)
Department of Housing and Urban Development, 11, 132 human participants research, regulations of, 157 human participants research, regulations of, statutory authority for, 205, 206 Secretary, 206
Department of Justice, 9, 11, 39, 85, 105, 132 Attorney General, 206 Bureau of Prisons, 9, 208 Federal Prison Industries, Inc., 206 human participants research, regulations of, 157 human participants research, regulations of, statutory authority for, 205, 206, 208
Department of Labor, 171
Department of Transportation, 11, 85, 132, 205
Federal Highway Administration, Human Factors Laboratory, 5 human participants research, regulations of, 157 U.S. Coast Guard, 5
Department of Veterans Affairs (VA), 5, 10, 11, 16, 49, 126, 132 Comprehensive Research Integrity Program, 53 human participants research, regulations of, 157 human participants research, regulations of, statutory authority for, 205, 207, 209
National Bioethics Advisory Commission
Multi-Assessment Program, 53
Office of Research Compliance and Assurance, 53 Secretary, 207 site inspections to ensure compliance with research standards, 53 Special Inquiry Force Teams, 53
DHEW. See Department of Health, Education, and Welfare (DHEW)
DHHS. See Department of Health and Human Services (DHHS)
Disclosure of information conflict of interest for investigators/IRBs, measures for managing, 59–60 human participants research and, ethical guidelines from
Belmont Report, 164–165 human participants research, requirements for, emphasis on, issues, iv, xvii, 98–100 human participants research, requirements for, listing, 99 See also Information; Informed consent
Diseases and disorders illness, serious, as vulnerability in research participants, ethical considerations, 89–90 See also Mental diseases and disorders; Syphilis
Documentation informed consent requirements for human participants research, discussion of, xviii, 103–104
DOD. See Department of Defense (DOD)
DOE. See Department of Energy (DOE)
Drug Abuse Office and Treatment Act of 1972, 207
Drugs and drug testing efficacy of, clinical trial on, risks/potential benefits analysis example, 82 vaccine, clinical trial on, risk/potential benefits analysis example, 82 research adverse events, FDA reporting requirements for, 113–114, 115, 116 See also Adverse reactions; Clinical trials; Vaccines and vaccination
DSMB. See Data and Safety Monitoring Board (DSMB)
Duke University, 133 research compliance investigations resulting in restrictions/actions, 56
Medical Center, research compliance investigations resulting in restrictions/actions, 56
E
Economics. See Investment; Personal finance
Edgar, H., 9
Editors scientific, oversight system proposed for human participants research, responsibilities of, 27
Education disadvantaged persons targeted for research, listing by sponsoring agency, 86
231
Index
research involving human participants, and IRB needs, 15 research involving human participants, measures needed for, vi–vii, xiii, 45–48 research involving human participants, oversight system proposal for, xii, 23
Eisenberg, J.M., 36
Ellis, Gary, 119
Embryos and embryonic materials research involving human participants, possible exemptions from oversight, 39 See also Fetus and fetal tissue; Pregnancy
Enforcement research involving human participants, protection of, oversight system proposal for, xii, 25–26 research involving human participants, protection of, oversight weaknesses, 12–13
Environmental Protection Agency (EPA), 5, 11, 132, 170, 197, 205 human participants research, regulations of, 157
EPA. See Environmental Protection Agency (EPA)
Equipoise research, explanation of, 77, 78 See also Clinical trials
Equivalent protections. See Protections for human participants
Ethical Advisory Board, 182 research involving pregnant women, fetuses, and in vitro fertilization, requirements for, 45 CFR 46, Subpart B, 180
Ethical and Policy Issues in International Research: Clinical Trials in Developing Countries, vii
Ethics accreditation of competence of research institutions/IRBs for, xiv, 49–50 benefits, potential, of research to others, 74 benefits, potential, of research to participants, 73–74 benefits, potential, of research to society, 73 benefits, potential, of research, types, 73–74 certification of competence of research individuals for, xiv, 46,
48–49 competence for research, measures for demonstrating, xiv,
48–50 compliance with research standards by institutions/IRBs/ investigators, mechanisms for ensuring, xiv, 50–58 research equipoise, explanation, 77, 78 risks of harms from research to nonparticipants, 72–73 risks of harms from research, types of, 71–73 risks, research, minimizing, 73 risks, research, v. potential benefits, analysis of, xvi–xvii,
69–85, 93 risks, research, v. potential benefits, balance between, assessment procedure, 78–80 risks, research, v. potential benefits, component-based framework for analysis, 76–77, 78, 81–82 risks, research, v. potential benefits, historical perspective,
75 –76
risks, research, v. potential benefits, relationship, assessment of,
74 –75
See also Beneficence; Conflict of interest; Ethics review and review committees; Justice; Protections for human participants; Respect for persons Ethics review and review committees adverse events and, local institutions/IRBs responsibilities, 114, 115–116 conflict of interest and IRB institutional role, measures for managing, xv, 61–63, 64 conflict of interest and IRB membership/composition, xv–xvi,
60–61, 63–64 continuing for ongoing research, local institutions responsibilities, xix, 111–112, 117 cooperative research, by IRBs, foreign models of, 121–122 cooperative research, by local IRBs, alternatives to, 120–122 cooperative research, by local IRBs, considerations, 118–120 cooperative research, current requirements and problems, xx, 118 cooperative research, role of local IRBs/institutions, xx, 122 human participants research, and IRB multi-site research studies, 14 human participants research, IRB burdens, 13–15 human participants research, oversight system proposal, xii,
23–24, 28, 30 independent IRBs, emergence of, 6 levels of, oversight system proposal for, vii–viii, xiii, 41–42 risk, minimal, research classification, use as sorting mechanism for, 84 risks v. potential benefits of human participants research, analysis in, ii–iv, 79 risks v. potential benefits, analysis in IRB process, 79 risks v. potential benefits, balance between, assessment procedure, 78–80 risks v. potential benefits, component-based framework for analysis, 76–77, 78, 81–82 risks v. potential benefits, component-based framework for analysis, examples, 81–82 See also Ethics; Evaluation; Institutional Review Boards (IRBs) Evaluation health services research and, determining oversight coverage,
36 –37
See also Ethics review and review committees Expert testimony. See Testimony and speakers
F
Family Education Rights and Privacy Act of 1974, 104
FDA. See Food and Drug Administration (FDA)
Federal Coordinators Council for Science, Engineering, and Technology, 154
Federal departments and agencies assurances of compliance, practices regarding, listing by agency, 51 human participants research, Common Rule and agency-specific regulations, 157
232
human participants research, inconsistencies among, 9, 10 human participants research, independent oversight office proposal for, vi, xii, 28, 30–32 human participants research, percent exempt, listing by agency, 11 human participants research, regulations of, statutory authority for, 205–211 human participants research, support for FY 1999, listing by agency, 132 human participants research, supported by, size and scope of, 5 vulnerability and human participants targeted in research, listing by agency/vulnerability, 86 See also Federal government; Federal regulation; specific departments and agencies
Federal Food, Drug, and Cosmetic Act, 29, 187, 190, 195 Kefauver-Harris amendments, 152, 153
Federal funding for research oversight system for, resources for, viii, xx–xxi, 131, 133 research investment trends, changes in, 4 research involving human participants supported by, size and scope of, 5 research involving human participants, resources for, xx–xxi, 131, 133 research involving human participants, support for FY 1999, listing by agency, 132 See also Federal government; Research funding
Federal government human research participants protection, activities of, 156 See also Federal departments and agencies; Federal funding for research; Federal legislation; Federal regulation; Public policy; U.S. Congress
Federal legislation human research participants protection, agency regulations and, statutory authority for, 205–211 See also Federal government; Federal regulation; specific legislation; U.S. Congress
Federal oversight. See Federal regulation; Oversight
Federal policy. See Federal government; Federal regulation; Public policy
Federal Policy for the Protection of Human Subjects (Common Rule), 2, 3, 5, 9, 11–12, 24, 26, 28, 29, 31, 35, 38, 51, 52, 70–71, 76, 113, 118, 135, 155–156 Common Rule definition, 154 Common Rule signatories, 159 human participants research, Common Rule and agency-specific regulations, 157 human participants research, inconsistencies among federal agencies, 10 See also Code of Federal Regulations, Title 45, Part 46 (45 CFR 46)
Federal Register, 10, 39, 154, 170, 174, 179, 182, 183, 184, 191, 199, 202, 205, 206
National Bioethics Advisory Commission
Federal regulation
accreditation of competence of research institutions/IRBs, vii, xiv, 49–50 adverse research events, FDA reporting requirements, 113–114, 115, 116, 213–216 adverse research events, reporting requirements for local institutions, xix–xx, 113–117 certification of competence of research individuals, vii, xiv,
48–49 compliance with research standards by institutions/IRBs/ investigators, mechanisms for ensuring, xiv, 50–58 conflict of interest for research IRBs, measures for managing,
60–63 cooperative research review, current requirements and problems, xx, 118 cooperative research review, requirements for IRBs, 122 education on human participants protection, measures needed for, vi–vii, xiii, 45–48 educational programs for human participants research, oversight system proposal for, xii, 23 human participants research, Common Rule and agency-specific regulations, 157 human participants research, consequences of flawed system, 16 human participants research, current oversight system, challenges for, 8–16 human participants research, current protection of, 29 human participants research, defining for oversight coverage determination, xii–xiii, 34–42 human participants research, description of, 154, 155–156, 157 human participants research, enforcement weaknesses, 12–13 human participants research, ethical issues for, future research on, xxi, 134 human participants research, history of, 151–154 human participants research, inconsistencies among agencies, 9, 10 human participants research, IRB burdens in, 13–15 human participants research, lack of adaptability in, 11–12 human participants research, limited scope of, 12 human participants research, oversight independent office proposed for, vi, xii, 28, 30–32 human participants research, oversight possible exemptions,
37–41 human participants research, oversight system proposal for, xi–xiii, 21–42 human participants research, oversight system for, purpose of, i–ii, xi, 2–4, 21–22 human participants research, oversight system for, resources for, viii, xx–xxi, 131, 133 human participants research, oversight system proposal for enforcement of, xii, 25–26 human participants research, oversight system proposal for ethical review of research, xii, 23–24 human participants research, oversight system proposal for monitoring, xii, 24–25 human participants research, oversight system proposal for National Office for Human Research Oversight for, xii, 28,
30 –32
233
Index
human participants research, oversight system proposal for policy development through, xii, 23 human participants research, oversight system proposal for public accountability, xii, 26 human participants research, oversight system proposal for unified/comprehensive policy for, v–vi, xii, 28, 30–32 human participants research, oversight system proposal for, as dynamic/responsive structure, 134–135 human participants research, oversight system proposal for, scope and structure, xii, 26–32 human participants research, oversight system proposed for, functions and responsibilities of, xi–xii, 23–27 human participants research, percent exempt from, listing by agency, 11 human participants research, procedural v. ethical emphasis, 13 human participants research, protection for, ACHRE recommendations, 158 human participants research, recent government interest in, 156 human participants research, recent initiatives for, 15–16 human participants research, report implications for IRBs/ investigators/institutions, 136–137 human participants research, report implications for participants, 137 human participants research, report recommendations for, general themes in, 135 informed consent requirements for research participants, deficiencies in, 98 informed consent requirements for research participants, scope/structure of, iv, xvii, 97–104 privacy and confidentiality protection for research participants, 104–105 research approval by IRBs, current regulations for, 69–71 research protocol changes and unanticipated problems, local institutions responsibilities, xix, 112–113, 117 research, ongoing, monitoring responsibilities of local institutions, xix, 117 research-related injuries, compensation for, current requirements, v, 123–124 review, continuing, of ongoing research, IRBs responsibilities, xix, 117 review, levels of, for human participants research, oversight system proposal for, vii–viii, xiii, 41–42 risk, minimal, research classification, definition in, xvi, 80,
83–84 risks, research, v. potential benefits, analysis in IRB review process, ii–iv, 79 vulnerability of research participants, current protections for,
85–92 vulnerable individuals protection in human participants research, oversight issues, iv–v, 9, 11
See also Federal departments and agencies; Federal government; Federal legislation; Public policy
Federalwide Assurance (FWA), 52
See also Assurances of compliance; Compliance
Fetus and fetal tissue research activities on, protections for, 45 CFR 46, Subpart B, in entirety, 179–182 research involving human participants, possible exemptions from oversight, 39 research targeting participants with vulnerabilities, listing by sponsoring agency, 86 See also Embryos and embryonic materials; Pregnancy
Finance. See Investments; Personal finance
Florida. See Florida State University; University of Florida; University of South Florida
Florida State University research compliance investigations resulting in restrictions/actions, 54
Food and Drug Administration (FDA), v, xiv, 2, 3, 4, 7, 9, 10, 11, 12, 15, 25, 26, 27, 28, 29, 32, 38, 50, 57, 59, 71, 98, 106, 120, 121, 126, 132, 152, 153, 154, 156, 170, 171, 187, 188, 191, 192, 193, 194, 203 Bioresearch Monitoring Program, 52, 53 Center for Biologics Evaluation and Research, 215, 216 Center for Drug Evaluation and Research, 215, 216 Commissioner, 202 human participants research, regulations of, 157 human participants research, regulations of, statutory authority for, 210–211 IRB functions/operations and, 21 CFR 56, Subpart C, in entirety, 198–201 IRB noncompliance with federal regulations and, administrative actions for, 21 CFR 56, Subpart E, in entirety, 201–203 IRB records/reports and, 21 CFR 56, Subpart D, in entirety, 201 IRBs and, general provisions for, 21 CFR 56, Subpart A, in entirety, 195–198 Office of Clinical Science, 15 research adverse events reporting requirements of, 113–114, 115, 116 research adverse events reporting requirements of, regulatory text, 213–216 site inspections by, to ensure IRBs compliance with research standards, xiv, 52–53 See also Department of Health and Human Services (DHHS)
Food, Drug, and Cosmetic Act. See Federal Food, Drug, and Cosmetic Act
Fordham University research compliance investigations resulting in restrictions/actions, 56
Foreign countries cooperative research, IRB review models in, 121–122 See also specific countries
France research involving human participants, terminology for, 33 research-related injuries, compensation for, 123
Francis, Leslie, 61
234
Friends Research Institute research compliance investigations resulting in restrictions/actions, 56 FWA. See Federalwide Assurance (FWA)
G
Gelsinger, Jesse, 3
General Accounting Office, 8
Germany. See Auschwitz, Germany
Government funding for research. See Federal funding for research
Great Britain human research participants, studies on abuses of, 153 See also United Kingdom
Groups research involving human participants, consideration of, 7–8
H
Harms
research involving human participants, oversight of, need for, 2–4 research participants vulnerability, current regulatory protections for, 85–92 research risks of, for nonparticipants, 72–73 research risks of, minimizing, 73 research risks of, types of, 71–73 research risks/potential benefits, analysis in IRB review process, 79 research risks/potential benefits, balance between, assessment procedure, 78–80 research risks/potential benefits, component-based framework for analysis, 76–77, 78, 81–82 research risks/potential benefits, historical perspective, 75–76 research risks/potential benefits, relationship, assessment of, 74–75 research-related, compensation for, current regulatory requirements, 123–124 research-related, compensation for, current situation, 125 research-related, compensation for, need for, v, 125–126 research-related, compensation for, previous recommendations for program, 124–125 research-related, compensation for, role of local institutions, xx, 123–126
Health and Human Services Department. See Department of Health and Human Services (DHHS)
Health and illness. See Diseases and disorders
Health care research v. practice of, distinction between, 161–162 research v. practice of, distinction between for determining oversight coverage, 34–37 See Diseases and disorders; Public health
Health Care Financing Administration, 5, 11, 132
See also Department of Health and Human Services (DHHS)
National Bioethics Advisory Commission
Health Education and Welfare Task Force, 71
Health Insurance Portability and Accountability Act of 1996, 105 Health Resources and Services Administration (HRSA), 10, 11, 132 Human Subjects Committee, 10 See also Department of Health and Human Services (DHHS) Health services research oversight system coverage determination, 36–37
See also Research Hippocratic oath, 163 Holm, Søren, 121
Hospitals. See Medical centers
HRSA. See Health Resources and Services Administration (HRSA) Human participants. See Protections for human participants Human Radiation Interagency Working Group, 158 Human Subjects Research Subcommittee, 154
I
ICH. See International Conference on Harmonization (ICH) IDE. See Investigational device exemption (IDE) Illinois. See University of Illinois at Chicago Illness. See Diseases and disorders Immunogenicity vaccine safety clinical trial, risk/potential benefits analysis example, 82
Implementing Human Research Regulations, 155
See also President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission) IND. See Investigational new drug application (IND) Independent bodies IRBs, emergence of, 6 human participants research, oversight office proposed for, vi, xii, 28, 30–32 India research-related injuries, compensation for, 123 Indian Health Service, 11, 132 See also Department of Health and Human Services (DHHS) Inducement. See Coercion Influence. See Coercion Information revelation of, about others, research involving human participants, 39 See also Disclosure of information; Informed consent Informed consent comprehension for, ethical guidelines from Belmont Report, 165 deficiencies in current requirements for, 98 documentation requirements for, discussion of, xviii, 103–104 human participants research and, ethical guidelines from
Belmont Report, 164–165
235
Index
human participants research, requirements for, 21 CFR 50, Subpart B, in entirety, 189–195 human participants research, requirements for, 45 CFR 46, Subpart A, 176–178 mentally impaired research participants and, requirements for, 101 process, emphasis on, discussion of, iv, xvii, 98–101 processes for various research settings, 100–101 recommendations of NBAC on, xvii, xviii, 98, 101, 102, 103, 104 voluntariness of, ethical guidelines from Belmont Report, 165 voluntary, requirements for research participants, scope/ structure of, 97–104 waiver of requirements for, discussion, xvii–xviii, 101–103 See also Disclosure of information; Information
Injuries. See Harms
Inspections. See Site inspections
Institute of Medicine (IOM), 36, 49, 107
Institutional Review Boards (IRBs), xi, xvi, xvii, xviii, 1, 10, 13, 22, 31, 32, 37, 39, 53, 58, 59, 71, 72, 73, 80, 83, 85, 87, 90, 91, 92, 97, 98, 99, 100, 101, 104, 105, 106, 107, 155, 158 accreditation of competence for research ethics, measures for demonstrating, vii, xiv, 49–50 adverse events reporting requirements for, xx, 117 adverse events review, requirements for, xix, xx, 114, 115–116, 117 adverse events, FDA reporting requirements for, regulatory text, 213–216 assurances of compliance for research standards by, xiv, 50–51, 52 audits, institutional, for research standards compliance by, xiv, 53 children as research subjects, protections for, requirements for, 45 CFR 46, Subpart D, 184 competence in research ethics, measures for demonstrating, xiv,
48–50 compliance oversight investigations resulting in restrictions/actions to MPAs, listing by institution, 54–56 compliance with research standards by, mechanisms for ensuring, xiv, 50–53 compliance with research standards of, by investigators, monitoring for verification, xiv, 57–58 composition of and conflict of interest, issues for, 63–64 conflict of interest and institutional role of, measures for managing, xv, 61–63, 64 conflict of interest and, measures for managing, xv–xvi, 64 cooperative research local review by, alternatives to, 120–122 cooperative research local review by, pros/cons, 118–119, 119–120 cooperative research review by, foreign models of, 121–122 cooperative research review role of, xx, 122 cooperative research review, current requirements and problems, xx, 118 education on human participants protection, measures needed for, vi–vii, xiii, 45–48 educational needs on human participants research, 15
functions/operations of, requirements for, 21 CFR 56, Subpart C, in entirety, 198–201 human participants research, current oversight burdens of,
13–15 human participants research, general provisions for, 21 CFR 56, Subpart A, in entirety, 195–198 human participants research, proposed oversight system, and ethical review, xii, 23–24 human participants research, proposed oversight system, and enforcement of requirements, xii, 25–26 human participants research, proposed oversight system, and functions/responsibilities of, xi–xii, 23–27 human participants research, proposed oversight system, and monitoring, xii, 24–25 human participants research, report implications for, 136–137 human participants research, requirements for, 45 CFR 46, Subpart A, 173–176 human participants research, resources for, 131, 133 human participants research, responsibilities of, issues concerning, xix–xx, 111–126 human participants research, revised oversight system, as dynamic/responsive structure, 134–135 independent, emergence of, 6 informed consent documentation requirements of, xviii, 104 informed consent requirements of, waiver of, xviii, 102, 103 membership and conflict of interest, measures for managing, xv–xvi, 60–61, 64 monitoring of ongoing research, responsibilities of, xix–xx, 111–117 multi-site research studies, and human participants, 14 noncompliance with federal regulations by, administrative actions for, 21 CFR 56, Subpart E, in entirety, 201–203 organization/personnel of, requirements for, 21 CFR 56, Subpart B, in entirety, 198 pregnant women, fetuses, and in vitro fertilization, research protections for, requirements for, 45 CFR 46, Subpart B, 180 prisoners in biomedical/behavioral research, protections for, requirements for, 45 CFR 46, Subpart C, 182–183 protocol changes and unanticipated problems, local institutions responsibilities, xix, 112–113, 117 records/reports of, requirements for, 21 CFR 56, Subpart D, in entirety, 201 research approval by, current regulations for, 69–71 research risks v. potential benefits analysis, xvi–xvii, 69–85, 93 research risks v. potential benefits, component-based framework for analysis, 76–77, 78, 81–82 research risks v. potential benefits, component-based framework for analysis, examples, 81–82 research risks v. potential benefits, relationship, assessment of,
74–75 review process for research of, 79 review, continuing, of ongoing research, responsibilities of, xix, 117 review, levels of, for human participants research, oversight system proposal for, xiii, 41–42
236
risk, minimal, for human research participants, review of, oversight proposal, 24 risk, minimal, research classification, use as sorting mechanism for review, 84 risks v. potential benefits of human participants research, analysis in review process, ii–iv, 79 site inspections by FDA to ensure compliance with research standards, xiv, 52–53 vulnerability of research participants, evaluation of, xvii, 85–93 See also Ethics review and review committees
Internal Revenue Service (IRS), 34, 40
International Conference on Harmonization (ICH), 123 Internet, 7 Interventions. See Health care
Interviews face-to-face, research risks/potential benefits analysis example, 81
Investigational device exemption (IDE), 193
Investigational new drug application (IND), 190, 191, 193 adverse research events, FDA reporting requirements for, regulatory text, 213–216
Investigators accreditation for competence of, measures for demonstrating, xiv, 49–50 certification of competence of research individuals, vii, xiv,
48–49 compliance with research standards by, monitoring for verification, xiv, 57–58 conflict of interest for, measures for managing, xiv–xv, 58–60 education on human participants research, measures needed for, vi–vii, xiii, 45–48 human participants research, report implications for IRBs/ investigators/institutions, 136–137 monitoring of ongoing research, local institutions responsibilities, xix, 117 oversight system, proposed, functions and responsibilities of, 26 oversight system, proposed, monitoring, xii, 24–25 oversight system, proposed, enforcement of requirements, xii,
25–26 recommendations of NBAC for, xiv, xv–xvi, xiii, xix, 47–48, 50, 64, 117
See also Research
Investment research, changes in trends in, 4
In Vitro fertilization (IVF) research activities involving, protections for, 45 CFR 46, Subpart B, in entirety, 179–182
IOM. See Institute of Medicine (IOM) IRBs. See Institutional Review Boards (IRBs) IRS. See Internal Revenue Service (IRS)
National Bioethics Advisory Commission
J
Joint United Nations Programme on HIV/AIDS (UNAIDS), 33, 123
Jonas, Hans, i, ii
Justice human research participants protection and, from Belmont Report, 163–164 human research participants selection and relevance, from
Belmont Report, 167 individual, and human research participants selection, 167 social, and human research participants selection, 167
L
Languages
German, interview, face-to-face, risks/potential benefits analysis example, 81 Hebrew, interview, face-to-face, risks/potential benefits analysis example, 81
Levine, Robert J., 75
Local entities adverse events reporting requirements for, xix–xx, 113–117 adverse events review, requirements for, 114, 115–116 compensation for research-related injuries, role of, xx, 123–126 cooperative research review, by, alternatives to, 120–122 cooperative research review, by, considerations, 118–120 cooperative research review, role of, 122 human participants research responsibilities of, issues concerning, xix–xx, 111–126 monitoring of ongoing research, responsibilities of, xix–xx, 111–117
Los Angeles, California, 119 See also California
M
Macon County, Alabama, 153
Massachusetts Eye and Ear Infirmary research compliance investigations resulting in restrictions/actions, 54
Maternity. See Pregnancy
Maryland, 56
See also University of Maryland, Baltimore Medicaid, 5 Medical care. See Health care
Medical centers academic, and research universities, stresses on, 4, 6
Medical devices research adverse events, FDA reporting requirements, 114, 115
Medical interventions/treatment. See Health care
Medical University of South Carolina research compliance investigations resulting in restrictions/actions, 54
237
Index
Medicare, 5, 7
Memorial-Sloan Kettering Cancer Center research compliance investigations resulting in restrictions/actions, 54
Mental diseases and disorders vulnerable persons targeted for research, listing by sponsoring agency, 86 See Diseases and disorders
Milgram, S., 71
Minnesota. See University of Minnesota
Monitoring human research participants protection, local institutions responsibilities for, xix–xx, 111–117 human research participants protection, oversight enforcement weaknesses, 12–13 human research participants protection, oversight proposal for, xii, 24–25 research standards compliance by investigators, verification of, xiv, 57–58
Moreno, Jonathan, 9, 22
MPAs. See Multiple Project Assurances (MPAs)
Mt. Sinai School of Medicine research compliance investigations resulting in restrictions/actions, 56
Multiple Project Assurances (MPAs), 8, 14, 46, 52, 118 compliance investigations by OPRR resulting in restrictions/actions to, listing by institution, 54–56 See also Assurances of compliance; Compliance; Office for Human Research Protections (OHRP)
Multi-site research. See Cooperative research
N
NAS. See National Academy of Sciences (NAS)
NASA. See National Aeronautics and Space Administration (NASA)
National Academy of Sciences (NAS), 47
National Aeronautics and Space Administration (NASA), 5, 11, 132, 205 human participants research, regulations of, 157
National Bioethics Advisory Commission (NBAC), ii, xi, xviii, 1, 5, 7, 16, 21, 22, 24, 26, 30, 31, 32, 33, 34, 35, 38, 39, 41, 45, 46, 47, 48, 52, 53, 60, 63, 71, 72, 75, 76, 84, 85, 92, 97, 99, 100, 101, 102, 103, 104, 106, 107, 116, 117, 119, 123, 125, 131, 133 accreditation of competence of research institutions/IRBs, recommendations, xiv, 50 adverse events reporting requirements for, recommendations, xx, 117 certification of competence of research individuals, recommendations, xiv, 48–49 compensation for research-related injuries, recommendations, xx, 126 compliance with research standards by institutions/IRBs/ investigators, ensuring, recommendations, xiv, 58
confidentiality for research participants, proposed definition for, 107 confidentiality for research participants, recommendation, xviii, 107 conflict of interest and investigators/IRBs/institutions, recommendations, xv–xvi, 64 cooperative research review, requirements for, recommendations, xx, 122 education on research ethics and human participants protection, education on, recommendations, xiii, 47–48 human participants research, defining for oversight coverage determination, recommendations, xii, 40 human participants research, ethical and policy issues in, recommendations summary, xi–xxi human participants research, ethical issues for, future research recommendations, xxi, 134 human participants research, report recommendations for, general themes in, 135 human participants research, resources for, recommendations, xx–xxi, 133 human participants research, revised oversight system for, scope/structure, recommendations, xii, 28, 30 human participants research, unified/comprehensive federal policy for, recommendations, xii, 30 informed consent documentation requirements for human participants, recommendation, xviii, 104 informed consent requirements for human participants, previous recommendations on, 98 informed consent requirements for human participants, process emphasis, recommendations, xvii, 101 informed consent requirements for human participants, waiver of, recommendations, xviii, 102, 103 IRB membership/composition and conflict of interest, recommendations, xv–xvi, 64 monitoring of ongoing research, local institutions responsibilities, recommendations, xix, 117 National Office for Human Research Oversight creation, recommendations, xii, 28, 30 privacy for research participants, proposed definition for, 106 privacy for research participants, recommendation, xviii, 107 research protocol changes and unanticipated problems, local institutions role, recommendations, xix, 117 research risks v. potential benefits, analysis of, recommendations, xvi–xvii, 77, 85 review, continuing, of ongoing research, IRBs responsibilities, recommendations, xix, 117 review, levels of, for human participants research, oversight proposal for, recommendations, xiii, 41–42 risk, minimal, of research, federal policy on, recommendation, xvi–xvii, 85 vulnerability of research participants, definition of, recommendation, xvii, 92
National Cancer Institute (NCI), 121
National Center for Educational Statistics Confidentiality Statute, 105, 107
National Center for Health Statistics, 39
238
National Commission. See National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission)
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission), i, 34, 35, 45, 50, 57, 58, 62, 83, 112, 118, 120, 124, 131, 153–154, 155, 161, 207
Institutional Review Boards, 76 Research Involving Children, 75 Research on the Fetus, 75 Research Involving Prisoners, 75 research risks/potential benefits, historical perspective, 75–76
See also Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research
National Heart, Lung, and Blood Institute, 7–8
National Human Experimentation Board, 153
National Institutes of Health (NIH), 3, 7, 10, 11, 15, 28, 30, 46, 48, 113, 116, 118, 131, 132, 133, 151, 152, 153, 155, 170, 172, 174 Clinical Center, 34, 151, 152 Director, 152, 208 Division of Research Grants, 50, 152 Institutional Relations Branch, 152 Office of Extramural Research, 8
Reducing Regulatory Burden, 133 research compliance investigations by OPRR resulting in restrictions/actions to MPAs, 54 See also Department of Health and Human Services (DHHS); Office for Protection from Research Risks (OPRR)
National Office for Human Research Oversight (NOHRO), 40, 84 advantages v. disadvantages of, 30–31 creation of, vi, xii, 28, 30–32 operating mode of, 31–32 private sector research, regulation of, 32
National Research Act. See Public Law 93–348
National Science Foundation (NSF), 5, 11, 53, 132, 205 Directorate of Social, Behavioral and Economic Sciences, 5 human participants research, regulations of, 157 Office of Inspector General, 53 site inspections to ensure compliance with research standards, 53
Nazis, 34, 81, 151, 164
NBAC. See National Bioethics Advisory Commission (NBAC)
NCI. See National Cancer Institute (NCI)
Netherlands research-related injuries, compensation for, 123
New York City, 3
See also City University of New York
New York State, 153
See also State University of New York
New York Times, 153
National Bioethics Advisory Commission
New Zealand research-related injuries, compensation for, 123
NIH. See National Institutes of Health (NIH)
NOHRO. See National Office for Human Research Oversight (NOHRO)
NRC. See Nuclear Regulatory Commission (NRC)
NSF. See National Science Foundation (NSF)
Nuclear Regulatory Commission (NRC), 31 See also Department of Energy (DOE)
Nuremberg Code, 151, 152, 161 Nuremberg Military Tribunal, 151 Nuremberg War Crime Trials, ii, 161
O
“Obedience to Authority” experiments, 71
Office for Human Research Protections (OHRP), 8, 9, 10, 12, 13,
15–16, 28, 30, 31–32, 49, 52, 58, 61, 62–63, 100, 121, 124, 133, 152 “Common Findings and Guidance,” 13 site inspections to ensure compliance with research standards, 53 See also Cooperative Project Assurance (CPA); Multiple Project Assurances (MPAs); Office for Protection from Research Risks (OPRR); Single Project Assurances (SPAs)
Office for Protection from Research Risks (OPRR), 3, 8, 10, 14, 15, 30, 46, 58, 83, 102, 119, 121, 133, 152, 154, 170, 172, 174 compliance investigations of, resulting in restrictions/actions to MPAs, listing by institution, 54–56
IRB Guidebook, 70 OPRR Reports, 154 site inspections to ensure compliance with research standards, 53 See also National Institutes of Health (NIH); Office for Human Research Protections (OHRP)
Office of Government Ethics (OGE), 31
See also Office of Personnel Management (OPM)
Office of Personnel Management (OPM), 31 See also Office of Government Ethics (OGE)
Office of Science and Technology Policy (OSTP), xi, 1, 155 human participants research, regulations of, 157 See also White House
Office of Special Education and Rehabilitative Services human participants research, regulations of, statutory authority for, 209
Office of the President
Committee on Science, National Science and Technology Council, 154 See also President of the U.S.
OGE. See Office of Government Ethics (OGE)
Opinion. See Public opinion
OHRP. See Office for Human Research Protections (OHRP)
239
Index
OPM. See Office of Personnel Management (OPM)
OPRR. See Office for Protection from Research Risks (OPRR)
OSTP. See Office of Science and Technology Policy (OSTP)
Oversight accreditation of competence of research institutions/IRBs, vii, xiv, 49–50 adverse research events, FDA reporting requirements, 113–114, 115, 116, 213–216 adverse research events, reporting requirements for local institutions, xix–xx, 113–117 biomedical/behavioral research, prisoners in, and, 45 CFR 46, Subpart C, in entirety, 182–184 certification of competence of research individuals, vii, xiv,
48–49 children as research subjects, 45 CFR 46, Subpart D, in entirety, 184–186 compliance with research standards by institutions/IRBs/ investigators, mechanisms for ensuring, xiv, 50–58 conflict of interest for research IRBs, measures for managing,
60–63 cooperative research review, current requirements and problems, xx, 118 cooperative research review, requirements for IRBs, 122 education on human participants protection, measures needed for, vi–vii, xiii, 45–48 educational programs for human participants research, proposal for, xii, 23 health services research/program evaluation, determining coverage, 36–37 human participants research, Common Rule and agency-specific regulations, 157 human participants research, consequences of flawed system, 16 human participants research, current protection of, 29 human participants research, current system, challenges for,
8–16 human participants research, current system, description of, 154, 155–156, 157 human participants research, current system, enforcement weaknesses, 12–13 human participants research, current system, IRB burdens,
13–15 human participants research, current system, lack of adaptability of, 11–12 human participants research, current system, limited scope of, 12 human participants research, current system, procedural v. ethical emphasis of, 13 human participants research, defining for coverage determination, xii–xiii, 34–42 human participants research, ethical issues for, future research on, xxi, 134 human participants research, exemptions possible, 37–41 human participants research, federal agency regulations and, statutory authority for, 205–211 human participants research, federal regulations on, 21 CFR 50, Subpart A, in entirety, 187–189
human participants research, federal regulations on, 45 CFR 46, Subpart A, in entirety, 169–179 human participants research, flawed, for, consequences, 16 human participants research, history of, 151–154 human participants research, inconsistencies among agencies, 9, 10 human participants research, percent exempt from, listing by agency, 11 human participants research, possible exemptions from, 37–41 human participants research, proposal for, independent office, vi, xii, 28, 30–32 human participants research, proposed system for, independent office for, vi, xii, 28, 30–32 human participants research, proposed system, functions/responsibilities of all parties, xi–xii, 23–27 human participants research, proposed system, as dynamic/responsive structure, 134–135 human participants research, proposed system, educational programs for all parties, xii, 23 human participants research, proposed system, enforcement of, xii, 25–26 human participants research, proposed system, ethical review of research, vii–viii, xii, 23–24 human participants research, proposed system, functions and responsibilities of, xi–xii, 23–27 human participants research, proposed system, monitoring for, xii, 24–25 human participants research, proposed system, National Office for Human Research Oversight for, xii, 28, 30–32 human participants research, proposed system, overview of, xi–xiii, 21–42 human participants research, proposed system, policy development through regulations/guidance, xii, 23 human participants research, proposed system, public accountability of, xii, 26 human participants research, proposed system, recommendations for, general themes in, 135 human participants research, proposed system, resources for, viii, xx–xxi, 131, 133 human participants research, proposed system, scope and structure, vii, xii, 26–32 human participants research, proposed system, unified/ comprehensive policy for, v–vi, xii, 28, 30–32 human participants research, protection for, ACHRE recommendations, 158 human participants research, purpose of, i–ii, xi, 2–4, 21–22 human participants research, recent government interest in, 156 human participants research, recent initiatives for, 15–16 human participants research, report implications for IRBs/ investigators/institutions, 136–137 human participants research, report implications for participants, 137 human participants research, report recommendations for, general themes in, 135 human participants research, resources for, viii, xx–xxi, 131, 133
240
informed consent for human participants research, 21 CFR 50, Subpart B, in entirety, 189–195 informed consent requirements for research participants, deficiencies in, 98 informed consent requirements for research participants, scope/structure of, iv, xvii, 97–104 IRB approval for research, current regulations for, 69–71 IRB functions/operations for human participants research, 21 CFR 56, Subpart C, in entirety, 198–201 IRB noncompliance for human participants research, 21 CFR 56, Subpart E, in entirety, 201–203 IRB organization/personnel and human participants research, 21 CFR 56, Subpart B, in entirety, 198 IRB records/reports and human participants research, 21 CFR 56, Subpart D, in entirety, 201 IRBs and, general provisions for, 21 CFR 56, Subpart A, in entirety, 195–198 medical practice v. research, distinction for determining coverage, 34–37 pregnant women, fetuses, and in vitro fertilization in research, 45 CFR 46, Subpart B, in entirety, 179–182 prisoners in biomedical/behavioral research and, 45 CFR 46, Subpart C, in entirety, 182–184 privacy and confidentiality protection for research participants, 104–105 research protocol changes and unanticipated problems, local institutions responsibilities, xix, 112–113, 117 research, ongoing, monitoring responsibilities of local institutions, xix, 117 research-related injuries, compensation for, current requirements, v, 123–124 review for, levels of, proposal for, vii–viii, xiii, 41–42 review, continuing, of ongoing research, IRBs responsibilities, xix, 117 review, levels of, for human participants research, proposal for, vii–viii, xiii, 41–42 risk, minimal, research classification, definition in, xvi, 80,
83–84 risks, research, v. potential benefits, analysis in IRB review process, ii–iv, 79 vulnerability and human participants research, issues, iv–v, 9, 11 vulnerability of research participants, current protections for,
85 –92
See also Federal regulation; Public policy
P
Papers commissioned human research participants protection, ethical and policy issues, 225–226
Participants. See Protections for human participants Peckman, Steven, 119 Pennsylvania. See University of Pennsylvania
National Bioethics Advisory Commission
Personal finance disadvantaged persons targeted for research, listing by sponsoring agency, 86 vulnerability in research participants, considerations, 90
Personnel human participants research, resources for, xx–xxi, 131, 133 human participants research, federal support for FY 1999, listing by agency, 132
Phillips, D.F., 9
PHS. See Public Health Service (PHS) P.L. 93–348. See Public Law 93–348 Policy. See Public policy
Postal service questionnaire, mail, for study of state law effects on behavior, risks/potential benefits analysis example, 81
Practice of health care/medicine. See Health care
Pregnancy research activities on, protections for, 45 CFR 46, Subpart B, in entirety, 179–182 vulnerable persons targeted for research, listing by sponsoring agency, 86 See also Embryos and embryonic materials; Fetus and fetal tissue
President of the U.S., 190, 191 Memorandum of, 11 See also Office of the President
President’s Commission. See President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission)
President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission), i, v, xx, 45–46, 50–51, 52, 57–58, 112, 124, 126, 154
See also Compensating for Research Injuries; Implementing Human Research Regulations
PRIM&R. See Public Responsibility in Medicine and Research (PRIM&R)
Prisoners biomedical/behavioral research subjects, protections for, 45 CFR 46, Subpart C, in entirety, 182–184 vulnerable persons targeted for research, listing by sponsoring agency, 86
Privacy definition for human research participants proposed by NBAC, 106 research participants protection, current regulations on, 104–105 research participants protection, issues for, xviii, 106–107 research participants protection, NBAC recommendation, xviii, 107 See also Confidentiality
241
Index
Private sector research involving human participants, recommendations of President’s Commission, 155 research involving human participants sponsored by, proposed regulation by NOHRO, 32
Professional societies human participants research, proposed oversight system for, responsibilities of, 27 research ethics and human participants research, programs in, measures needed for, xiii, 48
Protection of Pupil Rights Amendment, 104–105
Protections for human participants accreditation of competence for institutions/IRBs, vii, xiv, 49–50 adverse events FDA reporting requirements for, 113–114, 115, 213–216 adverse events reporting requirements for local institutions, xix–xx, 113–117 adverse events review, requirements for local institutions/IRBs, 114, 115–116 beneficence, relevance for, from Belmont Report, 162–163, 165–167 benefits, potential, to others, 74 benefits, potential, to participants, 73–74 benefits, potential, to society, 73 benefits, potential, types, 73–74 biomedical/behavioral, prisoners in, and, 45 CFR 46, Subpart C, in entirety, 182–184 certification of competence of individuals, vii, xiv, 48–49 children as subjects, 45 CFR 46, Subpart D, in entirety, 184–186 communities/groups and, consideration of, 7–8 compensation for research-related injuries, current situation, 123–125 compensation for research-related injuries, need for, v, 125–126 compensation for research-related injuries, previous recommendations for program, 124–125 compensation for research-related injuries, role of local institutions, xx, 123–126 competence for, measures for demonstrating, xiv, 48–50 compliance investigations by OPRR resulting in restrictions/actions to MPAs, listing by institution, 54–56 compliance with standards by institutions/IRBs/investigators, mechanisms for ensuring, xiv, 50–58 conflict of interest for investigators, measures for managing, xiv–xv, 58–60 conflict of interest for IRBs, measures for managing, xv, 60–64 cooperative research review, by IRBs, foreign models of, 121–122 cooperative research review, by local IRBs, alternatives to, 120–122 cooperative research review, by local IRBs, considerations, 118–120 cooperative research review, current requirements and problems, xx, 118 cooperative research review, role of local IRBs/institutions, 122
defining for oversight coverage determination, xii–xiii, 34–42 education on, measures needed for, vi–vii, xiii, 45–48 ethical and policy issues for, papers commissioned on, 225–226 ethical and policy issues for, public comments on, 217–220 ethical and policy issues for, testimony, public/expert, on, 221–223 ethical issues for, future research on, xxi, 133–134 ethical principles/guidelines for, Belmont Report text in entirety, 161–167 federal agency regulations for, statutory authority for, 205–211 federal agency support for FY 1999, listing by agency, 132 federal protection for, 29 federal regulations of Common Rule and specific agencies, 157 federal regulations on, 21 CFR 50, Subpart A, in entirety, 187–189 federal regulations on, 45 CFR 46, Subpart A, in entirety, 169–179 federal regulations on, exemptions from, percent listing by agency, 11 federal regulations on, inconsistent interpretation/implementation by agencies, 9, 10 federally supported research, size and scope of, 5 government recent interest in, 156 health services research/program evaluation, determining oversight coverage, 36–37 informed consent disclosure requirements for, emphasis on, issues, xvii, 98–100 informed consent disclosure requirements for, listing, 99 informed consent documentation requirements for, discussion of, xviii, 103–104 informed consent for, 21 CFR 50, Subpart B, in entirety, 189–195 informed consent for, ethical considerations, from Belmont Report, 164–165 informed consent process for, emphasis on, discussion of, iv, xvii, 98–101 informed consent requirements for, deficiencies in, 98 informed consent requirements for, scope/structure of, 97–104 informed consent requirements for, waiver of, discussion, xvii–xviii, 101–103 IRB functions/operations and, 21 CFR 56, Subpart C, in entirety, 198–201 IRB noncompliance with regulations, actions for, 21 CFR 56, Subpart E, in entirety, 201–203 IRB organization/personnel and, 21 CFR 56, Subpart B, in entirety, 198 IRB records/reports and, 21 CFR 56, Subpart D, in entirety, 201 IRB research review process, 79 IRB research studies approval, current regulations for, 69–71 IRBs and, general provisions for, 21 CFR 56, Subpart A, in entirety, 195–198 justice and participants selection, ethical guidelines from
Belmont Report, 167 justice, relevance for, from Belmont Report, 163–164, 167 local institutions and IRBs responsibilities for, issues for, xix–xx, 111–126
242
medical practice v., distinction for determining oversight coverage, 34–37 mentally impaired research participants and informed consent, requirements for, 101 moderate protectionism, explanation, 22 monitoring of ongoing research, local institutions responsibilities, xix–xx, 111–117 oversight of, need for, i–ii, 2–4 oversight office, independent, proposed for, vi, xii, 28, 30–32 oversight system for, purpose of, xi, 21–22 oversight system proposed for, as dynamic / responsive structure, 134–135 oversight system proposed for, educational programs for all parties, xii, 23 oversight system proposed for, enforcement of requirements, xii, 25–26 oversight system proposed for, ethical review of research, vii–viii, xii, 23–24 oversight system proposed for, functions/responsibilities of all parties, xi–xii, 23–27 oversight system proposed for, monitoring, xii, 24–25 oversight system proposed for, need for unified/comprehensive federal policy for, v–vi, xii, 28, 30–32 oversight system proposed for, overview of, xi–xiii, 21–42 oversight system proposed for, policy development through regulations/guidance, xii, 23 oversight system proposed for, public accountability for, xii, 26 oversight system proposed for, recommendations for, general themes in, 135 oversight system proposed for, resources for, viii, xx–xxi, 131, 133 oversight system proposed for, scope and structure, vii, xii,
26–32 oversight system, current, challenges for, 8–16 oversight system, current, description, 154, 155–156, 157 oversight system, current, enforcement weaknesses of, 12–13 oversight system, current, IRB burdens, 13–15 oversight system, current, lack of adaptability of, 11–12 oversight system, current, limited scope of, 12 oversight system, current, procedural v. ethical emphasis of, 13 oversight system, exemptions possible, 37–41 oversight system, flawed, for, consequences, 16 oversight system, history of, 151–154 oversight system, recent initiatives for, 15–16 pregnant women, fetuses, and in vitro fertilization and, 45 CFR 46, Subpart B, in entirety, 179–182 prisoners in biomedical/behavioral research and, 45 CFR 46, Subpart C, in entirety, 182–184 privacy and confidentiality for, current regulations on, 104–105 protocol changes and unanticipated problems, local institutions responsibilities, xix, 112–113, 117 public perspectives about research participation, changes in,
6–7 recommendations of ACHRE for, 158 recommendations of NBAC for, xi–xxi, 28, 30, 40, 41–42,
47–49, 50, 64, 77, 85, 92, 98, 101, 102, 103, 104, 117, 122, 124–126, 133, 134
National Bioethics Advisory Commission
report implications for, 137 respect for persons, relevance for, from Belmont Report, ii, 162, 164–165 review for, levels of, oversight system proposal for, vii–viii, xiii,
41–42 review, continuing, IRBs responsibilities, xix, 117 risk, minimal, classification, definition in federal regulations, xvi, 80, 83–84 risk, minimal, classification, use as sorting mechanism for IRB review, 84 risk, minimizing, 73 risks of harms, to nonparticipants, 72–73 risks of harms, types of, 71–73 risks v. potential benefits analysis for, ethical considerations, from Belmont Report, 165–167 risks v. potential benefits analysis for, new technologies effects on, 7 risks v. potential benefits analysis for, xvi–xvii, 69–85, 93 risks v. potential benefits analysis in IRB review process, ii–iv, 79 risks v. potential benefits, balance between, assessment procedure, 78–80 risks v. potential benefits, component-based framework for analysis, 76–77, 78, 81–82 risks v. potential benefits, component-based framework for analysis, examples, 81–82 risks v. potential benefits, historical perspective, 75–76 risks v. potential benefits, relationship, assessment of, 74–75 selection of, ethical considerations, from Belmont Report, 167 strong protectionism, explanation, 22 terminology for, 32–33 vulnerability and, current regulatory protections for, 85–92 vulnerability and, definition of, xvii, 92 vulnerability and, evaluation of, xvii, 85–93 vulnerability and, minimal risk and, 92 vulnerability and, oversight issues, iv–v, 9, 11 vulnerability, cognitive/communicative, and, considerations, 88 vulnerability, deferential, and, considerations, 89 vulnerability, economic, and, considerations, 90 vulnerability, institutional, and, considerations, 88–89 vulnerability, medical, and, considerations, 89–90 vulnerability, participants targeted in research, listing by sponsoring agency/vulnerability, 86 vulnerability, social, and, considerations, 90–91 weak protectionism, explanation, 22
See also Research
Psychology experiment to study human cognition/behavior, risk/potential benefits analysis example, 82
Public accountability to, for research involving human participants, oversight system proposal, xii, 26 See also Public comments; Public health; Public opinion; Public policy
243
Index
Public comments human participants research, ethical and policy issues, 217–220 See also Testimony and speakers
Public health activities, v. research involving human participants, distinction for determining oversight coverage, 35–36 See also Health care
Public Health Service (PHS), 124, 152
“Policy for Clinical Investigations with Human Subjects,” 152 See also Tuskegee Syphilis Study; Tuskegee Syphilis Study Ad Hoc Advisory Panel
Public Health Service Act, 29, 105, 106, 187, 188, 189, 196, 197
Public Law 93–348, 153–154, 207
Public opinion research participation and, changing perspectives, 6–7
Public policy
adverse research events, reporting requirements for, NBAC recommendations, xx, 117 conflict of interest and IRB membership/composition, NBAC recommendations, xv–xvi, 64 conflict of interest for research investigators/IRBs/institutions, NBAC recommendations, xv–xvi, 64 education for human participants research, NBAC recommendations, xiii, 47–48 human participants research, defining for oversight coverage, NBAC recommendations, xii, 40 human participants research, ethical principles/guidelines for, Belmont Report text in entirety, 161–167 human participants research, issues for, NBAC recommendations summary, xi–xxi human participants research, issues for, papers commissioned on, 225–226 human participants research, issues for, public comments on, 217–220 human participants research, issues for, testimony, public/expert, on, 221–223 human participants research, oversight system proposed for, development through regulations/guidance, xii, 23 human participants research, oversight system proposed for, independent office for, vi, xii, 28, 30–32 human participants research, oversight system proposed for, National Office for Human Research Oversight for, vi, xii, 28,
30–32 human participants research, oversight system proposed for, scope/structure of, NBAC recommendations, xii, 28, 30 human participants research, oversight system proposed for, unified/comprehensive policy for, v–vi, xii, 28, 30–32 human participants research, report implications for IRBs/ investigators/institutions, 136–137 human participants research, report implications for participants, 137 review, continuing, of ongoing research, IRBs responsibilities, NBAC recommendations, xix, 117 review, levels of, for human participants research, NBAC recommendations, xiii, 41–42
risk, minimal, of research, and, NBAC recommendation, xvi–xvii, 85 risks, research, v. potential benefits, analysis of, NBAC recommendations, xvi–xvii, 77, 85 vulnerability of research participants, definition of, NBAC recommendation, xvii, 92 See also Federal government; Federal regulation
Public Responsibility in Medicine and Research (PRIM&R), 15, 33, 46
Public testimony. See Testimony and speakers
Publishers scientific, and human participants research, oversight responsibilities proposed for, 27
Q
Questionnaires mail, study of effects of state law on behavior, risks/potential benefits analysis example, 81
R
Reactions. See Adverse reactions
Recommendations accreditation of competence of research institutions/IRBs, measures for demonstrating, xiv, 50 adverse events reporting requirements for, xx, 117 certification of competence of research individuals, measures for demonstrating, xiv, 48–49 compensation for research-related injuries, xx, 124–125, 126 compliance with research standards by institutions/IRBs/ investigators, mechanisms for ensuring, xiv, 58 confidentiality protection for research participants, xviii, 107 conflict of interest and investigators/IRBs/institutions, measures for managing, xv–xvi, 64 cooperative research review, requirements for, xx, 122 education on research ethics and human participants, measures needed for, xiii, 47–48 human participants research, ACHRE recommendations, 158 human participants research, defining for oversight coverage determination, xii, 40 human participants research, ethical and policy issues in, summary of, xi–xxi human participants research, ethical issues for, future research on, xxi, 134 human participants research, general themes in, 135 human participants research, oversight proposal for unified/comprehensive federal policy for, xii, 30 human participants research, oversight system proposal for, scope and structure, xii, 28, 30 human participants research, proposal for National Office for Human Research Oversight, xii, 28, 30 human participants research, resources for, xx–xxi, 133 informed consent documentation requirements for human participants, xviii, 104 informed consent requirements for human participants, waiver of, xviii, 102, 103
244
informed consent requirements for human participants, xvii, 98, 101 IRB membership/composition and conflict of interest, measures for managing, xv–xvi, 64 monitoring of ongoing research, local institutions responsibilities, xix, 117 privacy protection for research participants, xviii, 107 research protocol changes and unanticipated problems, local institutions responsibilities, xix, 117 research risks, v. potential benefits, analysis of, xvi–xvii, 77, 85 review, continuing, of ongoing research, IRBs responsibilities, xix, 117 review, levels of for human participants research, oversight system proposal for, xiii, 41–42 risk, minimal, of research, Federal policy on, xvi–xvii, 85 vulnerability of research participants, definition of, xvii, 92
Regulation. See Federal government; Federal regulation; Oversight; Research regulations
Reporting requirements adverse research events and, xix–xx, 113–117 adverse research events, FDA, 113–114, 115, 213–216 research protocol changes and unanticipated problems, local institutions responsibilities, xix, 112–113, 117
Research adverse events, FDA reporting requirements for, regulatory text, 213–216 assurances of compliance for institutions/IRBs, xiv, 50–51, 52 audits, institutional, for standards compliance by institutions/IRBs, xiv, 53 benefits, potential, to others, 74 benefits, potential, to participants, 73–74 benefits, potential, to society, 73 benefits, potential, types, 73–74 communities/groups consideration for, 7–8 compliance with standards by institutions/IRBs/investigators, mechanisms for ensuring, xiv, 50–58 conduct of, current factors influencing, 4, 6–8 equipoise of, explanation, 77, 78 ethical issues for, future research on, xxi, 133–134 informed consent processes for various settings, 100–101 injuries from, compensation for, current regulatory requirements, 123–124 injuries from, compensation for, current situation, 125 injuries from, compensation for, need for, v, 125–126 injuries from, compensation for, previous recommendations for program, 124–125 injuries from, compensation for, role of local institutions, xx, 123–126 IRB approval for studies, current regulations for, 69–71 IRB review process, 79 observational study of effects of state law on behavior, risks/potential benefits analysis example, 81 participation in, changing public perspectives about, 6–7 practice v., distinction between, from Belmont Report, 161–162
National Bioethics Advisory Commission
pregnant women, fetuses, and in vitro fertilization in, protections for, 45 CFR 46, Subpart B, in entirety, 179–182 risk of harms to nonparticipants, 72–73 risk of harms, minimizing, 73 risk of harms, types of, 71–73 risks v. potential benefits, analysis in IRB review process, 79 risks v. potential benefits, balance between, assessment procedure, 78–80 risks v. potential benefits, component-based framework for analysis, 76–77, 78, 81–82 risks v. potential benefits, component-based framework for analysis, examples, 81–82 risks v. potential benefits, historical perspective, 75–76 risks v. potential benefits, relationship, assessment of, 74–75 site inspections by FDA to ensure IRBs compliance with standards, xiv, 52–53 site inspections to ensure compliance with standards, xiv, 53 universities for, and academic medical centers, stresses on, 4, 6 value of, summary, 1–2 See also Behavioral research; Biomedical research; Clinical trials; Cooperative research; Health services research; Investigators; Protections for human participants; Research documents; Research funding; Research institutions; Research regulations; Science and technology; Sponsors
Research design. See Equipoise; Research
Research documents
IRB records/reports requirements, 21 CFR 56, Subpart D, in entirety, 201
See also Research
Researchers. See Investigators
Research funding academic medical centers and research universities, stresses on, 4, 6 human participants protection, federal support for FY 1999, listing by agency, 132 human participants protection, resources for, viii, xx–xxi, 131, 133 investment trends for, changes in, 4 See also Federal funding for research; Research
Research institutions accreditation for research ethics competence, measures for demonstrating, vii, xiv, 49–50 adverse events FDA reporting requirements, 113–114, 115 adverse events reporting requirements for, xix–xx, 113–117 adverse events review, requirements for, 114, 115–116 assurances of compliance for research standards, xiv, 50–51, 52 audits, institutional, for research standards compliance by, xiv, 53 compensation for research-related injuries, role of, xx, 123–126 compliance oversight investigations resulting in restrictions/actions to MPAs, listing by institution, 54–56 compliance with research standards by, mechanisms for ensuring, xiv, 50–53 conflict of interest for, measures for managing, xiv–xv, 58–64
245
Index
cooperative research review by local IRBs, alternatives to, 120–122 cooperative research review by local IRBs, considerations, 118–120 cooperative research review, current requirements and problems, xx, 118 cooperative research review, role of local IRBs/institutions, 122 education on human participants protection, measures needed for, vi–vii, xiii, 45–48 human participants research responsibilities of local institutions, issues concerning, xix–xx, 111–126 human participants research, report implications for IRBs/ investigators/institutions, 136–137 monitoring of ongoing research, local institutions responsibilities, xix–xx, 111–117 oversight system, proposed, for human participants research, functions and responsibilities of, 26–27 oversight system, proposed, for human participants research, monitoring, xii, 24–25 oversight system, proposed, for human participants research, enforcement of requirements, xii, 25–26 protocol changes and unanticipated problems, reporting requirements for, xix, 112–113, 117 site inspections to ensure compliance with research standards, xiv, 53
See also Research
Research Involving Persons with Mental Disorders That May Affect Decisionmaking Capacity, vii
Research regulations accreditation of competence of institutions/IRBs, vii, xiv, 49–50 adverse events, FDA reporting requirements, 113–114, 115, 116, 213–216 adverse events, reporting requirements for local institutions, xix–xx, 113–117 biomedical/behavioral, prisoners in, and, 45 CFR 46, Subpart C, in entirety, 182–184 certification of competence of individuals, vii, xiv, 48–49 children as subjects, 45 CFR 46, Subpart D, in entirety, 184–186 compliance with standards by institutions/IRBs/investigators, mechanisms for ensuring, xiv, 50–58 conflict of interest for IRBs, measures for managing, 60–63 cooperative review, current requirements and problems, xx, 118 cooperative review, requirements for IRBs, 122 education on human participants protection, measures needed for, vi–vii, xiii, 45–48 educational programs on human participants protection, oversight system proposal for, xii, 23 human participants, Common Rule and agency-specific regulations, 157 human participants, consequences of flawed system, 16 human participants, current oversight system, challenges for,
8–16 human participants, current protection of, 29 human participants, defining for oversight coverage determination, xii–xiii, 34–42
human participants, description of, 154, 155–156, 157 human participants, enforcement weaknesses, 12–13 human participants, ethical issues for, future research on, xxi, 134 human participants, federal regulations on, 21 CFR 50, Subpart A, in entirety, 187–189 human participants, federal regulations on, 45 CFR 46, Subpart A, in entirety, 169–179 human participants, history of, 151–154 human participants, inconsistencies among agencies, 9, 10 human participants, IRB burdens in, 13–15 human participants, lack of adaptability in, 11–12 human participants, limited scope of, 12 human participants, oversight independent office proposed for, vi, xii, 28, 30–32 human participants, oversight possible exemptions, 37–41 human participants, oversight system proposal for, xi–xiii,
21–42 human participants, oversight system for, purpose of, i–ii, xi,
2–4, 21–22 human participants, oversight system for, resources for, viii, xx–xxi, 131, 133 human participants, oversight system proposal for enforcement of, xii, 25–26 human participants, oversight system proposal for ethical review of research, xii, 23–24 human participants, oversight system proposal for monitoring, xii, 24–25 human participants, oversight system proposal for National Office for Human Research Oversight for, xii, 28, 30–32 human participants, oversight system proposal for policy development through, xii, 23 human participants, oversight system proposal for public accountability, xii, 26 human participants, oversight system proposal for unified/ comprehensive policy for, v–vi, xii, 28, 30–32 human participants, oversight system proposal for, as dynamic/responsive structure, 134–135 human participants, oversight system proposal for, scope and structure, xii, 26–32 human participants, oversight system proposed for, functions and responsibilities of, xi–xii, 23–27 human participants, percent exempt from, listing by agency, 11 human participants, procedural v. ethical emphasis, 13 human participants, protection for, ACHRE recommendations, 158 human participants, recent government interest in, 156 human participants, recent initiatives for, 15–16 human participants, report implications for IRBs/investigators/ institutions, 136–137 human participants, report implications for participants, 137 human participants, report recommendations for, general themes in, 135 informed consent for human participants, 21 CFR 50, Subpart B, in entirety, 189–195 informed consent requirements for participants, deficiencies in, 98
246
informed consent requirements for participants, scope/structure of, iv, xvii, 97–104 injuries, compensation for, current requirements, v, 123–124 IRB approval, current regulations for, 69–71 IRB functions/operations for human participants, 21 CFR 56, Subpart C, in entirety, 198–201 IRB noncompliance for human participants, actions for, 21 CFR 56, Subpart E, in entirety, 201–203 IRB organization/personnel and human participants, 21 CFR 56, Subpart B, in entirety, 198 IRB records/reports and human participants, 21 CFR 56, Subpart D, in entirety, 201 IRBs and, general provisions for, 21 CFR 56, Subpart A, in entirety, 195–198 ongoing, monitoring responsibilities of local institutions, xix, 117 pregnant women, fetuses, and in vitro fertilization and, 45 CFR 46, Subpart B, in entirety, 179–182 prisoners in biomedical/behavioral research and, 45 CFR 46, Subpart C, in entirety, 182–184 privacy and confidentiality protection for participants, 104–105 protocol changes and unanticipated problems, local institutions responsibilities, xix, 112–113, 117 review, continuing, IRBs responsibilities, xix, 117 review, levels of, for human participants, oversight system proposal for, vii–viii, xiii, 41–42 risk, minimal, classification, definition in, xvi, 80, 83–84 risks v. potential benefits, analysis in IRB review process, ii–iv, 79 vulnerability of participants, current protections for, 85–92 vulnerable participants protection, oversight issues, iv–v, 9, 11
See also Research
Research review. See Ethics review and review committees; Institutional Review Boards (IRBs)
Resources human participants research, federal support for FY 1999, listing by agency, 132 human participants research oversight system and, viii, xx–xxi, 131, 133
Respect for persons research participants informed consent and, ethical guidelines from Belmont Report, 164–165 research participants protection and, from Belmont Report, ii, 162, 164–165
See also Ethics
Review bodies. See Ethics review and review committees; Institutional Review Boards (IRBs)
Risks informed consent requirements for research participants and, xviii, 102, 103 levels of, review for, oversight system proposal for, vii–viii, xiii,
41–42 minimal, of research, Federal policy on, NBAC recommendation, xvi–xvii, 85 minimal, research classification, definition in federal regulations, xvi, 80, 83–84
National Bioethics Advisory Commission
minimal, research classification, use as sorting mechanism for IRB review, 84 minimal, research involving no more than for human participants, IRB review of, oversight proposal, 24 minimal, use related to vulnerability of research participants, 92 research, for nonparticipants, 72–73 research, for participants, minimizing, 73 research, for participants, types of, 71–73 research, v. potential benefits, analysis in IRB review process, ii–iv, 79 research, v. potential benefits, analysis of, new technologies effects on, 7 research, v. potential benefits, analysis of, xvi–xvii, 69–85, 93 research, v. potential benefits, assessment for protection of participants, ethical guidelines, 165–167 research, v. potential benefits, balance between, assessment procedure, 78–80 research, v. potential benefits, component-based framework for analysis, 76–77, 78, 81–82 research, v. potential benefits, component-based framework for analysis, examples, 81–82 research, v. potential benefits, historical perspective, 75–76 research, v. potential benefits, relationship, assessment of,
74–75 Rothman, D.J., 9
Rush Presbyterian St. Lukes Medical Center research compliance investigations resulting in restrictions/actions, 56
S
Safeguards. See Protections for human participants
Sanctions administrative action, for IRB noncompliance, 21 CFR 56, Subpart E, in entirety, 201–203 human research participants protection, oversight enforcement weaknesses, 12–13 research compliance investigations resulting in restrictions/actions to MPAs, listing by institution, 54–56
Science and technology
IRB scientists membership and conflict of interest, measures for managing, xv, 62–63, 64 technologies, new, challenges to research risk/potential benefit analysis, 7
See also Research
Scripps Clinic and Research Foundation, Scripps Research Institute research compliance investigations resulting in restrictions/actions, 56
Senate Committee on Governmental Affairs, 8 Senate Labor and Public Welfare Committee, 153 Shannon, James, 152
Single Project Assurances (SPAs), 14, 52, 55, 56
See also Assurances of compliance; Compliance; Office for Human Research Protections (OHRP)
247
Index
Site inspections compliance investigations by OPRR resulting in restrictions/actions to MPAs, listing by institution, 54–56 compliance with research standards by IRBs, FDA, xiv, 52–53 compliance with research standards, site inspections to ensure, xiv, 53 directed (for cause), of FDA, 53 surveillance (routine), of FDA, 53
Site Management Organizations (SMOs), 6
SMOs. See Site Management Organizations (SMOs)
Social position vulnerability of research participants, considerations, 90–91
Social Security Act, 10
Social Security Administration (SSA), 9, 10, 11, 85, 132, 156 human participants research, regulations of, 157
South Africa research-related injuries, compensation for, 123
South Carolina. See Medical University of South Carolina
SPAs. See Single Project Assurances (SPAs)
Sponsors accreditation of competence in research ethics, NBAC recommendations, xiv, 50 education on research ethics and human participants protection, NBAC recommendations, xiii, 47–48 human participants research, proposed oversight system, functions and responsibilities of, 27 human participants research, proposed oversight system, monitoring, xii, 24–25 research conflict of interest for institutions/IRBs/investigators, NBAC recommendations, xv, 64 research, ongoing, monitoring of, local institutions responsibilities, NBAC recommendations, xix, 117 vulnerability, participants targeted in research, listing by sponsoring agency, 86
See also Research
SSA. See Social Security Administration (SSA)
State Children’s Health Insurance Program, 5
State laws behavior effects of state law, observational study of, risks/ potential benefits analysis example, 81
State regulation human participants research, oversight system proposed for, responsibilities of, 27
State University of New York
College of Optometry, research compliance investigations resulting in restrictions/actions, 54
St. Jude Children’s Research Hospital research compliance investigations resulting in restrictions/actions, 56
Subordination deferential vulnerability in research participants, considerations, 89 institutional vulnerability in research participants, considerations,
88 –89
Substance Abuse and Mental Health Services Administration, 5, 11, 132 See also Department of Health and Human Services (DHHS)
Substantive standard of informed consent. See Informed consent
Syphilis, 46, 153, 164
T
Technology. See Science and technology
Terminology research involving human participants, discussion, 32–33
Testimony and speakers human research participants protection, issues for, testimony, public/expert, on, 221–223 See also Public comments
Translation. See Languages Treatment. See Health care Trials. See Clinical trials Tuskegee, Alabama, 153, 164
Tuskegee Syphilis Study, 46, 153, 164
Tuskegee Syphilis Study Ad Hoc Advisory Panel, 125, 153
Final Report, 124
See also Public Health Service (PHS)
U
Uganda
human participants research, terminology for, 33 research-related injuries, compensation for, 123
UNAIDS. See Joint United Nations Programme on HIV/AIDS (UNAIDS)
Undue influence. See Coercion
United Kingdom cooperative research, IRB review models in, 121 human participants research, terminology for, 33 research-related injuries, compensation for, 123 See also Great Britain
United Nations (UN). See Joint United Nations Programme on HIV/AIDS (UNAIDS)
United Nations Programme on HIV/AIDS. See Joint United Nations Programme on HIV/AIDS (UNAIDS)
Universities and colleges research, and academic medical centers, stresses on, 4, 6 research ethics and human participants protection, programs in, measures needed for, xiii, 48 See also specific universities and colleges
248
University of Alabama at Birmingham research compliance investigations resulting in restrictions/actions, 56
University of California, Irvine research compliance investigations resulting in restrictions/actions, 55
University of California, Los Angeles research compliance investigations resulting in restrictions/actions, 54, 55
University of California, San Diego research compliance investigations resulting in restrictions/actions, 54
University of California, San Francisco
research compliance investigations resulting in restrictions/actions, 54
University of Florida research compliance investigations resulting in restrictions/actions, 54
University of Illinois at Chicago research compliance investigations resulting in restrictions/actions, 56
University of Maryland, Baltimore research compliance investigations resulting in restrictions/actions, 55
University of Minnesota
Department of Surgery, research compliance investigations resulting in restrictions/actions, 54
University of Pennsylvania, 3
University of Rochester, 3 research compliance investigations resulting in restrictions/actions, 55
University of South Florida, 3–4
University of Virginia research compliance investigations resulting in restrictions/actions, 55
USAID. See Agency for International Development (USAID)
U.S. Congress, 7, 27, 31, 42, 52, 125, 134, 155, 156 human participants research, interest in, 156 human participants research, oversight system proposed for, responsibilities of, 27
U.S. Department of Agriculture (USDA), 205 Food Safety and Inspection Service, 170, 197 human participants research, regulations of, 157
U.S. district courts, 11
U.S. government. See Federal government; Federal regulation; Public policy; U.S. Congress
U.S. Public Health Service Act. See Public Health Service Act
National Bioethics Advisory Commission
V
VA. See Department of Veterans Affairs (VA)
Vaccines and vaccination safety/immunogenicity, clinical trial on, risks/potential benefits analysis example, 82 See also Drugs and drug testing
Vanderpool, Harold, Y., 22, 97
Verification compliance with research standards by investigators, monitoring for, xiv, 57–58
Veterans Affairs, Greater Los Angeles Health Care System research compliance investigations resulting in restrictions/actions, 56 See also Veterans Affairs Medical Center, West Los Angeles
Veterans Affairs Medical Center, West Los Angeles research compliance investigations resulting in restrictions/actions, 55
See also Veterans Affairs, Greater Los Angeles Health Care System
Virginia. See University of Virginia; Virginia Commonwealth University
Virginia Commonwealth University research compliance investigations resulting in restrictions/actions, 56
Voluntary consent. See Coercion; Informed consent
Vulnerability cognitive/communicative, in research participants, considerations, 88 deferential, in research participants, considerations, 89 economic, in research participants, considerations, 90 institutional, in research participants, considerations, 88–89 medical, in research participants, considerations, 89–90 research participants protection and, current regulatory protections for, 85–92 research participants protection and, definition of, NBAC recommendation, xvii, 92 research participants protection and, evaluation of, xvii, 85–93 research participants protection and, oversight issues for, iv–v, 9, 11 research participants protection and, use of minimal risk and, 92 research participants targeted for, listing by sponsoring agency/vulnerability, 86 research participants, aligning protections with, considerations,
91–92 social, in research participants, considerations, 90–91
249
Index
W
Waivers informed consent requirements and, discussion, xvii–xviii, 101–103 informed consent requirements and, NBAC previous recommendations, 102 informed consent requirements and, NBAC recommendation, xviii, 103
Wayne State University research compliance investigations resulting in restrictions/actions, 55
Weijer, Charles, 76
Western Carolina Center research compliance investigations resulting in restrictions/actions, 55
West Virginia University research compliance investigations resulting in restrictions/actions, 54
White House, xi, 1, 154
See also Office of Science and Technology Policy (OSTP) Willowbrook State School for the Retarded, 153 Wilson, Charles, 151 Women. See Pregnancy World Medical Assembly Declaration, 170 World War II, 161
250
